

Abstract
Cell-extracellular matrix interactions play major roles in controlling progenitor cell fate and differentiation. The receptor tyrosine kinase, discoidin receptor 2 (DDR2), is an important mediator of interactions between cells and fibrillar collagens. DDR2 signals through both ERK1/2 and p38 MAP kinase, which stimulate osteoblast differentiation and bone formation. Here we show that DDR2 is critical for skeletal development and differentiation of marrow progenitor cells to osteoblasts while suppressing marrow adipogenesis. Smallie mice (Ddr2slie/slie), which contain a non-functional Ddr2 allele, have multiple skeletal defects. A progressive decrease in tibial trabecular BV/TV was observed when wild type, Ddr2wt/slie and Ddr2slie/slie mice were compared. These changes were associated with reduced trabecular number and thickness and increased trabecular spacing in both males and females, but reduced cortical thickness only in Ddr2slie/slie females. Bone changes were attributed to decreased bone formation rather than increased osteoclast activity. Significantly, marrow fat and adipocyte-specific mRNA expression were significantly elevated in Ddr2slie/slie animals. Additional skeletal defects include widened calvarial sutures and reduced vertebral trabecular bone. To examine the role of DDR2 signaling in cell differentiation, bone marrow stromal cells (BMSCs) were grown under osteogenic and adipogenic conditions. Ddr2slie/slie cells exhibited defective osteoblast differentiation and accelerated adipogenesis. Changes in differentiation were related to activity of RUNX2 and PPARγ, transcription factors that are both controlled by MAPK-dependent phosphorylation. Specifically, the defective osteoblast differentiation in calvarial cells from Ddr2slie/slie mice was associated with reduced ERK/MAP kinase and RUNX2-S319 phosphorylation and could be rescued with a constitutively-active phosphomimetic Runx2 mutant. Also, DDR2 was shown to increase RUNX2-S319 phosphorylation and transcriptional activity while also increasing PPARγ-S112 phosphorylation, but reducing its activity. DDR2 is, therefore, important for maintenance of osteoblast activity and suppression of marrow adipogenesis in vivo and these actions are related to changes in MAPK-dependent RUNX2 and PPARγ phosphorylation.
Keywords: genetic animal models, transcription factors, osteoblasts, bone-fat interactions
Introduction
Extracellular matrix (ECM) signals have profound effects on the differentiation of many cell types. In bone, differentiation of mesenchymal progenitor cells to osteoblasts requires interaction of progenitor cells with a type I collagen-containing ECM, a response that is in part mediated by α1β1 and α2β1 integrins(1–4). However, the relatively mild bone phenotypes observed with knockout or inactivation of collagen-binding integrins in vivo suggests involvement of other collagen-binding molecules(5, 6).
Discoidin domain receptors (DDRs) are a second class of collagen receptors that differ from integrins in having intrinsic tyrosine kinase activity and selective affinity for triple-helical, native fibrillar and non-fibrillar collagens. DDR1 is of epithelial origin and has a broad ligand specificity, which includes all known collagens, while DDR2 is expressed by mesenchymal cells and principally binds collagens I, II, III and X(7, 8). Both DDR1 and DDR2 participate in a broad range of cell functions related to development, ECM turnover, growth regulation and cancer (for reviews, see (7, 9)). Two lines of genetic evidence suggest a role for DDR2 in bone. First, human DDR2 mutations cause spondylo-meta-epiphyseal dysplasia (SMED), a skeletal disorder associated with dwarfism, short fingers, bowing of long bones, abnormal calcifications and craniofacial abnormalities(10, 11). In addition, polymorphisms in DDR2 are associated with low BMD and fracture risk in a Han Chinese population(12). Mice harboring deletions in the Ddr2 locus have a SMED-like phenotype characterized by dwarfism and reduction in total bone mineral density(13, 14). However, the detailed bone phenotype of these animals has not been reported. Transgenic manipulation of Ddr2 also affects body mass index (BMI) and adiposity(14, 15), suggesting possible additional effects of this receptor on energy metabolism.
Significantly, after collagen activation, DDR2 propagates down-stream signals using the ERK1/2 and p38 MAP kinase pathways(16, 17), which are also required for normal skeletal development(18–20). The ERK1/2 MAP kinase pathway affects osteoblast function by selectively phosphorylating RUNX2, a key transcription factor controlling differentiation of osteoblast from mesenchymal progenitor cells. ERK1/2 binds and phosphorylates RUNX2 on several serine residues including S301 and S319, that are both required for RUNX2-dependent transcription(21). These phosphorylation events are necessary for the response of bone to several important stimuli including ECM synthesis, mechanical loading, FGF2 and BMP treatment(21–23). Similarly, PPARγ undergoes ERK-dependent phosphorylation at S112, resulting in decreased PPARγ transcriptional activity(24). As we recently showed, activation of MAPK activity in mesenchymal cells both increases osteoblast differentiation and suppresses adipogenesis, actions that require intact MAPK-dependent phosphorylation of RUNX2 and PPARγ(25). These findings let us to hypothesize that DDR2 could also alter osteoblast and adipocyte differentiation via MAPK-dependent phosphorylation. To test this hypothesis, we examined the bone and marrow fat phenotypes of Ddr2-deficient mice and show that actions of DDR2 on osteoblast and adipocyte differentiation can be explained by the selective regulation of RUNX2 and PPARγ.
Materials and Methods
Animals
Smallie mice, which contain a spontaneous deletion in Ddr2, were initially obtained from the Jackson Laboratory on a C57BLS/J (BKS) background (BKS(HRS-Ddr2slie/JngJ). Mice were bred with C57BL/J6 (B6) mice for at least 10 generations. Unlike mice on the BKS background, which are sterile when homozygoic (Ddr2slie/slie), homozygotes on the B6 background bred normally with normal litter sizes (results not shown). Nevertheless, mutant mice of both sexes retained the dwarf phenotype described on the BKS background (Suppl. Fig. 1A–C). Female and male mice were genotyped using previously defined PCR primers and maintained on a normal chow diet until sacrifice at 3 or 5 months for analysis of skeletal and marrow fat phenotypes. Mice were used for skeletal analysis, RNA isolation and as a source of calvarial osteoblasts, marrow stromal cells (MSC) and marrow macrophage cultures. All animal studies were approved by the University of Michigan Committee on the Use and Care of Animals (UCUCA) and conformed to all guidelines and regulations for the protection of animal subjects. Mice were housed in specific pathogen-free AAALAC-certified facilities. After genotyping, littermates were assigned to the experimental groups indicated.
Micro-computed tomography (μCT) analysis of bone
Trabecular and cortical bone parameters were measured by micro-computed tomography using a Scanco Model 100 (Scanco Medical, Bassersdorf, Switzerland). Scan settings were: voxel size 12 μm, 70 kVp, 114 μA, 0.5 mm AL filter, and integration time 500 ms. All scans were analyzed using fixed thresholds (180 for trabecular bone and 280 for cortical bone). Trabecular parameters were collected from 50 sections (8μm/section) under growth plates of proximal tibia and the anterior end of L3 vertebrae. Cortical data were collected from 30 sections above trabecular bone of tibia and fibula junction.
