

SUMMARY
Mitochondrial movements are tightly controlled to maintain energy homeostasis and prevent oxidative stress. Miro is an outer mitochondrial membrane protein that anchors mitochondria to microtubule motors, and is removed to stop mitochondrial motility as an early step in clearance of dysfunctional mitochondria. Here, using human iPSC-derived neurons and other complementary models, we build on a previous connection of Parkinson’s disease (PD)-linked PINK1 and Parkin to Miro, by showing that a third PD-related protein, LRRK2, promotes Miro removal via forming a complex with Miro. Pathogenic LRRK2G2019S disrupts this function, delaying the arrest of damaged mitochondria and consequently slowing the initiation of mitophagy. Remarkably, partial reduction of Miro levels in LRRK2G2019S human neuron and Drosophila PD models rescues neurodegeneration. Miro degradation and mitochondrial motility are also impaired in sporadic PD patients. We reveal that prolonged retention of Miro, and the downstream consequences that ensue, may constitute a central component of PD pathogenesis.
eTOC SUMMARY
Using iPSC-derived neurons and other models, Hsieh et al uncover a defect in clearance of damaged mitochondria in Parkinson’s Disease. They show that in mutant cells the mitochondrial outer membrane protein Miro is stabilized and remains on damaged mitochondria for longer than normal, prolonging active transport and inhibiting mitochondrial degradation.
INTRODUCTION
Parkinson’s disease (PD) is the second most common neurodegenerative disease, and is characterized by selective loss of dopaminergic neurons in the substantia nigra. The most common genetic mutation in PD is the G2019S mutation in the Ser/Thr kinase domain of LRRK2 (leucine-rich repeat kinase 2), which represents about 1–2% of total cases (Berg et al., 2005; Bonifati, 2002), and 5–6% of total familial cases (Bonifati, 2002). Intriguingly, LRRK2 is also a risk locus for sporadic PD (Cookson, 2010), and the clinical manifestations of patients bearing LRRK2G2019S are indistinguishable from those of idiopathic PD (Gilks et al., 2005). Thus, understanding the pathogenic pathway of mutant LRRK2 will yield insights into the disease mechanisms that are broadly applicable to both familial and sporadic forms of PD.
Mitochondria are highly mobile organelles and their movements are tightly controlled (Glater et al., 2006; Wang and Schwarz, 2009b; Wang et al., 2011). Mitochondrial motility ceases prior to the initiation of mitophagy (Ashrafi et al., 2014; Liu et al., 2012; Wang et al., 2011), a crucial cellular mechanism by which depolarized mitochondria are degraded through autophagosomes and lysosomes (Ashrafi et al., 2014; Wang et al., 2011; Whitworth and Pallanck, 2009; Youle and Narendra, 2011). The arrest of motility may sequester damaged mitochondria, preventing them from moving and from reintroducing damage to other healthy mitochondria. Miro is an outer mitochondrial membrane (OMM) protein that anchors the microtubule motors kinesin and dynein to mitochondria (Glater et al., 2006; Koutsopoulos et al., 2010; Wang and Schwarz, 2009b). This depolarization-triggered mitochondrial arrest is achieved by removal of Miro from the damaged mitochondrial surface (Ashrafi et al., 2014; Liu et al., 2012; Wang et al., 2011). Miro is subsequently degraded by proteasomes (Figure 1A) (Wang et al., 2011). Evidence has shown that two PD-linked proteins, PINK1 (PTEN-induced putative kinase 1) and Parkin, act in concert to target Miro for degradation (Ashrafi et al., 2014; Liu et al., 2012; Wang et al., 2011). Mutations in PINK1 or Parkin cause the rare forms of recessive early-onset PD (Kitada et al., 1998; Valente et al., 2004). However, whether Miro is connected to other more common forms of PD remains unkown. In this study, we discover that a third PD-linked protein, LRRK2, promotes Miro removal from damaged mitochondria, by forming a complex with Miro. Pathogenic LRRK2G2019S disrupts this complex, and thus slows Miro removal and mitochondrial arrest and delays the subsequent mitophagy. Miro is also misregulated in sporadic PD patients, and targeting Miro may have therapeutic benefits. Because LRRK2 mutations are inherited in a dominant manner and are the most common genetic cause for PD, and sporadic cases account for the majority of PD cases, our results reveal that Miro may be a common denominator of multiple types of PD.
Figure 1. Miro Is Resistant to Degradation in PD Patients.

(A) Schematic representation of Miro removal from the OMM of damaged mitochondria causing mitochondrial arrest prior to proteasomal degradation of Miro. KHC, kinesin heavy chain. Milton, a mitochondrial adaptor linking Miro to KHC. (B) Fibroblasts from PD patients and healthy controls were incubated with 40 μM CCCP prior to being lysed. Immunoblots of lysates were probed with antibodies as indicated. (C) The intensity of each band is normalized to the loading control Actin, and expressed as a fraction of the mean of “Control, 0 hr”. “Control” was included in every independent experiment. n=4–27 independent experiments. Fluorescent western blotting was performed to ensure the linear correlation between protein levels and band intensities. The band intensities of Actin are not significantly different among all genotypes and conditions (P=0.1470). * P<0.05, ** P<0.01, *** P<0.001, and error bars represent mean±S.E.M. here and for all figures unless otherwise stated. See also Figures S1–S2.
RESULTS
Miro Is Resistant to Degradation in Both Familial and Sporadic PD Patients
Although Miro is targeted for proteasomal degradation by two PD-associated proteins, PINK1 and Parkin, it is unknown whether Miro protein levels are altered in PD patients. We thus immunoblotted Miro1 in whole cell lysates of skin fibroblasts from PD patients. We used 20 independent lines (Figure S1): 4 healthy control subjects, 5 apparently sporadic patients with no known mutations, 6 familial patients with G2019S, R1441G, R1441C or Y1699C in LRRK2, respectively, 2 familial patients with I368N in PINK1, and 3 familial patients with small deletions, R42P, or R275W in Parkin, respectively. We found that the basal levels of Miro1 protein were normal in PD fibroblasts, although the mitochondrial membrane potential (ΔΨ) as detected by tetramethylrhodamine (TMRM) staining was slightly lower (Figure S1 and S2A–B). We applied a well-established mitochondrial depolarizer for non-neuronal cells, carbonyl cyanide m-chlorophenyl hydrazone (CCCP), to fibroblasts. CCCP treatment for just 15 min significantly depolarized the ΔΨ (Figure S2A). CCCP-triggered proteasomal degradation of Miro precedes mitophagy, which occurs through autophagosomes and lysosomes (Wang et al., 2011). Therefore, we lysed cells at 6 hrs after CCCP treatment to observe Miro1 degradation, and at 14 hrs to observe mitophagy, which is characterized by clearance of multiple mitochondrial proteins including the matrix protein ATP5β (ATP Synthase subunit β) and the OMM protein VDAC (voltage-dependent anion channel). We found that Miro1 was degraded within 6 hrs after CCCP treatment, and ATP5β and VDAC were degraded within 14 hrs, in all 4 lines of wildtype control fibroblasts (Figure 1). Remarkably, these sequential actions of Miro1 degradation and the subsequent mitochondrial clearance induced by CCCP were significantly impaired in fibroblasts from all 16 familial and sporadic PD patients. Miro1 and multiple mitochondrial proteins showed no pronounced degradation for up to 14 hrs after CCCP treatment (Figure 1). Application of the solvent of CCCP – dimethyl sulfoxide (DMSO) – alone, did not affect mitochondrial protein levels for up to 14 hrs in control or patient lines (Figure S2C). Therefore, we have unexpectedly uncovered a cellular phenotype (the striking impairment in Miro degradation and the subsequent damaged mitochondrial clearance) in skin fibroblasts associated with a wide spectrum of PD backgrounds, linking Miro to LRRK2 and sporadic PD.
The Arrest of Damaged Mitochondria Is Delayed in LRRK2G2019S IPSC-Derived Axons
Because Miro anchors mitochondria to microtubule motors, retention of Miro on damaged mitochondria is predicted to prolong active mitochondrial transport. To determine whether mitochondrial motility is impaired in PD, we live imaged mitochondria in axons of induced pluripotent stem cell (iPSC)-derived neurons from PD patients with LRRK2G2019S, the most common mutation in PD. Neuronal axons represent the ideal model for studying bidirectional microtubule-based mitochondrial movement owing to their uniform microtubule polarity (Wang and Schwarz, 2009a, b; Wang et al., 2011). We used iPSCs reprogrammed from 3 PD patients harboring the LRRK2G2019S mutation – LRRK2G2019S-I: homozygous (Nguyen et al., 2011); LRRK2G2019S-II: heterozygous (Sanders et al., 2014); LRRK2G2019S-III: homozygous (Cooper et al., 2012). We also included 3 control iPSC lines – Wildtype-I: healthy control (Nguyen et al., 2011); Wildtype-II: the G2019S mutation in LRRK2G2019S-II was genetically corrected as an isogenic control for LRRK2G2019S-II (Sanders et al., 2014); Wildtype-III: healthy control (ND41866, NINDS Human Cell and Data Repository). We differentiated iPSCs to dopaminergic neurons that express tyrosine hydroxylase (TH), the rate-limiting enzyme in dopamine biosynthesis, as previously described (Nguyen et al., 2011; Sanders et al., 2014). The iPSCs and derived neurons have been fully validated and described in detail (Cooper et al., 2012; Nguyen et al., 2011; Sanders et al., 2014). To label mitochondria, we transiently transfected iPSC-derived neurons with mito-dsRed, and imaged axons at day 22–23 when they were sufficiently mature to express TH (Figure S2D–E). The ΔΨ detected by TMRM staining was slightly lower in LRRK2G2019S neurons, but this lowered ΔΨ was insufficient to trigger Miro degradation (Figure S3A–B). We chose the distal segment of the axon for analysis and the parameters of mitochondrial transport under basal conditions did not significantly vary between wildtype and PD LRRK2G2019S neurons (Figure 2, Table S1).
Figure 2. LRRK2G2019S Delays Mitochondrial Arrest and Clearance in IPSC-Derived Axons.

(A) Mitochondrial movement in representative axons transfected with mito-dsRed before and after treatment with 100 μM Antimycin A. The first frame of each live-imaging series is shown above a kymograph generated from the movie. The x-axis is mitochondrial position and the y-axis corresponds to time (moving from top to bottom). Vertical white lines represent stationary mitochondria and diagonal lines are moving mitochondria. Scale bars: 10 μm. (B) From kymographs as in (A), the percent of time each mitochondrion is in motion is determined and averaged. Comparisons with “Wildtype-I, before treatment”. n=30–110 mitochondria from 8 axons from 8 separate transfections per genotype. (C) The mitochondrial intensity normalized to that of the same axonal region at “0 min” is expressed as a percent of the mean of “Wildtype-I, 0 min”. Comparisons with “Wildtype-I, 0 min”. n=8 axons per genotype. (D) The Mitochondrial (Mito) and cytosolic (Cyto) fractions were purified from iPSC-derived neurons with 100 μM Antimycin A treatment and blotted with antibodies indicated. Loading ratio is 2:1:1 for “Mito:Cyto:Total”. (E) Quantification of the band intensities as in (D), of ATP5β, MCAD, VDAC, Miro1, or Mitofusion2 in the mitochondrial fraction. Their band intensities are normalized to those of Actin in “Total”, and then expressed as a fraction of the mean of “Wildtype-I, 0 min”. n=3 independent experiments. The band intensities of Actin are not significantly different among all genotypes and conditions (P=0.9). See also Figures S2–S4, Table S1.
We next applied the Complex III inhibitor Antimycin A, which has been shown to successfully trigger mitophagy in rodent neurons (Ashrafi et al., 2014; Wang et al., 2011), to iPSC-derived neurons. We confirmed that application of Antimycin A for 10 min significantly depolarized the ΔΨ in human iPSC-derived neurons (Figure S3A). In wildtype neurons from 2 healthy control subjects and 1 isogenic control line, we observed a sequence of mitochondrial events after Antimycin A treatment. Within 15 min, mitochondrial length was significantly shortened; within 25 min, mitochondrial motility in both anterograde and retrograde directions was significantly reduced; finally within 70 min, mitochondrial clearance was induced (Figure 2A–C, Figure S3C, Table S1). The mitochondrial clearance was not due to laser damage, because mito-dsRed was not degraded even after 70 min of laser scanning when only the solvent of Antimycin A, ethanol, was applied (Figure S4A–B). Nor was it caused by non-mitochondrial stress, because mito-dsRed was not degraded after 70 min treatment of dithiothreitol (DTT), an endoplasmic reticulum (ER) stressor (Figure S4A–B). Antimycin A treatment resulted in a specific clearance of mitochondria because lysosomes, labeled by Lamp1-RFP, were unaffected (Figure S4C–D). The clearance of the entire mitochondria is consistent with macroautophagic degradation in lysosomes during mitophagy (Ashrafi and Schwarz, 2013; Klionsky et al., 2012). In contrast, in LRRK2G2019S neurons damage-induced mitochondrial arrest and clearance were impaired. Damaged mitochondria did not stop moving until 70 min after treatment as compared to 25 min in wildtype, and mitochondrial intensity was not significantly affected up to 70 min after treatment (Figure 2A–C, Table S1). In summary, LRRK2G2019S delays, although does not completely block, mitochondrial arrest in response to mitochondrial depolarization.
Miro Removal from Damaged Mitochondria Is Delayed in LRRK2G2019S IPSC-Derived Neurons
The delayed mitochondrial arrest in LRRK2G2019S neurons (Figure 2A–B) suggests that Miro removal from the OMM of damaged mitochondria is also delayed. To test this hypothesis, we separated the mitochondrial fraction, which contains proteins bound to the OMM, from the cytosolic fraction of iPSC-derived neurons 21 days after differentiation. Application of the solvent of Antimycin A, ethanol, did not affect mitochondrial protein levels for up to 70 min in either wildtype or LRRK2G2019S iPSC-derived neurons (Figure S4E). In wildtype neurons at 25 min after Antimycin A treatment, the same time when damaged mitochondria stop moving in the live videoscopic experiments (Figure 2A–B), Miro1 was significantly reduced from the mitochondrial fraction, while other mitochondrial markers including the matrix proteins ATP5β and MCAD (medium chain acyl dehydrogenase), and the OMM protein VDAC remained intact (Figure 2D–E). Additionally, at 70 min after treatment, multiple mitochondrial matrix and OMM proteins were significantly degraded, demonstrating the occurrence of mitophagy (Figure 2D–E). This is consistent with the imaging results showing that mitochondria are cleared at 70 min in wildtype iPSC-derived neurons (Figure 2A–C). However, in LRRK2G2019S neurons, Miro1 was not significantly reduced from the mitochondrial fraction until 70 min after treatment, and multiple mitochondrial proteins were not affected by Antimycin A up to 70 min after treatment (Figure 2D–E). Thus, LRRK2G2019S delays depolarization-triggered Miro removal from mitochondria. These biochemical results coincide with our imaging results showing that mitochondria do not stop moving until 70 min after Antimycin A treatment, and mitochondria are not cleared after 70 min treatment in LRRK2G2019S neurons (Figure 2A–C). In contrast to Miro1, the OMM protein Mitofusin2 was significantly degraded after 25 min in the mitochondrial fractions of both wildtype and LRRK2G2019S neurons (Figure 2D–E), confirming that Mitofusin2 also undergoes depolarization-induced degradation as previously reported (Poole et al., 2010; Tanaka et al., 2010) and that it is not affected by LRRK2G2019S. Taken together, LRRK2G2019S selectively delays Miro1 removal from the OMM and slows the subsequent mitochondrial clearance in iPSC-derived neurons.