Analysis of bone marrow fat accumulation
After microCT scanning and calculation of osseous parameters and an accurate marrow volume, samples were decalcified with 10% ethylenediaminetetraacetic acid. After fixation with 10% neutral buffered formalin for 24 hours, the samples were incubated with 1% osmium tetroxide at room temperature for 2 hour to stain marrow fat. Osmium staining was measured by microCT analysis(26).
Dynamic histomorphometry and osteoclast analysis
For dynamic bone histomorphometry, 12 week old mice were intraperitoneally injected with 30 mg/kg calcein 9 days prior to sacrifice and then with 10 mg/kg Alizarin Red at 2 days prior to sacrifice. Tibia were harvested and embedded with methyl methacrylate. Six micron sections were cut using Leica SM2500 metallurgical cutting system. For the measurement of in vivo osteoclast activity, tibea were harvested from 12 week old mice. TRAP staining was performed on histological sections using the Sigma Trap staining kit (387-A). The fluorescence and TRAP stained images were photographed using a Nikon 50i microscope. Histomorphometric parameters for double labels and osteoclast activity were assessed using an OsteoMeasurexp system (OsteoMetrics Inc, GA, USA).
Cell cultures and in vitro differentiation
Primary calvaria osteoblasts and bone marrow stromal cells (BMSCs) were isolated from 12 week old mice as previously described (27–29). Osteoblast differentiation was induced by growth in α-MEM/10%FBS containing 50μg/ml ascorbic acid and 10 mM β-glycerophosphate. For adipogenesis, cells were grown for 2 days in α-MEM containing 10% FBS, insulin (5 μg/ml), dexamethasone (1 μM), IBMX (500 μM) and troglitazone (5 μM), followed by growth in medium containing troglitazone (5 μM) for 9 days. To stain mineral, cells were incubated at room temperature in 2% Alizarin Red S (pH 4.2 with 10% ammonium hydroxide) for 0.5 hour. For measurement of fat droplet accumulation, fixed cells were incubated with 60% isopropyl alcohol for 10 min, then stained with 2 mg/ml Oil Red O for 1 hour. For in vitro osteoclast differentiation, bone marrow macrophages (BMM) were isolated from tibiae and femurs of 12 week-old mice using the method of Mizoguchi and coworkers(30) and treated with 10 ηg/ml M-CSF and 50 ηg/ml RANKL for 3 days(31). For measurement of osteoclast induction, cells were stained for TRAP (Sigma staining kit) or grown on BioCoat Osteologic dishes (BD Biosciences) for 3 days before measurement of pit formation. Cell images were taken using an inverted phase contrast microscope (Nikon D300). Osteoblast and adipocyte-specific mRNA expression was assessed by real-time RT/PCR and normalized to GAPDH as previously described(25). The following mRNAs were measured: Bglap2, Ibsp and Runx2 mRNAs for osteoblasts and Pparg, Cebpa, Adipoq and Fabp4 for adipocytes. Western blot analysis was performed using standard procedures. Sources for antibodies were as follows: total RUNX2 antibody, MBL; PPARγ phospho-S112-specific antibody and total PPARγ antibody, Millipore; phospho-ERK1/2 and total ERK1/2 antibody, Cell Signaling. RUNX2 phospho-S319 -specific antibody was previously described(22). Phosphorylation at this site is closely correlated with overall levels of RUNX2 transcriptional activity(21, 22).
Transfections
The 6OSE2-luc reporter, pCMV wild type RUNX2 (RUNX2-WT), S301,319A RUNX2 (RUNX2-SA) and S301,319E RUNX2 (RUNX2-SE) expression vectors were previously described (21) as were ARE-luc, RXR, wild type PPARγ (PPARγ-WT) and S112A PPARγ vectors (PPARγ-SA)(32, 33). Wild type, dominant-negative and constitutively-active DDR2 expression vectors were a generous gift from Dr. Scott Friedman, Mt Sinai School of Medicine(34). For lentivirus production, cDNAs encoding wild type RUNX2, RUNX2-SA or RUNX2-SE were subcloned into pLenti-GFP-puro (Addgene #17448) and packaged by the University of Michigan Vector Core. Calvarial cells were isolated from 12 week-old wild type and Ddr2slie/slie mice, transduced with lentivirus vectors and stable cell lines were developed by selection in 100 μg/mL puromycin for 4 weeks. To measure effects of DDR2 on RUNX2 and PPARγ transcriptional activity, COS7 cells were transfected with the indicated expression plasmids and either 6OSE2-Luc (RUNX2 reporter) or PPRE-luc (PPARγ reporter) and pRL-SV40 (encodes Renilla reformis luciferase to control for transfection efficiency) as previously described(25).
Statistical analysis
All statistical analyses were performed using SPSS 16.0 Software. Unless indicated otherwise, each reported value is the mean ± S.D. of triplicate independent samples for in vitro cultures or at least 6 animals per group for in vivo studies. Statistical significance was assessed using a one-way analysis of variance.
Results
Reduced trabecular bone in the appendicular and axial skeletons of Ddr2slie/slie mice is explained by a selective defect in bone formation
Smallie mice contain a 150 kb deletion in the Ddr2 locus that includes most of the coding sequence (17 out of 18 exons deleted) resulting in a loss of function allele(14). Like SMED patients, these animals are dwarf and have reduced total bone mass, but otherwise, their bone phenotype has not been characterized. To further define the skeletal phenotype of Smallie mice, microCT was used to examine tibiae from wild type, Ddr2wt/slie and Ddr2slie/slie animals. Tibiae of 3 and 5 month-old mice showed major decreases in trabecular bone that was proportional to gene dose in both sexes (females, Suppl. Fig. 2 and Fig. 1; males, Suppl Fig. 3). For example, in 5 month females Ddr2wt/slie mice exhibited a 35 percent decrease in BV/TV relative to wild type controls while this parameter was decreased nearly 70 percent in Ddr2slie/slie animals (Fig. 1B). These changes were accompanied by expected reductions in trabecular number and thickness and increased trabecular spacing (Fig 1C–E). In addition, a 7 percent decrease in cortical bone was observed in females, but not males (compare Fig. 1F with Suppl Fig. 3F). Changes in bone mass were accompanied by a gene dosage-related decline in mineral apposition and bone formation rate (Fig 1G–I), osteoblast marker (Runx2, Bglap2 and Ibsp) mRNA levels (Fig 1J–L) and serum osteocalcin when Ddr2wt/slie and Ddr2slie/slie mice were compared with wild type (Fig 1M). Ddr2slie/slie mice also had large reductions in vertebral trabecular bone. MicroCT analysis of the 3rd lumbar vertebrae revealed 20 and 60 percent declines in BV/TV in Ddr2wt/slie and Ddr2slie/slie mice, respectively (Suppl. Fig. 4).
Figure 1. Decreased trabecular and cortical bone, bone formation and osteoblast gene expression in Ddr2slie/slie mice.