Axonal Mitophagy Is Delayed in LRRK2G2019S IPSC-Derived Neurons
To validate that mitochondrial degradation induced by Antimycin A treatment (Figure 2) occurs via mitophagy, we first immunostained optineurin – an early mitophagy marker for recruiting autophagosomes (Lazarou et al., 2015; Wong and Holzbaur, 2014) – in iPSC-derived neurons. In both wildtype and LRRK2G2019S neuronal axons, optineurin appeared undetectable prior to Antimycin A treatment. However after treatment, optineurin became punctate substantially colocalizing with mitochondria labeled by ATP5β, 50 min later in wildtype and 210 min later in LRRK2G2019S neurons (Figure 3A), showing a delay in the initiation of mitophagy in LRRK2G2019S neurons. We next cotransfected neurons with LC3-GFP, an autophagosome marker, and live-recorded individual axons before and after treatment. The accumulation of LC3-GFP on damaged mitochondria is a functional reporter of mitophagy (Klionsky et al., 2012). In both wildtype and LRRK2G2019S neuronal axons, LC3-GFP was undetectable prior to Antimycin A treatment (Figure 3B–C), demonstrating that mitophagy was not activated. However, after treatment, LC3-GFP formed puncta that significantly colocalized with mito-dsRed 60 min later in wildtype axons, indicating autophagosome formation on damaged mitochondria (Figure 3B–C). We verified that formation of autophagosomes on mitochondria was a specific response to the mitochondrial stressor Antimycin A, as it did not occur when the ER stressor DTT was applied for the same period of time (Figure S4F–G). LC3-GFP also formed puncta significantly colocalizing with mito-dsRed in LRRK2G2019S axons, but at a much later time point – 220 min, as compared to 60 min in wildtype (Figure 3B–C). In conclusion, the kinetics of autophagosome formation is disrupted in LRRK2G2019S neurons; autophagosomes can still form on damaged mitochondria, but formation is delayed.
Figure 3. LRRK2G2019S Delays Mitophagy in IPSC-Derived Axons.

(A) Neurons were transfected with GFP and immunostained as indicated. The percentage of optineurin-positive neurons is quantified. Comparisons with “Wildtype-I, 0 min”. n=186–241 neurons from 3 independent transfections. (B) Neurons were transfected with mito-dsRed and LC3-GFP. The intensity profile is generated using the ImageJ “Plot Profile” function. (C) Quantification of the colocalization coefficient (the correlation coefficient of the intensities in each color of each individual pixel) of LC3-GFP and mito-dsRed in each axon before and after Antimycin A treatment as shown in (B) using the ImageJ “colocalization” function. “1” represents complete colocalization and “0” represents no colocalization. The values of 8 axons of each line are averaged, and the mean values of three lines (Wildtype-I, II and III; LRRK2G2019S-I, II and III) are grouped. Comparisons with “Wildtype, 0 min”. (D) Neurons were transfected with mito-mkeima. 488 nm (red) excites neutral mitochondria, and 561 nm (green) excites acidic mitochondria. (E) Quantification of the fluorescent ratio as shown in (D), expressed as a fraction of the mean of “Wildtype-I, 0 min”. Comparisons with “Wildtype-I, 0 min”. n=8–9 axons from 8–9 independent transfections. (F) GFP-expressing neurons were treated with Antimycin A for the indicated time durations, and immunostained. Yellow arrowheads show LC3 puncta in TH-positive axons after treatment. In Wildtype neurons at 60 min after Antimycin A treatment LC3-positive axons accounted for 90.90% of total dopaminergic (n=11) and 89.47% of total non-dopaminergic axons (n=57); in LRRK2G2019S, LC3-positive dopaminergic and non-dopaminergic axons were 0% and 0% at 120 min after treatment, but increased to 100% (n=20) and 88.76% (n=89) at 220 min after treatment, respectively. For all panels, 100 μM Antimycin A was applied. Scale bars: 10 μm. See also Figures S2–S4.
Furthermore, we measured fusion of mitochondria-containing autophagosomes with lysosomes by mito-mkeima, a mitochondrial targeted ratiometric pH-sensitive biosensor (Bingol et al., 2014). High-ratio fluorescence (Excitation: 561nm/488nm) reflects fusion of autophagosomes with acidic lysosomes. In wildtype neuronal axons transfected mito-mkeima displayed a significant increase in the fluorescence ratio 65 min after Antimycin A treatment, whereas in LRRK2G2019S neuronal axons mito-mkeima did not elevate the fluorescence ratio until 230 min after treatment (Figure 3D–E). These results show that fusion of mitochondria-containing autophagosomes with lysosomes is consequently delayed in LRRK2G2019S neurons.
Midbrain dopaminergic neurodegeneration is a hallmark of PD. Although we found that axonal mitophagy was delayed in LRRK2G2019S iPSC-derived neurons by live imaging mitochondria (Figure 2, 3B–E), we were not able to distinguish whether these neurons were dopaminergic, due to lack of available fluorescent dopaminergic markers for live cells. We applied an alterative strategy by immunostaining GFP-labeled neuronal cultures with anti-TH, along with anti-LC3 to recognize autophagosomes. We found that autophagosome formation was delayed in dopaminergic neurons derived from LRRK2G2019S iPSCs. In wildtype neurons, LC3 formed puncta at 60 min after Antimycin A treatment in TH-positive dopaminergic axons indicating autophagosome formation; in LRRK2G2019S neurons, however, LC3 did not form puncta until 220 min after treatment in dopaminergic axons (Figure 3F). This cellular phenotype of delayed mitophagy also occurred in TH-negative (non-dopaminergic) axons (Figure 3F). Our results now link LRRK2 to the regulation of Miro and mitochondrial motility, which consequently influences mitophagy.
Partial Reduction of Miro Arrests Damaged Mitochondria and Restores Mitophagy in LRRK2G2019S IPSC-Derived Neurons
Our human neuron model demonstrates that removal of Miro from damaged mitochondria is slowed by LRRK2G2019S, delaying mitochondrial arrest (Figure 2). Because Miro anchors mitochondria to motors and microtubules, it is likely that a minimum amount of Miro is required on the OMM in order to enable movement. In this case, while it takes wildtype neurons 25 min to reach that amount, it will take LRRK2G2019S neurons 70 min (Figure 2, 4A). Based on this model (Figure 4A), we reasoned that one way to correct this slower speed is to start with less Miro, by mildly lowering the basal level of Miro in LRRK2G2019S neurons. To test this possibility, we performed Miro1 RNAi to knock down Miro1 expression and examined whether this could rescue the mitochondrial phenotypes in LRRK2G2019S neurons. We used non-targeting RNAi as a negative control. We transfected neurons with mito-dsRed, EGFP, and RNA duplexes at day 20 after neuronal induction, and live imaged mito-dsRed from EGFP-positive axons 3 days later. Neurons positive for both mito-dsRed and EGFP have also likely taken up RNA duplexes (Wang and Schwarz, 2009b; Wang et al., 2011). Endogenous Miro1 protein levels did not appear to significantly differ in neurons positive for both mito-dsRed and EGFP between wildtype and LRRK2G2019S with control RNAi, and were reduced to 70.88±6.26% and 71.61±8.00% by Miro1 RNAi in wildtype and LRRK2G2019S, respectively (Figure S5A–B). We first tested our hypothetical model presented in Figure 4A directly. While there was no significant difference in Miro1 levels at baseline between wildtype (100%) and LRRK2G2019S neurons with control RNAi, at 25 min after Antimycin A treatment, Miro1 was reduced to 23.61±4.19% in wildtype and only to 72.15±9.44% in LRRK2G2019S neurons; at 70 min after treatment, Miro1 was reduced to 29.14±3.26% in LRRK2G2019S neurons, revealing a slower rate in degrading Miro by LRRK2G2019S. With Miro1 RNAi in LRRK2G2019S neurons, Miro1 was reduced to 62.46±9.89% before treatment, and to 26.40±4.87% at 25 min after treatment, showing a correction of the slower degradation speed (Figure 4A). Miro1 RNAi in LRRK2G2019S iPSC-derived neurons did not significantly affect mitochondrial transport before Antimycin A treatment (Figure 4B, S5C, Table S2), confirming that the partially reduced Miro protein levels by RNAi at the steady state are above the minimum amount required for movement. Intriguingly, after Antimycin A treatment, Miro1 RNAi in LRRK2G2019S iPSC-derived neurons halted mitochondria and restored mitophagy: the percentage of mitochondria in motion was significantly reduced in both anterograde and retrograde directions 25 min after Antimycin A treatment (Figure 4B, S5C, Table S2), and mitochondria were significantly degraded 70 min after treatment (Figure 4B, C). Control RNAi in LRRK2G2019S iPSC-derived neurons, in contrast, did not influence their phenotypes (Figure 2, 4B–C, S5C, Table S2). We validated that Miro1 RNAi specifically targeted Miro1 mRNA, because the restorative effect was abolished when RNAi-resistant Drosophila Miro (DMiro) with no homology to Miro1 siRNA was co-expressed (Figure S6A–B). To summarize, we have discovered not only a novel cellular phenotype associated with LRRK2 mutations in PD (failure to promptly arrest mitochondrial movement upon depolarization and to clear damaged mitochondria), but also a way to rescue this defect (by lowering the basal level of Miro1 protein).
Figure 4. Miro RNAi Rescues Phenotypes of LRRK2G2019S IPSC-Derived Neurons and Flies.

(A) Schematic representation of Miro removal rates. The y-axis is the percent of total Miro protein levels on the OMM, and the x-axis is the time following depolarization (yellow flash sign). There is a hypothetical minimum amount of Miro on the OMM required for successfully anchoring mitochondria to motors and MT (microtubules) to enable movement. Note that the number of Miro protein on one mitochondrion drawn represents the relative protein amount rather than the actual number of the protein. In the experimental model, the Miro1 protein level is quantified in each condition using immunostaining as in Figure S5 expressed as a percentage of the mean of “Wildtype-I, control RNAi, 0 min”, or the degradation rate of Miro1 (%/min) is calculated within the first 25 min compared to “Wildtype-I, control RNAi”. n=93–151 neurons from 3 independent transfections. (B) Mitochondrial movement in representative axons transfected with mito-dsRed before and after treatment with 100 μM Antimycin A. The types of RNAi and genotypes are indicated. Quantification is in Figure S5C. (C) The normalized mitochondrial intensity is quantified as in Figure 2, expressed as a percent of the mean of “Wildtype, control RNAi, 0 min”. Comparisons with “Wildtype, control RNAi, 0 min”. n=10 axons from 10 separate transfections (pooled from Wildtype-I and II or from LRRK2G2019S-I and II). (D) Quantification of the number of surviving neurons as shown in Figure S6C. Each data point is from 60 fields from 3 independent transfections, expressed as a fraction of the mean of “Wildtype-I, control RNAi, 0 μM”. The densities of neurons before treatment were not significantly different among 4 genotypes (P=0.1071). (E) The crawling ability of third instar larvae with different genotypes is quantified, expressed as a fraction of the mean of the control “Da-GAL4”. n=17–30. (F) The climbing and jumping abilities of adult flies 20 days after eclosion are quantified, expressed as a fraction of the mean of “Da-GAL4”. n=44–60. (G) The PPM1/2 clusters of dopaminergic (DA) neurons visualized by anti-TH in adult brains 35 days after eclosion are shown. The number of DA neurons is quantified. n=11–20 brains. Comparisons with “Da-GAL4” unless otherwise indicated. Scale bars: (B) 10 μm; (G) 5 μm. See also Figures S5–S6, Table S2.
Partial Reduction of Miro Protects LRRK2G2019S IPSC-Derived Neurons against Mitochondrial Stress
Accumulation of dysfunctional mitochondria may cause neuronal cell death in vulnerable neurons. With partial reduction of Miro we have a way to clear damaged mitochondria. Can this approach rescue neuronal cell death? To answer this question, we performed Miro1 RNAi in wildtype and LRRK2G2019S iPSC-derived neurons and examined if it could protect LRRK2G2019S neurons during oxidative stress. We transfected neuronal cultures with EGFP, and applied Antimycin A with a series of concentrations for 6 hrs to induce oxidative stress. We identified EGFP-positive neurons by morphology (Wang et al., 2011). We found that both wildtype and LRRK2G2019S neurons showed an Antimycin A dose-dependency of the loss of EGFP-positive neurons, but LRRK2G2019S neurons exhibited a significantly higher sensitivity (Figure 4D, S6C), consistent with the previous findings showing that these neurons are more vulnerable to stress (Nguyen et al., 2011). Interestingly, Miro1 RNAi did not affect the neuronal sensitivity to oxidative stress of wildtype, yet significantly rescued the loss of EGFP-positive neurons of LRRK2G2019S caused by 1 or 10 μM Antimycin A (Figure 4D, S6C). Thus, partial reduction of Miro protects PD LRRK2G2019S neurons against mitochondrial stress.
Partial Reduction of Miro Rescues Locomotor Deficits and Dopaminergic Neurodegeneration in Drosophila Expressing LRRK2G2019S
We next tested the neuroprotective effect of lowering Miro protein levels in vivo. Rodent models expressing mutant LRRK2 do not produce consistent locomotion and neurodegenerative phenotypes (Garcia-Miralles et al., 2015; Li et al., 2010; Sloan et al., 2012). However, Drosophila with overexpressed human LRRK2G2019S, exhibits locomotor defects and dopaminergic neurodegeneration (Liu et al., 2008; Martin et al., 2014), making the fruit fly a powerful in vivo model for studying PD mechanisms. We knocked down DMiro by RNAi in flies ubiquitously expressing human LRRK2 or LRRK2G2019S (Figure S6D). Third instar larvae expressing LRRK2G2019S showed significantly compromised crawling ability as compared to control larvae, and larvae bearing wildtype LRRK2; DMiro RNAi completely restored their crawling ability (Figure 4E). Adult flies expressing LRRK2G2019S displayed defects in the climbing and jumping abilities, and dopaminergic neurodegeneration in the clusters of paired posterior medial 1 and 2 (PPM1/2), in an age-dependent manner (Figure 4F–G, S6E–F) (Liu et al., 2008; Martin et al., 2014). These behavioral deficits and dopaminergic neurodegeneration in adults were also fully rescued by DMiro RNAi (Figure 4F–G). Collectively, partial inhibition of DMiro rescues the locomotor defects and dopaminergic neurodegeneration by LRRK2G2019S in vivo.
LRRK2G2019S Disrupts Interaction with Miro on Damaged Mitochondria
To explore the mechanism by which LRRK2G2019S slows removal of Miro from the OMM (Figure 2), we considered the possibility that wildtype LRRK2 directly promotes Miro removal and LRRK2G2019S loses this function. This scenario may require LRRK2 to form a complex with Miro. To determine whether LRRK2 and Miro interact before Miro removal, we co-immunoprecipitated endogenous LRRK2 and Miro1 from iPSC-derived neurons 21 days after differentiation. We detected significant interaction of Miro1 with wildtype LRRK2 but not with LRRK2G2019S after Antimycin A treatment for only 10 min (Figure 5A). We next tested whether mitochondrial depolarization was sufficient to recruit LRRK2 to mitochondria to allow its binding to Miro, since LRRK2 mainly localizes to the cytosol. Indeed, by mitochondrial fractionation we found that LRRK2 in the mitochondrial fraction was significantly upregulated after Antimycin A treatment for 10 min in wildtype but not in LRRK2G2019S iPSC-derived neurons (Figure 5B). Hence, at 10 min after depolarization, prior to Miro removal (25 min, Figure 2), LRRK2 is recruited to mitochondria and forms a complex with Miro in iPSC-derived neurons; on the contrary, LRRK2G2019S loses this function. This evidence indicates that wildtype LRRK2 promotes Miro removal from the OMM by associating in a complex with Miro; by disrupting this complex, pathogenic LRRK2G2019S slows Miro removal from the OMM (Figure 2) and delays the following initiation of mitophagy (Figure 3).
Figure 5. LRRK2G2019S Impairs Interaction with Miro on Damaged Mitochondria.