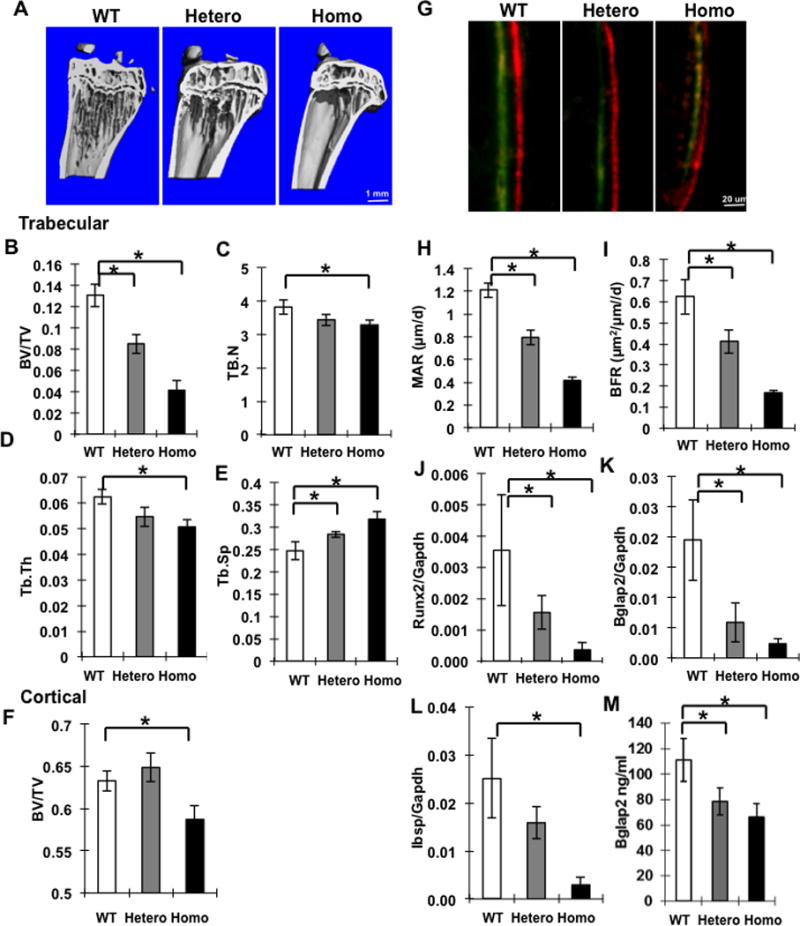
Trabecular and cortical bone parameters were measured by μCT in 5 month old Ddr2wt/wt (WT), Ddr2slie/wt (hetero) or Ddr2slie/slie (Homo) female mice. (A) Representative micro-CT images of tibia showing trabecular and cortical bone. (B–E) Measurements are shown for trabecular bone volume over total volume (BV/TV), trabecular number (Tb.N), trabecular thickness (Tb.Th., mm) and trabecular separation (Tb.Sp, mm). (F) Cortical BV/TV. (G–I) Dynamic bone histomorphometry. For measurement of bone formation parameters, 12 week-old female mice were injected with calcein and alizarin red followed by measurement of mineral apposition rate (MAR) and bone formation rate (BFR). Representive fluorescent images are shown in (G). (J–L) Osteoblast marker mRNAs. RNA was extracted from whole bones of 5 month-old females and the following mRNAs were measured: Runx2, Bglap2 and Ibsp. (M) Serum levels of total osteocalcin (Bglap2). Statistics: * p < 0.01, n = 8/group.
Two approaches were used to determine if the decreased bone in Ddr2slie/slie mice is in part explained by changes in bone resorption; i) TRAP staining was used to measure osteoclast surface and numbers in tibia and ii) the ability of bone marrow macrophage cultures to undergo osteoclast differentiation was measured. No differences were seen in osteoclast surface, osteoclast number or osteoclast number/osteoclast surface. In addition, the serum bone resorption markers, CTX-I and TRAP were not affected by Ddr2 status (Suppl Fig. 5A–F). Furthermore, after growth of bone marrow macrophages in medium containing MCSF and RANKL to induce osteoclast formation, no differences were observed between groups in terms of TRAP-positive cells/dish or mineral resorption measured using BioCoat™ dishes (Suppl. Fig. 6A–D). Lastly, equivalent levels of two osteoclast marker mRNAs, Ctsk and Nfatc1, and culture medium levels of CTX-I and TRAP were detected in all groups (Suppl. Fig. 6, E–H). Thus, Ddr2 deletion in vivo preferentially affects bone formation without noticeably altering osteoclast activity.
The reduced bone in Ddr2slie/slie mice is associated with increased marrow fat
Histological examination of bones from Ddr2wt/slie and Ddr2slie/slie mice often showed regions containing apparent adipocytes (Suppl Fig. 5A). To quantify this marrow fat, tibia from 5 month old female mice were stained with the lipophilic compound, osmium tetroxide, and tibial marrow fat was visualized by microCT(26). As shown in Figure 2, marrow fat increased in proximal and distal regions. Specific quantitation of osmium density in the proximal tibia revealed a more than five-fold increase in marrow adipose tissue when wild type, Ddr2wt/slie and Ddr2slie/slie mice were compared (Fig. 2B–D). Gene expression profiles of whole bone also confirmed a shift from osteoblast (Runx2, Bglap2, Ibsp) to adipocyte (Pparg, Cebpa, Fabp4 and Adipoq) marker mRNAs (compare Fig. 1J–L with Fig. 2E–H). In addition, serum adiponectin levels increased in parallel with Adipoq mRNA while serum leptin decreased (Fig. 2I,J).
Figure 2. Increased marrow adipose tissue in tibiae from 5 month female Ddr2slie/slie mice.
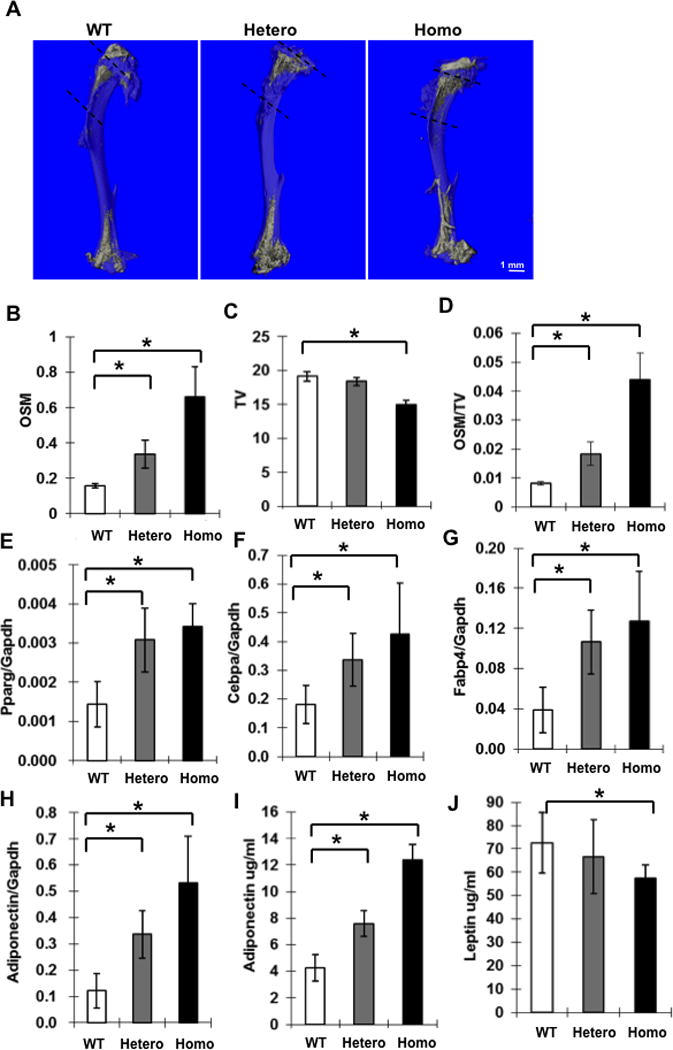
Analysis used the same samples as Fig. 1A–F. (A) Micro-CT images of osmium-stained demineralized tibiae from wild type, heterozygous and homozygous Ddr2slie/slie mice showing landmarks used to calculate MAT in the proximal tibiae (dotted lines). (B–D) Measurement of total osmium-positive volume (OSM, mm3), total bone volume (TV, mm3) and osmium-positive volume/total bone volume(OSM/TV). (E–H) Adipocyte marker mRNA levels from whole bone. Pparg mRNA (E), Cebpa mRNA (F), Fabp4 mRNA (G) and Adipoq mRNA (H). (I, J) Measurement of serum adipocyte marker proteins by ELISA: adiponectin (I) and leptin (J). Statistics: * p < 0.01, n = 8.
Impaired osteoblast differentiation and increased adipogenesis in BMSCs from Ddr2slie/slie mice
To determine the requirement for DDR2 in the differentiation of marrow progenitors to osteoblasts or adipocytes, BMSC from wild type and Ddr2slie/slie mice were grown in osteoblast or adipocyte induction conditions (Fig 3). Cells from Ddr2slie/slie mice formed fewer mineralized nodules and expressed lower levels of osteoblast marker mRNAs (Fig. 3A–D). In contrast, they formed increased numbers of lipid droplet-containing cells and expressed higher levels of adipocyte markers (Fig. 3E–I). Although DDR2 has been reported to affect cell proliferation in other cell types such as fibroblasts, no major differences in proliferation rates were observed when BMSCs from wild type and Ddr2slie/slie mice were compared (Suppl Fig. 1D).
Figure 3. Decreased osteoblast and increased adipocyte differentiation in BMSC from Ddr2slie/slie mice.
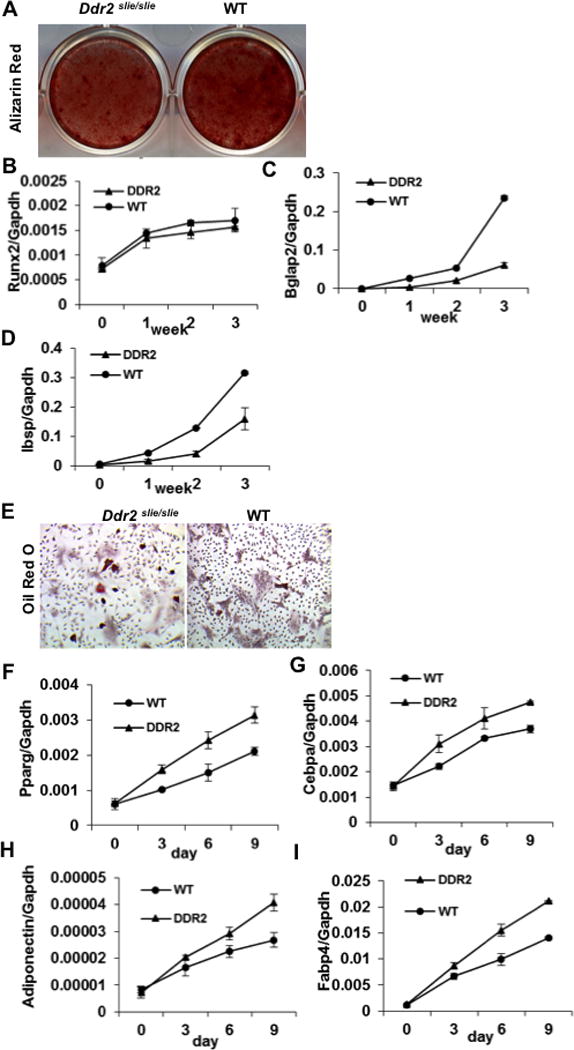
BMSCs were isolated from 12 week-old wild type (WT) and homozygous Ddr2slie/slie female mice (DDR2) and grown in osteogenic (A–D) or adipogenic conditions (E–I). (A) Mineralization. Cells were stained with Alizarin red after 3 weeks in culture. (B–D) Osteoblast differentiation marker mRNAs. (B) Runx2 mRNA, (C) Bglap2 mRNA and (D) Ibsp mRNA. (E) Lipid droplet accumulation. Cells were stained with Oil red O after 9 days in culture. (F–I) Adipocyte differentiation marker mRNAs. (F) Pparg mRNA, (G) Cebpa mRNA, (H) Adipoq mRNA and (I) Fabp4 mRNA.
Defective calvarial mineralization and osteoblasts differentiation in Ddr2slie/slie mice
In addition to the defects in the appendicular and axial skeleton noted above, skulls from newborn Ddr2slie/slie mice had widened sutures and were poorly mineralized such that total cranial bone volume was reduced by approximately 40 percent (Fig. 4AB). In addition, calvarial osteoblasts from these mice, like BMSCs, formed few mineralized nodules and expressed lower levels of osteoblast marker mRNAs than cells from wild type littermates (Fig. 4C–F).
Figure 4. Defective calvarial mineralization and osteoblast differentiation in Ddr2slie/slie mice is related to reduced RUNX2 phosphorylation.