(A, C, G) IPSC-derived neurons (A), fibroblasts (C) or HEK293T cells transfected with indicated constructs (G), were incubated with 100 μM Antimycin A or 40 μM CCCP prior to immunoprecipitation with anti-Miro1 or anti-HA. Co-immunoprecipitated LRRK2 or KHC represented in (A) is quantified by normalizing its band intensity to that of immunoprecipitated Miro1 from the same experiment, and expressed as a fraction of the mean of “Wildtype-I, 0 min”. n=3 independent experiments. For (C, G), representative results were repeated for at least three times. (B) Mito and Cyto fractions were purified from iPSC-derived neurons with 100 μM Antimycin A treatment and blotted with antibodies indicated. Loading ratio is 2:1:1 for “Mito:Cyto:Total”. The LRRK2 band intensity in Mito is quantified by normalizing it to that of the mitochondrial loading control VDAC, and expressed as a fraction of the mean of “Wildtype-I, 0 min”. n=3 independent experiments. (D) Mito and Cyto fractions were extracted from fibroblasts, loaded as 2:1, and blotted with antibodies indicated. Mitochondrial LRRK2 band intensity is normalized to that of VDAC, and expressed as a fraction of the mean of “Control, 0 hr”. n=3–5 independent experiments. (E) Wildtype isogenic control and LRRK2 knockout (KO) HAP cells were treated with CCCP, lysed, blotted as indicated, and quantified as in Figure 1. n=3 independent experiments. (F) Fibroblasts or iPSC-derived neurons were incubated with 40 μM CCCP or 100 μM Antimycin A prior to immunoprecipitation with anti-Miro1. The immunoprecipitates were run either in a regular SDS-PAGE, or in a phos-tag SDS-PAGE, and then blotted with anti-Miro1. For quantification, the intensity above 75Kd (shifted bands of phosphorylated Miro1) is divided by the total Miro1 intensity, expressed as a fraction of the mean of wildtype control without treatment. n=3 independent experiments. (H) Fibroblasts were treated with CCCP with or without LRRK2-IN-1, lysed, and blotted as indicated. n=3 independent experiments. Quantification is in Figure S7D. See also Figure S7.
Similar to neurons, we detected depolarization-triggered interaction between endogenous Miro1 and LRRK2 in wildtype control but not in LRRK2G2019S fibroblasts (Figure 5C). We also observed LRRK2 recruitment to the mitochondrial fraction after CCCP treatment in wildtype but not in LRRK2G2019S fibroblasts (Figure 5D). LRRK2 recruitment to damaged mitochondria and interaction with Miro1 upon CCCP depolarization occurred earlier (1 hr, Figure 5C–D) than Miro1 removal in wildtype fibroblasts (6 hrs, Figure S7A). LRRK2G2019S fibroblasts failed to remove Miro1 from the OMM of damaged mitochondria, but not Mitofusin2 (Figure S7A), consistent with the results from neurons (Figure 2D–E). Because in LRRK2G2019S fibroblasts and neurons where no LRRK2 is present on damaged mitochondria (Figure 5A–D), Mitofusin2 is removed as efficiently as in wildtype (Figure 2D–E, S7A), this demonstrates that LRRK2 specifically targets Miro1, but not Mitofusin2, for removal.
These results from patients’ neurons and fibroblasts suggest that LRRK2G2019S impairs Miro removal from damaged mitochondria by a loss-of-function. This predicts that cells lacking LRRK2 should also display Miro accumulation following mitochondrial depolarization similar to cells harboring LRRK2G2019S. Indeed, Miro1 and multiple mitochondrial proteins were resistant to degradation up to 14 hrs after CCCP treatment in LRRK2 knockout HAP1 (human haploid cells), derived from the CML (male chronic myelogenous leukemia) cell line KBM-7 (Carette et al., 2011) (Figure 5E), just as in LRRK2G2019S fibroblasts (Figure 1). Therefore, LRRK2G2019S disrupts Miro removal in a loss-of-function manner.
It is possible that the purpose for LRRK2 to interact with Miro on damaged mitochondria is to phosphorylate Miro. If this hypothesis were true, preventing LRRK2 from getting to damaged mitochondria and Miro in LRRK2G2019S fibroblasts and neurons (Figure 5A–D) should abolish phosphorylation of Miro. To test this hypothesis, we examined phosphorylation of Miro1 in fibroblasts and iPSC-derived neurons using phos-tag SDS-PAGE, where phosphorylated proteins are trapped by phos-tag ligands resulting in slower migration. We detected mitochondrial depolarization-induced phsophorylation of Miro1 in both neurons and fibroblasts, but found that the degree of Miro1 phosphorylation upon depolarization was indistinguishable between wildtype and LRRK2G2019S (Figure 5F). This indicates that LRRK2 does not cause substantial phosphorylation of Miro when present on damaged mitochondria. The depolarization-triggered phosphorylation of Miro is likely mediated by PINK1 (Lai et al., 2015; Wang et al., 2011).
Since the G2019S mutation resides in the kinase domain, we examined whether the kinase activity of LRRK2 is required for LRRK2/Miro interaction. We consistently observed CCCP-triggered association of exogenously expressed wildtype LRRK2, but not LRRK2G2019S, with Miro1 in HEK293T cells (Figure 5G, S7B), and thus used HEK293T cells for these experiments. Notably, a kinase-inactive form of LRRK2 (LRRK2KD) was still able to significantly bind to Miro1 after depolarization, just like wildtype LRRK2 (Figure 5G, S7B). However, LRRK2R1441C, with a mutation outside the kinase domain, failed to bind to Miro1 upon CCCP treatment, similar to LRRK2G2019S (Figure S7B–C). Therefore, the LRRK2 kinase activity is dispensable for forming the LRRK2/Miro complex. We further determined whether the kinase activity is required for CCCP-triggered degradation of Miro. We applied an LRRK2 Inhibitor – LRRK2-IN-1 (Deng et al., 2011; Skibinski et al., 2014) – to control and LRRK2G2019S fibroblasts. The LRRK2 kinase activity was efficiently inhibited by LRRK2-IN-1, as demonstrated by the elimination of phosphorylation of LRRK2 at Ser935 (Figure 5H) (Deng et al., 2011; Skibinski et al., 2014). Importantly, inhibition of the kinase activity had no influence on CCCP-triggered degradation of Miro1 and the subsequent mitochondrial clearance in control fibroblasts, or on retention of Miro1 and multiple mitochondrial proteins after CCCP treatment in LRRK2G2019S fibroblasts (Figure 5H, S7D). These results reveal that the kinase activity of LRRK2 is not required for LRRK2/Miro interaction or for Miro removal from damaged mitochondria.
Taken together, our studies show that LRRK2 promotes Miro removal by forming a complex with Miro, independent of LRRK2-mediated phosphorylation; mutant LRRK2 loses this function.
LRRK2 and the PINK1/Parkin Pathway Function in Parallel and Converge on Miro
It remains possible that mutant LRRK2G2019S slows Miro removal from mitochondria (Figure 2) indirectly by disrupting the PINK1/Parkin pathway. We investigated this possibility. The signature of activating the PINK1/Parkin pathway is translocation of cytosolic Parkin to damaged mitochondria following depolarization (Youle and Narendra, 2011). We found that this process was not affected by LRRK2G2019S: Parkin was significantly upregulated in the mitochondrial fractions upon depolarization both in iPSC-derived neurons (Figure 6A) and in fibroblasts (Figure 6B) with LRRK2G2019S. Therefore, LRRK2G2019S does not affect the PINK1/Parkin pathway.
Figure 6. The PINK1/Parkin Pathway and LRRK2 Function in Parallel.

(A) Mito and Cyto fractions were purified from iPSC-derived neurons with 100 μM Antimycin A treatment and blotted with antibodies indicated. Loading ratio is 2:1:1 for “Mito:Cyto:Total”. The intensity of mitochondrial Parkin is normalized to that of VDAC, expressed as a fraction of the mean of “Wildtype-I, 0 min”. Comparisons with “Wildtype-I, 0 min”. n=3 independent experiments. (B, D) Mito and Cyto fractions (2:1 loading ratio) from fibroblasts were probed as indicated. The mitochondrial Parkin or LRRK2 band intensity normalized to that of VDAC, is quantified and expressed as a fraction of the mean of “Control, 0 hr” (in B or Figure 5D). n=3–5 independent experiments. (C) Fibroblasts from PD patients, or (E) HEK293T or Hela cells transfected with indicated constructs, were incubated with 40 μM CCCP prior to immunoprecipitation with anti-Miro1 or anti-Myc. Representative results were repeated for at least three times. (F) Fibroblasts were transfected with mCherry-Parkin, and immunostained with anti-Miro1. One mCherry-Parkin-transfected cell is labeled by a white-dashed circle, adjacent to a few untransfected cells in the same field. Note: at 6 hrs after CCCP treatment, mCherry-Parkin is cytosolic. Parkin recruitment to damaged mitochondria is an earlier step during mitophagy (Narendra et al., 2008), and it occurs at 1 hr after treatment in fibroblasts (B). Scale bar: 50 μm. (G) Quantification of the mean intensity of Miro1 in fibroblasts transfected with mCherry-Parkin or Control DNA (pcDNA3.1), expressed as a percentage of the mean of “Control, Control DNA, 0 hr”. Comparisons with “0 hr” within the same genotype. Overexpression of mCherry-Parkin mildly promoted Miro1 degradation at baseline without CCCP treatment (0 hr, P<0.001), probably because mCherry-Parkin had been expressed for 2 days before the cells were analyzed. n=143–151 cells from 3 independent transfections. See also Figure S7.