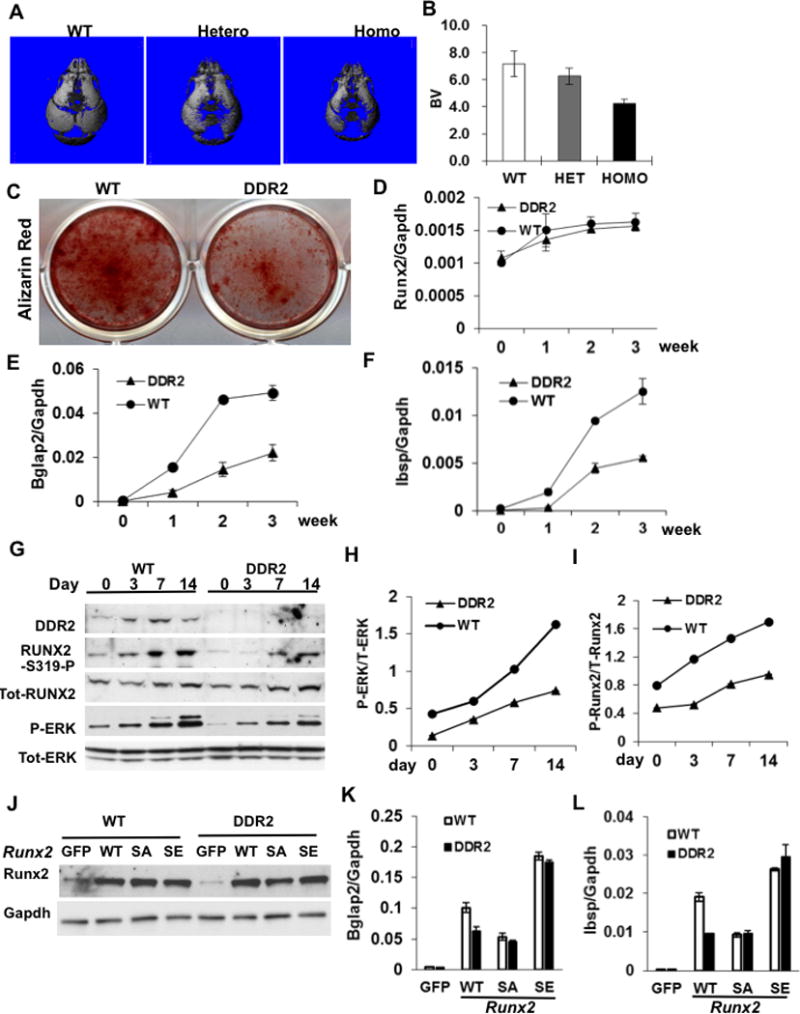
(A,B) Micro-CT analysis of calvaria from newborn mice. (A) Representative micro-CT images, (B) calculation of total bone volume (BV, mm3). (C–I) Osteoblast differentiation. Calvarial osteoblasts were isolated from 12 week-old wild type or homozygous Ddr2slie/slie female mice and grown in osteogenic medium for the times indicated. (C) Alizarin red staining of 2 week calvaria cell cultures. (D–F) Osteoblast differentiation marker mRNAs. (D) Runx2 mRNA, (E) Bglap2 mRNA and (F) Ibsp mRNA. (G–I) Western blot quantitation of DDR2, phosphorylated RUNX2, total RUNX2, phosphorylated ERK1/2 and total ERK1/2. (J–L) Analysis of stable cell lines from wild type and Ddr2slie/slie mice expressing control vector (GFP), wild type RUNX2 (WT), phosphorylation site mutant RUNX2 (RUNX2 S301,319A; SA) or phosphorylation site mimetic RUNX2 (RUNX2 S301,319E; SE). Stable cell lines were established using lentivirus transduction as described in Methods and RUNX2 protein levels were measured by Western blotting (J). Bglap2 (K) and Ibsp (L) mRNAs were measured after growth in osteoblast differentiation conditons. Statistics: * p < 0.01, n = 8/group.
Basis for DDR2 effects on osteoblast and adipocyte differentiation
MAPK activation in mesenchymal cells both increases osteoblast differentiation and inhibits adipogenesis via phosphorylation of RUNX2 and PPARγ(22). Since MAPK is a major DDR2-activated downstream signal(16, 17), we hypothesized that reduced MAPK signaling in DDR2-deficient cells could explain the observed decrease in osteoblast differentiation and increase in adipogenesis. This hypothesis was evaluated in two ways: 1) We examined the role of RUNX2 phosphorylation in the differentiation of calvarial cells from wild type and DDR2-deficient mice. 2) The ability of DDR2 to regulate RUNX2 and PPARγ phosphorylation and transcriptional activity was directly measured.
In agreement with our previous report(22), MAP kinase activity (P-ERK/total ERK) and RUNX2 phosphorylation (P-RUNX2/total RUNX2) gradually increased during calvarial osteoblast differentiation. This increase was accompanied by a parallel increase in DDR2 protein (Fig. 4G–I). Consistent with their reduced differentiation potential, DDR2-deficient cells had much lower levels of MAP kinase activity and RUNX2 phosphorylation. To determine if this could explain the observed suppression of osteoblast differentiation in cells from Ddr2slie/slie mice, we compared the activity of wild type (RUNX2-WT), phosphorylation site mutant (RUNX2 S301,S319A; RUNX2-SA) and phosphorylation site mimetic (RUNX2 S301,S319E; RUNX2-SE) forms of RUNX2 in wild type or DDR2-deficient cells (Fig. 4J–L). Lentiviral transduction was used to generate stable cell lines that were then grown in osteoblast differentiation conditions for 2 weeks before measurement of osteoblast mRNAs. Consistent with previous findings(21), overexpression of wild type RUNX2 in DDR2-sufficient (WT) cells strongly induced Ibsp and Bglap2 mRNAs while levels were reduced by approximately 50 percent in cells expressing the phosphorylation-deficient mutant (RUNX2-SA). As would be expected if RUNX2 phosphorylation was necessary for DDR2 effects on differentiation, mRNA levels were reduced to similar levels in DDR2-deficient (DDR2) cells regardless of whether they were expressing wild type or phosphorylation-deficient RUNX2. Similarly, in cells expressing the phosphorylation mimetic RUNX2 (RUNX2-SE), which does not require phosphorylation for full activity, both Bgalp2 and Ibsp mRNAs were expressed at high levels regardless of DDR2 status.
Effects of DDR2 on RUNX2 and PPARγ transcriptional activity measured using luciferase reporters are shown in Figure 5. In cells transfected with wild type RUNX2, wild type DDR2 and, to a greater extent, a constitutively active DDR2 mutant increased RUNX2 phosphorylation (P-RUNX2/total RUNX2) and transcriptional activity while a dominant-negative DDR2 mutant had no effect (Fig. 5A,B). As expected, DDR2 also increased MAPK activity (P-ERK) without affecting either total RUNX2 or ERK levels. The RUNX2 response required MAPK-dependent phosphorylation since the RUNX2-SA mutant was resistant to DDR2 regulation. Similarly, DDR2 actions on PPARγ transcriptional activity are explained by a mechanism involving MAPK-dependent phosphorylation (Fig. 5C,D). In this case, wild type or constitutively-active DDR2 increased PPARγ S112 phosphorylation with concomitant inhibition of reporter activity while a dominant-negative mutant was inactive. In contrast, PPARγ containing an S112A mutation rendering it resistant to MAPK-dependent inhibition had higher basal activity and was not inhibited by DDR2 overexpression.
Figure 5. Reciprocal control of Runx2 and PPARγ transcriptional activity by DDR2-dependent phosphorylation.
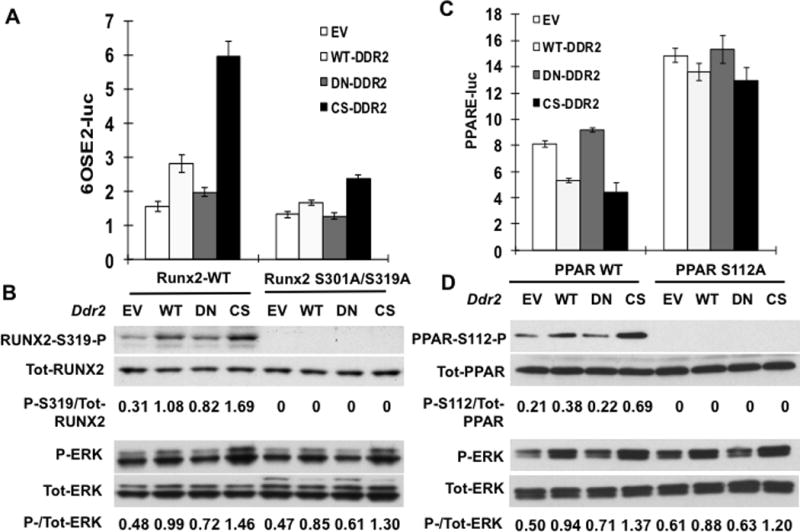
(A,B) Runx2 regulation. COS7 cells were transfected with a RUNX2 reporter plasmid (6OSE2-luc) and expression vectors encoding wild type (Runx2-WT) or phosphorylation site mutant RUNX2 (Runx2 S301A/S319A) and empty vector (EV), wild type (WT) or constitutively-active (CS) DDR2. (A) Normalized luciferase activity. (B) Immunoblot of RUNX2-S319-P, total RUNX2, P-ERK and total ERK. Values under each lane indicate ratios or P-RUNX2/total RUNX2 and P-ERK/total ERK as measured by densitometry. (C,D) PPARɣ regulation. Cells were transfected with PPARγ reporter/RXR combination (PPRE-luc) as described in Methods together with expression vectors encoding wild type (PPAR-WT) or phosphorylation site mutant PPARγ (PPAR-S112AA) and empty vector (EV), wild type (WT) or constitutively-active (CS) DDR2. (C) Normalized luciferase activity. (D) Immunoblot of PPARγ-S112-P, total PPARγ, P-ERK and total ERK.
Discussion
Here we describe the skeletal and marrow fat phenotypes of Ddr2-deficient mice and relate these changes to defects in MAP kinase signaling and phosphorylation of RUNX2 and PPARγ. Although absence of DDR2 inhibits growth and body size, effects previously attributed to reduced proliferation of growth plate chondrocytes(13), our studies clearly show that DDR2 is also required for normal trabecular bone formation in the absence of changes in resorption. Furthermore, loss of DDR2 increases marrow adipogenesis and alters serum levels of the adipocyte-related hormones, adiponectin and leptin.
This study provides the first in vivo evidence that DDR2 increases bone mass largely by increasing osteoblast differentiation and bone formation. Specifically, bone formation in Ddr2slie/slie mice was shown to be dramatically reduced leading to decreased trabeculae in long bones and vertebrae. Furthermore, osteoblast differentiation was reduced in BMSC and calvarial cells from Ddr2-deficient mice due to a reduction in ERK/MAP kinase signaling and RUNX2 phosphorylation. The concept that DDR2 stimulates bone formation through MAP kinase and RUNX2 was originally proposed by Zhang and coworkers on the basis of studies with osteoblast and chondrocyte cell lines(35). In their study, DDR2 was shown to increase MAP kinase activity and phosphorylation of RUNX2 at Ser 301 and Ser 319(35), sites previously identified by us as being essential for MAPK-dependent RUNX2 transcriptional activity(21). More recently, the same group proposed, largely on the basis of cell culture studies, that DDR2 can also suppress osteoclastogenesis and showed that overexpression of DDR2 in bone marrow could reverse ovarectomy-induced bone loss(36). While our studies provide in vivo validation of the former concept, we failed to obtain evidence for defective osteoclast formation in Ddr2slie/slie mice. These animals had normal levels of bone-associated osteoclasts and serum resorption markers. Furthermore, osteoclast differentiation from bone marrow macrophages as measured by induction of osteoclast markers or bone resorbing activity (resorptive pit formation) was normal, indicating that the principal in vivo effects of DDR2 on bone are at the level of osteoblast differentiation and bone formation. Although it is difficult to directly compare our in vivo results with this previous work, a possible reason for this apparent discrepancy might be compensation for loss of DDR2 when it is absent during all stages of osteoclast development in vivo that does not occur when DDR2 is knocked down with shRNA during differentiation of osteoclasts in vitro.
In the initial report describing Ddr2slie/slie mice, homozygous animals were found to be sterile due to gonadal insufficiency in both males and females(14). Although all pituitary and hypothalamic hormones and releasing factors were normal as was circulating IGF-1, secreted steroid hormones were reduced in both sexes. Clearly, reduced steroid hormone levels would be expected to affect bone metabolism as well as reproduction. However, we do not think this explains our results. As noted in Methods, when originally characterized, smallie mice were on a BKS background while all our studies used B6 mice, which bred normally even as Ddr2slie/slie homozygotes (see Methods). Although we have not measured circulating steroid hormone levels, they are clearly sufficient for sexual maturation and function. Furthermore, our failure to observe defects in osteoclastogenesis in females is clearly incompatible with a major reduction in sex steroids. Thus, the reproductive defects associated with the absence of DDR2 are likely mouse line-specific. However, a more detailed understanding of the functions of DDR2 in bone will require the development of a tissue-specific knockout model that is currently under development in our laboratory.
Our observation that marrow adipose tissue is dramatically increased in Ddr2slie/slie mice is of particular interest in that marrow adiposity is associated with many skeletal diseases including osteoporosis and disuse osteopenia(37–39). Previous analysis of Ddr2slie/slie mice detected a modest decrease in total body fat in males and females and significantly elevated blood glucose levels. However, marrow fat was not examined(14). Paradoxically, transgenic overexpression of Ddr2 in all tissues also decreased body mass index (BMI) and epididymal fat pad weight while decreasing serum LDL-cholesterol, albumin and uric acid, but, again, marrow fat was not analyzed(15).
As we recently showed, differentiation of mesenchymal cells into osteoblasts or adipocytes is reciprocally controlled by the relative activity of RUNX2 and PPARγ, transcription factors that are both regulated by MAP kinase-dependent phosphorylation(25). Specifically, phosphorylation of RUNX2 at Ser 301 and Ser 319 increases transcriptional activity and osteoblast differentiation while phosphorylation of PPARγ at Ser 112 inhibits its activity and blocks adipogenesis. This pathway provides a plausible mechanism to explain how DDR2, acting through MAP kinase, increases bone formation and suppresses marrow adipogenesis. As shown in Figure 5, DDR2 stimulates phosphorylation of both RUNX2 and PPARγ leading to increased RUNX2-dependent transcriptional activity and decreased PPARγ-dependent transcription. Also, as would be predicted, MAP kinase activity, RUNX2 phosphorylation and osteoblast differentiation are reduced in calvarial cultures from Ddr2slie/slie mice. The decreased osteoblast differentiation in DDR2-deficient cells was explained by a defect in RUNX2 phosphorylation that was likely a consequence of the reduced MAPK activity in these cells; differences in RUNX2-dependent gene expression in wild-type versus DDR2-deficient cells were not seen in cells containing a phosphorylation-deficient RUNX2 mutant (RUNX2 S301,319A) while a phosphomimetic mutant (RUNX2 S301,319E) restored transcriptional activity to the same levels in wild type or DDR2-deficient cells (Fig. 4J–L).