To further dissect the relationship between LRRK2 and PINK1/Parkin, we examined LRRK2 function in two PD fibroblast lines with PINK1/Parkin mutations: PINK1I368N and Parkin255A EX3-4Del (Figure S1). Both mutant lines failed to degrade Miro1 and Mitofusin2 – two targets of the PINK1/Parkin pathway for degradation, and both disrupted Parkin recruitment to damaged mitochondria (Figure S7E). However, these lines did not affect CCCP-triggered binding of LRRK2 to Miro1 (Figure 6C), or LRRK2 recruitment to damaged mitochondria (Figure 6D). Furthermore, in Hela cells that lack endogenous Parkin, LRRK2 still interacted with Miro1 following depolarization (Figure 6E). Thus, LRRK2 binding to Miro and recruitment to damaged mitochondria are not affected by PINK1/Parkin mutations. Lastly, we found that overexpression of mCherry-Parkin in LRRK2G2019S fibroblasts did not rescue their phenotype – impaired Miro1 degradation after CCCP treatment (Figure 6F, G). Altogether, these results reveal that the PINK1/Parkin pathway and LRRK2 function in parallel and converge on Miro.
Mitochondrial Motility and Mitophagy Are Impaired in Sporadic PD Patients
Because Miro is resistant to degradation after mitochondrial depolarization in fibroblasts also from sporadic PD patients (Figure 1), we tested the possibility that recruitment of LRRK2 or Parkin to damaged mitochondria was disrupted leading to retention of Miro in these cells. Strikingly, after CCCP treatment for 1 hr relocation of both LRRK2 and Parkin to the mitochondrial fractions was compromised in all 5 independent sporadic cell lines, although the degree of severity varied from line to line (Figure 7A). This indicates that impairments in the PINK1/Parkin and LRRK2 pathways contribute to the inefficiency of removing Miro from damaged mitochondria in sporadic cases.
Figure 7. Mitochondrial Motility and Mitophagy Are Impaired in Sporadic Patients.

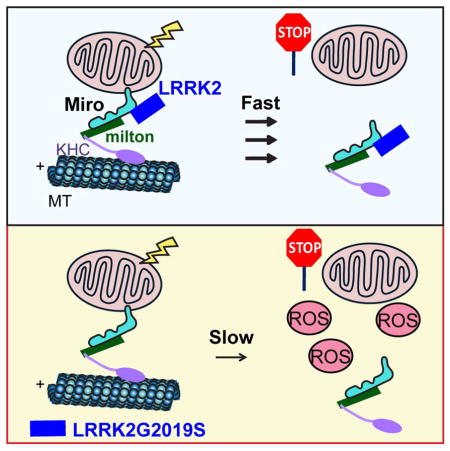
(A) Mito and Cyto fractions (2:1 loading ratio) from fibroblasts were probed with antibodies indicated. The Mitochondrial Parkin or LRRK2 band intensity is normalized to that of VDAC, expressed as a fraction of the mean of “Control, 0 hr”. Comparisons with “Control, 0 hr”. n=3 independent experiments. (B–E) Mitochondrial motility (B–C), intensity (D), and LC3-GFP recruitment to mito-dsRed (E) are analyzed as in Figure 2–3. n=8 axons per line. For (E), the values of all axons are pooled (Wildtype: 24; Sporadic: 16). Comparisons with “Wildtype, 0 min/before treatment”. The same Wildtype values as in Figure 2–3 are used. (F) Schematic representation of the mechanism by which LRRK2G2019S slows Miro removal from damaged mitochondria. ROS: reactive oxygen species. Scale bars: 10 μm.
The accumulation of Miro in sporadic patients (Figure 1) should also delay damaged mitochondrial arrest, the same phenotype we observed in LRRK2-related familiar cases (Figure 2–4). Indeed, in iPSC-derived neuronal axons from 2 independent sporadic patients (Sporadic-I: ND39896, NINDS Human Cell and Data Repository; Sporadic-II: ASE-9028, AppliedStemCell), mitochondria did not stop moving until 70 min after Antimycin A treatment, compared to 25 min in wildtype axons (Figure 2, 7B–C). The following mitophagy was consequently delayed: damaged mitochondria were not cleared at 70 min after treatment when they were efficiently degraded in wildtype neurons, and LC3-GFP was not recruited to mito-dsRed until 190 min after treatment compared to 60 min in wildtype neurons (Figure 2, 3, 7B–E). In conclusion, the halt of damaged mitochondria and subsequent mitophagy are delayed in axons from sporadic patients.
DISCUSSION
In this study, using multiple independent in vivo and cellular models, we have provided novel insights into the molecular pathological mechanisms widely relevant to both sporadic and LRRK2-linked familial PD. LRRK2 forms a complex with Miro on damaged mitochondria and targets it for removal, which leads to the initiation of mitophagy. Pathogenic LRRK2 disrupts this complex, slows Miro removal, and consequently delays the onset of mitophagy (Figure 7F). Delayed mitophagy may accumulate reactive oxygen species and cause cell death in vulnerable neurons.
PD is a debilitating disease, but the cellular mechanisms underlying PD’s disastrous path remain elusive. There is an urgent need for more effective treatments and more reliable diagnoses. Here we have shown a strikingly apparent phenotype (retention of Miro) in skin fibroblasts from PD patients with a wide range of backgrounds (Figure 1). Because these cells are notably easy to obtain and culture, this raises the possibility that these lines and this novel phenotypic readout can be used to screen for therapeutic interventions, pharmacodynamics biomarkers, and diagnostic innovations.
Our studies suggest that basal levels of Miro protein are normal but Miro degradation upon mitochondrial depolarization is compromised in various PD models. Accordingly, axonal mitochondrial motility at baseline is normal in both sporadic and LRRK2-related familial PD patients (Figures 2, 7). A recent study has shown that basal bidirectional movements of mitochondria are increased in LRRK2G2019S or R1441C iPSC-derived neurons (Cooper et al., 2012). Here we have measured mitochondrial motility using several parameters including the percentage of time each mitochondrion is in motion (Table S1 and S2) (Wang and Schwarz, 2009a, b). Cooper and colleagues used a different parameter, the percentage of mobile mitochondria out of total mitochondria over a certain period of time. In addition, we analyzed the distal segment of the axons, whereas Cooper and colleagues examined the proximal part. The different methods used may account for the different conclusions drawn on baseline motility, but both studies agree that basal mitochondrial motility is not reduced suggesting that Miro does not lose its function in human neuron PD models. Miro loss-of-function mutations in humans may cause prenatal death or early developmental defects, since Miro null mutations are lethal to Drosophila and mice (Guo et al., 2005; Nguyen et al., 2014). Mutations in Miro have so far not been found linked to PD. But our results suggest that the Miro gene needs not be altered, and instead the Miro protein is misregulated in PD patients.
Previous studies have shown contrasting results regarding how LRRK2G2019S changes mitochondrial morphology in fibroblasts. Some have shown elongation (Mortiboys et al., 2010), fragmentation (Su et al., 2015; Wang et al., 2012), or no change (Papkovskaia et al., 2012). In our hands, we have observed no significant morphological change of mitochondria in LRRK2G2019S fibroblasts in the steady state. The differing results highlight the complexity of measuring mitochondria morphology, and the confounding factors may include quantifying methods, age of cells, culturing conditions, and variations from line to line.
We have shown that LRRK2 and the PINK1/Parkin pathway function in parallel but converge on Miro (Figure 6). Unexpectedly, both pathways are compromised in sporadic cases, leading to Miro accumulation similar as in familial cases (Figure 1 and 7). These results reveal a shared molecular signature in clinically and genetically distinct patients. Miro serves as a converging nexus to relay many upstream cellular signals to mitochondria (Pekkurnaz et al., 2014; Wang and Schwarz, 2009b; Wang et al., 2011), which are yet to be finalized. Impairments in these signals might contribute to neurodegeneration via a common, Miro-dependent mechanism.