Notably, MAPK activity, RUNX2 phosphorylation and osteoblast differentiation is only partially inhibited in cells from Ddr2slie/slie mice (Fig. 4G–I). This indicates that DDR2 is not the only mediator of the collagen/MAPK/RUNX2 pathway in bone cells. As mentioned in the Introduction, collagen binding integrins can also stimulate osteoblast differentiation and they likely account for the residual osteoblast differentiation and bone formation in the absence of DDR2.
The observation that DDR2 can affect peripheral and marrow fat as well as the adipokines, adiponectin and leptin, suggest that DDR2 may also participate in the regulation of energy metabolism, possibly through its action on marrow adipose tissue. Recent studies showed that marrow fat is a major source of circulating adiponectin and manipulations that alter marrow adiposity can also affect adiponectin levels(40). Thus, it is possible that the increased serum adiponectin we observed in Ddr2slie/slie mice may be related to the increased marrow adipose tissue in these animals and that changes in adiponectin may explain the reported decrease in BMI in the absence of DDR2(14). However, further studies will be required to establish a direct role for DDR2 in energy metabolism.
Supplementary Material
Acknowledgments
This work was supported by NIDCR Grants DE11723 (RTF) and K12 DE023574 (Junior Faculty Training Award, CG) and NIDDK Grant P30 DK092926 (Michigan Center for Diabetes Translational Research).
Footnotes
Disclosure
All authors state that they have no conflicts of interest
Authors’ roles: GC and RTF designed the research. GC, ZW, GZ, BL, JL and HS conducted all experiments. RTF and GC wrote the manuscript, which was proofread and approved by all authors.
References
- 1.Xiao G, Wang D, Benson MD, Karsenty G, Franceschi RT. Role of the alpha2-integrin in osteoblast-specific gene expression and activation of the Osf2 transcription factor. J Biol Chem. 1998;273(49):32988–94. doi: 10.1074/jbc.273.49.32988. [DOI] [PubMed] [Google Scholar]
- 2.Xiao G, Gopalakrishnan R, Jiang D, Reith E, Benson MD, Franceschi RT. Bone morphogenetic proteins, extracellular matrix, and mitogen-activated protein kinase signaling pathways are required for osteoblast-specific gene expression and differentiation in MC3T3-E1 cells. J Bone Miner Res. 2002;17(1):101–10. doi: 10.1359/jbmr.2002.17.1.101. [DOI] [PubMed] [Google Scholar]
- 3.Takeuchi Y, Suzawa M, Kikuchi T, Nishida E, Fujita T, Matsumoto T. Differentiation and transforming growth factor-beta receptor down-regulation by collagen-alpha2beta1 integrin interaction is mediated by focal adhesion kinase and its downstream signals in murine osteoblastic cells. J Biol Chem. 1997;272(46):29309–16. doi: 10.1074/jbc.272.46.29309. [DOI] [PubMed] [Google Scholar]
- 4.Reyes CD, Garcia AJ. Alpha2beta1 integrin-specific collagen-mimetic surfaces supporting osteoblastic differentiation. J Biomed Mater Res A. 2004;69(4):591–600. doi: 10.1002/jbm.a.30034. [DOI] [PubMed] [Google Scholar]
- 5.Zimmerman D, Jin F, Leboy P, Hardy S, Damsky C. Impaired bone formation in transgenic mice resulting from altered integrin function in osteoblasts. Dev Biol. 2000;220(1):2–15. doi: 10.1006/dbio.2000.9633. [DOI] [PubMed] [Google Scholar]
- 6.Gardner H, Broberg A, Pozzi A, Laato M, Heino J. Absence of integrin alpha1beta1 in the mouse causes loss of feedback regulation of collagen synthesis in normal and wounded dermis. J Cell Sci. 1999;112(Pt 3):263–72. doi: 10.1242/jcs.112.3.263. [DOI] [PubMed] [Google Scholar]
- 7.Fu HL, Valiathan RR, Arkwright R, Sohail A, Mihai C, Kumarasiri M, et al. Discoidin domain receptors: unique receptor tyrosine kinases in collagen-mediated signaling. J Biol Chem. 2013;288(11):7430–7. doi: 10.1074/jbc.R112.444158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leitinger B, Kwan AP. The discoidin domain receptor DDR2 is a receptor for type X collagen. Matrix Biol. 2006;25(6):355–64. doi: 10.1016/j.matbio.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 9.Valiathan RR, Marco M, Leitinger B, Kleer CG, Fridman R. Discoidin domain receptor tyrosine kinases: new players in cancer progression. Cancer Metastasis Rev. 2012;31(1–2):295–321. doi: 10.1007/s10555-012-9346-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bargal R, Cormier-Daire V, Ben-Neriah Z, Le Merrer M, Sosna J, Melki J, et al. Mutations in DDR2 gene cause SMED with short limbs and abnormal calcifications. Am J Hum Genet. 2009;84(1):80–4. doi: 10.1016/j.ajhg.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Al-Kindi A, Kizhakkedath P, Xu H, John A, Sayegh AA, Ganesh A, et al. A novel mutation in DDR2 causing spondylo-meta-epiphyseal dysplasia with short limbs and abnormal calcifications (SMED-SL) results in defective intra-cellular trafficking. BMC Med Genet. 2014;15:42. doi: 10.1186/1471-2350-15-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo Y, Yang TL, Dong SS, Yan H, Hao RH, Chen XF, et al. Genetic analysis identifies DDR2 as a novel gene affecting bone mineral density and osteoporotic fractures in Chinese population. PLoS One. 2015;10(2):e0117102. doi: 10.1371/journal.pone.0117102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Labrador JP, Azcoitia V, Tuckermann J, Lin C, Olaso E, Manes S, et al. The collagen receptor DDR2 regulates proliferation and its elimination leads to dwarfism. EMBO Rep. 2001;2(5):446–52. doi: 10.1093/embo-reports/kve094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kano K, Marin de Evsikova C, Young J, Wnek C, Maddatu TP, Nishina PM, et al. A novel dwarfism with gonadal dysfunction due to loss-of-function allele of the collagen receptor gene, Ddr2, in the mouse. Mol Endocrinol. 2008;22(8):1866–80. doi: 10.1210/me.2007-0310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kawai I, Matsumura H, Fujii W, Naito K, Kusakabe K, Kiso Y, et al. Discoidin domain receptor 2 (DDR2) regulates body size and fat metabolism in mice. Transgenic Res. 2014;23(1):165–75. doi: 10.1007/s11248-013-9751-2. [DOI] [PubMed] [Google Scholar]