The presence of the defect in Miro and mitophagy in skin fibroblasts, non-dopaminergic, and dopaminergic neurons is consistent with other work indicating that several fundamental cellular pathologies associated with PD involve impairments in highly conserved cellular pathways (Gitler et al., 2009; Ryan et al., 2015). PD-causing genes such as LRRK2, PINK1 or Parkin are all ubiquitously expressed, and their mutations should affect cellular functions in cells not limited to dopaminergic neurons. In human patients, their cellular impairments may exist ubiquitously but lead to cell death only in the most vulnerable neurons, for example, aging dopaminergic neurons in the substantia nigra. These neurons have unusually elevated cellular stress caused by intense neuronal activities, dopamine metabolism, and elaborate axonal networks, and may contain more damaged mitochondria (Pacelli et al., 2015; Surmeier et al., 2010) and be more susceptible to impaired mitophagy.
We have not only identified this cellular defect, but also found a way to correct this defect by partial reduction of Miro levels. Remarkably, this approach is neuronal protective both in cultured iPSC-derived neurons and in vivo. In human LRRK2G2019S neurons it alleviates the neuronal sensitivity to stress, and in LRRK2G2019S flies it rescues dopaminergic neurodegeneration and the locomotor deficits. This indicates that this Miro-dependent pathway may constitute a major part of LRRK2-linked PD pathogenesis. Our results warrant closer examination of Miro as a potential target for PD, especially since its partial inhibition could alleviate defects of both familial and sporadic PD patients.
EXPERIMENTAL PROCEDURES
Neuronal Derivation from IPSCs
IPSCs were derived to midbrain dopaminergic neurons using an adaptation of the dual-smad inhibition method with the use of smad inhibitors dorsormorphin (Sigma) and SB431542 (Tokris), and the addition of GSK3β inhibitor CHIR99021 (Stemgent). Shh was replaced with the smoothened agonist SAG. Please see Supplementary Experimental Procedures for details.
Cell Culture
Human dermal primary fibroblasts isolated from PD patients and healthy controls, HEK293T, and Hela cells were cultured in high glucose Dulbecco’s Modified Eagle’s Medium (Invitrogen) supplemented with 10% fetal bovine serum, and maintained in a 37°C, 5% CO2 incubator with humidified atmosphere. Wildtype and LRRK2 knockout HAP1 cell lines (https://www.horizondiscovery.com/human-lrrk2-knockout-cell-line-hzghc002500c005) were cultured as instructed.
Immunoprecipitation and Western Blotting
CCCP was applied at 40 μM for different time periods, or LRRK2-IN-1 (R&D systems) was applied at 500 nM for 18 hrs prior to CCCP treatment, to fibroblasts or HAP1 cells before the cells were lysed in a RIPA lysis buffer with 0.25 mM PMSF and protease inhibitors. Lysates were cleaned by centrifugation at 17,000 g for 10 min at 4°C, and the supernatants were run in an SDS-PAGE for western blotting. For each preparation of immunoprecipitation, 1.1×106 cells were lysed in 300 μl Triton X-100 lysis buffer. Please see Supplementary Experimental Procedures for more details.
Live Image Acquisition and Quantification, Mitochondrial Isolation, Immunocytochemistry and Confocal Microscopy, Fly Behavior Assay, Fly stocks, Constructs
Statistical Analysis
Throughout the paper, the distribution of data points is expressed as mean ± standard error of the mean (Mean±S.E.M.). The One-Way ANOVA Post-Hoc Tukey test was performed for comparisons unless otherwise stated. Statistical analyses were performed using the Prism 5 software (GraphPad). Manual quantification was performed in a blind fashion for all figures.
Supplementary Material
HIGHLIGHTS.
Damaged mitochondria continue movements in LRRK2 mutant iPSC-derived neurons
Miro is stabilized and remains on damaged mitochondria in LRRK2 mutant neurons
Partial reduction of Miro rescues functional neuronal defects in vitro and in vivo
Similar Miro accumulation and mitochondrial defects also occur in sporadic PD
Acknowledgments
We thank the NINDS Human Cell and Data Repository and Dr. R.R. Pera for cell lines, Drs. T. Schwarz, D. Selkoe, M. Cookson, S. Finkbeiner, R. Youle, and A.M. Craig for constructs, Dr. C.T. Chien for flies, M.M. Course and Dr. A. Gitler for critical reading the manuscript. This work was supported by the William N. and Bernice E. Bumpus Foundation (X.W.), the Alfred P. Sloan Foundation (X.W.), the Klingenstein Foundation (X.W.), the Shurl and Kay Curci Foundation (X.W.), the California Institute of Regenerative Medicine (X.W.), the National Institute of Health (X.W. RO1NS089583), the Michael J. Fox Foundation (X.W. and T.D.P.), and the Blume Foundation (T.D.P.).
Footnotes
AUTHOR CONTRIBUTIONS
C.H. designed and performed experiments, and made figures. A.S., A.C., L.B., E.L., B.S., D.K., and T.P. provided and culured iPSCs and derived neurons. A.G. performed fly experiments. T.P helped iniation of the project. X.W. conceived and supervised the project, designed experiments, and wrote the paper with assistance from all authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ashrafi G, Schlehe JS, LaVoie MJ, Schwarz TL. Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. The Journal of cell biology. 2014;206:655–670. doi: 10.1083/jcb.201401070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashrafi G, Schwarz TL. The pathways of mitophagy for quality control and clearance of mitochondria. Cell death and differentiation. 2013;20:31–42. doi: 10.1038/cdd.2012.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg D, Schweitzer KJ, Leitner P, Zimprich A, Lichtner P, Belcredi P, Brussel T, Schulte C, Maass S, Nagele T, et al. Type and frequency of mutations in the LRRK2 gene in familial and sporadic Parkinson’s disease*. Brain: a journal of neurology. 2005;128:3000–3011. doi: 10.1093/brain/awh666. [DOI] [PubMed] [Google Scholar]
- Bingol B, Tea JS, Phu L, Reichelt M, Bakalarski CE, Song Q, Foreman O, Kirkpatrick DS, Sheng M. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature. 2014;510:370–375. doi: 10.1038/nature13418. [DOI] [PubMed] [Google Scholar]
- Bonifati V. Deciphering Parkinson’s disease--PARK8. The Lancet Neurology. 2002;1:83. doi: 10.1016/s1474-4422(02)00036-4. [DOI] [PubMed] [Google Scholar]
- Carette JE, Raaben M, Wong AC, Herbert AS, Obernosterer G, Mulherkar N, Kuehne AI, Kranzusch PJ, Griffin AM, Ruthel G, et al. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature. 2011;477:340–343. doi: 10.1038/nature10348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cookson MR. The role of leucine-rich repeat kinase 2 (LRRK2) in Parkinson’s disease. Nature reviews Neuroscience. 2010;11:791–797. doi: 10.1038/nrn2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper O, Seo H, Andrabi S, Guardia-Laguarta C, Graziotto J, Sundberg M, McLean JR, Carrillo-Reid L, Xie Z, Osborn T, et al. Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson’s disease. Science translational medicine. 2012;4:141ra190. doi: 10.1126/scitranslmed.3003985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X, Dzamko N, Prescott A, Davies P, Liu Q, Yang Q, Lee JD, Patricelli MP, Nomanbhoy TK, Alessi DR, et al. Characterization of a selective inhibitor of the Parkinson’s disease kinase LRRK2. Nature chemical biology. 2011;7:203–205. doi: 10.1038/nchembio.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Miralles M, Coomaraswamy J, Habig K, Herzig MC, Funk N, Gillardon F, Maisel M, Jucker M, Gasser T, Galter D, et al. No dopamine cell loss or changes in cytoskeleton function in transgenic mice expressing physiological levels of wild type or G2019S mutant LRRK2 and in human fibroblasts. PloS one. 2015;10:e0118947. doi: 10.1371/journal.pone.0118947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilks WP, Abou-Sleiman PM, Gandhi S, Jain S, Singleton A, Lees AJ, Shaw K, Bhatia KP, Bonifati V, Quinn NP, et al. A common LRRK2 mutation in idiopathic Parkinson’s disease. Lancet. 2005;365:415–416. doi: 10.1016/S0140-6736(05)17830-1. [DOI] [PubMed] [Google Scholar]