- 16.Ruiz PA, Jarai G. Collagen I induces discoidin domain receptor (DDR) 1 expression through DDR2 and a JAK2-ERK1/2-mediated mechanism in primary human lung fibroblasts. J Biol Chem. 2011;286(15):12912–23. doi: 10.1074/jbc.M110.143693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang K, Corsa CA, Ponik SM, Prior JL, Piwnica-Worms D, Eliceiri KW, et al. The collagen receptor discoidin domain receptor 2 stabilizes SNAIL1 to facilitate breast cancer metastasis. Nat Cell Biol. 2013;15(6):677–87. doi: 10.1038/ncb2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ge C, Xiao G, Jiang D, Franceschi RT. Critical role of the extracellular signal-regulated kinase-MAPK pathway in osteoblast differentiation and skeletal development. J Cell Biol. 2007;176(5):709–18. doi: 10.1083/jcb.200610046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matsushita T, Chan YY, Kawanami A, Balmes G, Landreth GE, Murakami S. Extracellular signal-regulated kinase 1 (ERK1) and ERK2 play essential roles in osteoblast differentiation and in supporting osteoclastogenesis. Mol Cell Biol. 2009;29(21):5843–57. doi: 10.1128/MCB.01549-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Greenblatt MB, Shim JH, Zou W, Sitara D, Schweitzer M, Hu D, et al. The p38 MAPK pathway is essential for skeletogenesis and bone homeostasis in mice. J Clin Invest. 2010;120(7):2457–73. doi: 10.1172/JCI42285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ge C, Xiao G, Jiang D, Yang Q, Hatch NE, Roca H, et al. Identification and functional characterization of ERK/MAPK phosphorylation sites in the Runx2 transcription factor. J Biol Chem. 2009;284(47):32533–43. doi: 10.1074/jbc.M109.040980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ge C, Yang Q, Zhao G, Yu H, Kirkwood KL, Franceschi RT. Interactions between extracellular signal-regulated kinase 1/2 and p38 MAP kinase pathways in the control of RUNX2 phosphorylation and transcriptional activity. J Bone Miner Res. 2012;27(3):538–51. doi: 10.1002/jbmr.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Y, Ge C, Long JP, Begun DL, Rodriguez JA, Goldstein SA, et al. Biomechanical stimulation of osteoblast gene expression requires phosphorylation of the RUNX2 transcription factor. J Bone Miner Res. 2012;27:1263–74. doi: 10.1002/jbmr.1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu E, Kim JB, Sarraf P, Spiegelman BM. Inhibition of adipogenesis through MAP kinase-mediated phosphorylation of PPARgamma. Science. 1996;274(5295):2100–3. doi: 10.1126/science.274.5295.2100. [DOI] [PubMed] [Google Scholar]
- 25.Ge C, Cawthorn WP, Li Y, Zhao G, MacDougald OA, Franceschi RT. Reciprocal Control of Osteogenic and Adipogenic Differentiation by ERK/MAP Kinase Phosphorylation of Runx2 and PPARgamma Transcription Factors. J Cell Physiol. 2016;231(3):587–96. doi: 10.1002/jcp.25102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fretz JA, Nelson T, Xi Y, Adams DJ, Rosen CJ, Horowitz MC. Altered metabolism and lipodystrophy in the early B-cell factor 1-deficient mouse. Endocrinology. 2010;151(4):1611–21. doi: 10.1210/en.2009-0987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ducy P, Karsenty G. Two distinct osteoblast-specific cis-acting elements control expression of a mouse osteocalcin gene. Mol Cell Biol. 1995;15(4):1858–69. doi: 10.1128/mcb.15.4.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krebsbach PH, Mankani MH, Satomura K, Kuznetsov SA, Robey PG. Repair of craniotomy defects using bone marrow stromal cells. Transplantation. 1998;66(10):1272–8. doi: 10.1097/00007890-199811270-00002. [DOI] [PubMed] [Google Scholar]
- 29.Rim JS, Mynatt RL, Gawronska-Kozak B. Mesenchymal stem cells from the outer ear: a novel adult stem cell model system for the study of adipogenesis. FASEB J. 2005;19(9):1205–7. doi: 10.1096/fj.04-3204fje. [DOI] [PubMed] [Google Scholar]
- 30.Mizoguchi T, Muto A, Udagawa N, Arai A, Yamashita T, Hosoya A, et al. Identification of cell cycle-arrested quiescent osteoclast precursors in vivo. J Cell Biol. 2009;184(4):541–54. doi: 10.1083/jcb.200806139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yamashita J, Datta NS, Chun YH, Yang DY, Carey AA, Kreider JM, et al. Role of Bcl2 in osteoclastogenesis and PTH anabolic actions in bone. J Bone Miner Res. 2008;23(5):621–32. doi: 10.1359/JBMR.071211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Camp HS, Tafuri SR. Regulation of peroxisome proliferator-activated receptor gamma activity by mitogen-activated protein kinase. J Biol Chem. 1997;272(16):10811–6. doi: 10.1074/jbc.272.16.10811. [DOI] [PubMed] [Google Scholar]
- 33.Perez A, Kastner P, Sethi S, Lutz Y, Reibel C, Chambon P. PMLRAR homodimers: distinct DNA binding properties and heteromeric interactions with RXR. EMBO J. 1993;12(8):3171–82. doi: 10.1002/j.1460-2075.1993.tb05986.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Olaso E, Labrador JP, Wang L, Ikeda K, Eng FJ, Klein R, et al. Discoidin domain receptor 2 regulates fibroblast proliferation and migration through the extracellular matrix in association with transcriptional activation of matrix metalloproteinase-2. J Biol Chem. 2002;277(5):3606–13. doi: 10.1074/jbc.M107571200. [DOI] [PubMed] [Google Scholar]
- 35.Zhang Y, Su J, Yu J, Bu X, Ren T, Liu X, et al. An essential role of discoidin domain receptor 2 (DDR2) in osteoblast differentiation and chondrocyte maturation via modulation of Runx2 activation. J Bone Miner Res. 2011;26(3):604–17. doi: 10.1002/jbmr.225. [DOI] [PubMed] [Google Scholar]
- 36.Zhang Y, Su J, Wu S, Teng Y, Yin Z, Guo Y, et al. DDR2 (discoidin domain receptor 2) suppresses osteoclastogenesis and is a potential therapeutic target in osteoporosis. Science signaling. 2015;8(369):ra31. doi: 10.1126/scisignal.2005835. [DOI] [PubMed] [Google Scholar]
- 37.Justesen J, Stenderup K, Ebbesen EN, Mosekilde L, Steiniche T, Kassem M. Adipocyte tissue volume in bone marrow is increased with aging and in patients with osteoporosis. Biogerontology. 2001;2(3):165–71. doi: 10.1023/a:1011513223894. [DOI] [PubMed] [Google Scholar]
- 38.Meunier P, Aaron J, Edouard C, Vignon G. Osteoporosis and the replacement of cell populations of the marrow by adipose tissue. A quantitative study of 84 iliac bone biopsies. Clin Orthop Relat Res. 1971;80:147–54. doi: 10.1097/00003086-197110000-00021. [DOI] [PubMed] [Google Scholar]
- 39.Minaire P, Edouard C, Arlot M, Meunier PJ. Marrow changes in paraplegic patients. Calcif Tissue Int. 1984;36(3):338–40. doi: 10.1007/BF02405340. [DOI] [PubMed] [Google Scholar]
- 40.Cawthorn WP, Scheller EL, Learman BS, Parlee SD, Simon BR, Mori H, et al. Bone marrow adipose tissue is an endocrine organ that contributes to increased circulating adiponectin during caloric restriction. Cell Metab. 2014;20(2):368–75. doi: 10.1016/j.cmet.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.