- Gitler AD, Chesi A, Geddie ML, Strathearn KE, Hamamichi S, Hill KJ, Caldwell KA, Caldwell GA, Cooper AA, Rochet JC, et al. Alpha-synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nature genetics. 2009;41:308–315. doi: 10.1038/ng.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glater EE, Megeath LJ, Stowers RS, Schwarz TL. Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. The Journal of cell biology. 2006;173:545–557. doi: 10.1083/jcb.200601067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Macleod GT, Wellington A, Hu F, Panchumarthi S, Schoenfield M, Marin L, Charlton MP, Atwood HL, Zinsmaier KE. The GTPase dMiro is required for axonal transport of mitochondria to Drosophila synapses. Neuron. 2005;47:379–393. doi: 10.1016/j.neuron.2005.06.027. [DOI] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutsopoulos OS, Laine D, Osellame L, Chudakov DM, Parton RG, Frazier AE, Ryan MT. Human Miltons associate with mitochondria and induce microtubule-dependent remodeling of mitochondrial networks. Biochimica et biophysica acta. 2010;1803:564–574. doi: 10.1016/j.bbamcr.2010.03.006. [DOI] [PubMed] [Google Scholar]
- Lai YC, Kondapalli C, Lehneck R, Procter JB, Dill BD, Woodroof HI, Gourlay R, Peggie M, Macartney TJ, Corti O, et al. Phosphoproteomic screening identifies Rab GTPases as novel downstream targets of PINK1. The EMBO journal. 2015;34:2840–2861. doi: 10.15252/embj.201591593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524:309–314. doi: 10.1038/nature14893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Patel JC, Wang J, Avshalumov MV, Nicholson C, Buxbaum JD, Elder GA, Rice ME, Yue Z. Enhanced striatal dopamine transmission and motor performance with LRRK2 overexpression in mice is eliminated by familial Parkinson’s disease mutation G2019S. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2010;30:1788–1797. doi: 10.1523/JNEUROSCI.5604-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Sawada T, Lee S, Yu W, Silverio G, Alapatt P, Millan I, Shen A, Saxton W, Kanao T, et al. Parkinson’s disease-associated kinase PINK1 regulates Miro protein level and axonal transport of mitochondria. PLoS genetics. 2012;8:e1002537. doi: 10.1371/journal.pgen.1002537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Wang X, Yu Y, Li X, Wang T, Jiang H, Ren Q, Jiao Y, Sawa A, Moran T, et al. A Drosophila model for LRRK2-linked parkinsonism. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:2693–2698. doi: 10.1073/pnas.0708452105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin I, Kim JW, Lee BD, Kang HC, Xu JC, Jia H, Stankowski J, Kim MS, Zhong J, Kumar M, et al. Ribosomal protein s15 phosphorylation mediates LRRK2 neurodegeneration in Parkinson’s disease. Cell. 2014;157:472–485. doi: 10.1016/j.cell.2014.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortiboys H, Johansen KK, Aasly JO, Bandmann O. Mitochondrial impairment in patients with Parkinson disease with the G2019S mutation in LRRK2. Neurology. 2010;75:2017–2020. doi: 10.1212/WNL.0b013e3181ff9685. [DOI] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. The Journal of cell biology. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HN, Byers B, Cord B, Shcheglovitov A, Byrne J, Gujar P, Kee K, Schule B, Dolmetsch RE, Langston W, et al. LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell stem cell. 2011;8:267–280. doi: 10.1016/j.stem.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TT, Oh SS, Weaver D, Lewandowska A, Maxfield D, Schuler MH, Smith NK, Macfarlane J, Saunders G, Palmer CA, et al. Loss of Miro1-directed mitochondrial movement results in a novel murine model for neuron disease. Proc Natl Acad Sci U S A. 2014;111:E3631–3640. doi: 10.1073/pnas.1402449111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacelli C, Giguere N, Bourque MJ, Levesque M, Slack RS, Trudeau LE. Elevated Mitochondrial Bioenergetics and Axonal Arborization Size Are Key Contributors to the Vulnerability of Dopamine Neurons. Current biology: CB. 2015;25:2349–2360. doi: 10.1016/j.cub.2015.07.050. [DOI] [PubMed] [Google Scholar]
- Papkovskaia TD, Chau KY, Inesta-Vaquera F, Papkovsky DB, Healy DG, Nishio K, Staddon J, Duchen MR, Hardy J, Schapira AH, et al. G2019S leucine-rich repeat kinase 2 causes uncoupling protein-mediated mitochondrial depolarization. Human molecular genetics. 2012;21:4201–4213. doi: 10.1093/hmg/dds244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekkurnaz G, Trinidad JC, Wang X, Kong D, Schwarz TL. Glucose regulates mitochondrial motility via Milton modification by O-GlcNAc transferase. Cell. 2014;158:54–68. doi: 10.1016/j.cell.2014.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole AC, Thomas RE, Yu S, Vincow ES, Pallanck L. The mitochondrial fusion-promoting factor mitofusin is a substrate of the PINK1/parkin pathway. PloS one. 2010;5:e10054. doi: 10.1371/journal.pone.0010054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan BJ, Hoek S, Fon EA, Wade-Martins R. Mitochondrial dysfunction and mitophagy in Parkinson’s: from familial to sporadic disease. Trends in biochemical sciences. 2015;40:200–210. doi: 10.1016/j.tibs.2015.02.003. [DOI] [PubMed] [Google Scholar]
- Sanders LH, Laganiere J, Cooper O, Mak SK, Vu BJ, Huang YA, Paschon DE, Vangipuram M, Sundararajan R, Urnov FD, et al. LRRK2 mutations cause mitochondrial DNA damage in iPSC-derived neural cells from Parkinson’s disease patients: reversal by gene correction. Neurobiology of disease. 2014;62:381–386. doi: 10.1016/j.nbd.2013.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skibinski G, Nakamura K, Cookson MR, Finkbeiner S. Mutant LRRK2 toxicity in neurons depends on LRRK2 levels and synuclein but not kinase activity or inclusion bodies. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2014;34:418–433. doi: 10.1523/JNEUROSCI.2712-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan M, Alegre-Abarrategui J, Wade-Martins R. Insights into LRRK2 function and dysfunction from transgenic and knockout rodent models. Biochemical Society transactions. 2012;40:1080–1085. doi: 10.1042/BST20120151. [DOI] [PubMed] [Google Scholar]
- Su YC, Guo X, Qi X. Threonine 56 phosphorylation of Bcl-2 is required for LRRK2 G2019S-induced mitochondrial depolarization and autophagy. Biochimica et biophysica acta. 2015;1852:12–21. doi: 10.1016/j.bbadis.2014.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, Guzman JN, Sanchez-Padilla J, Goldberg JA. What causes the death of dopaminergic neurons in Parkinson’s disease? Progress in brain research. 2010;183:59–77. doi: 10.1016/S0079-6123(10)83004-3. [DOI] [PubMed] [Google Scholar]
- Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, Youle RJ. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. The Journal of cell biology. 2010;191:1367–1380. doi: 10.1083/jcb.201007013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304:1158–1160. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- Wang X, Schwarz TL. Imaging axonal transport of mitochondria. Methods in enzymology. 2009a;457:319–333. doi: 10.1016/S0076-6879(09)05018-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Schwarz TL. The mechanism of Ca2+-dependent regulation of kinesin-mediated mitochondrial motility. Cell. 2009b;136:163–174. doi: 10.1016/j.cell.2008.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, Rice S, Steen J, LaVoie MJ, Schwarz TL. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell. 2011;147:893–906. doi: 10.1016/j.cell.2011.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Yan MH, Fujioka H, Liu J, Wilson-Delfosse A, Chen SG, Perry G, Casadesus G, Zhu X. LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Human molecular genetics. 2012;21:1931–1944. doi: 10.1093/hmg/dds003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitworth AJ, Pallanck LJ. The PINK1/Parkin pathway: a mitochondrial quality control system? Journal of bioenergetics and biomembranes. 2009;41:499–503. doi: 10.1007/s10863-009-9253-3. [DOI] [PubMed] [Google Scholar]
- Wong YC, Holzbaur EL. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:E4439–4448. doi: 10.1073/pnas.1405752111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, Narendra DP. Mechanisms of mitophagy. Nature reviews Molecular cell biology. 2011;12:9–14. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.