

Abstract
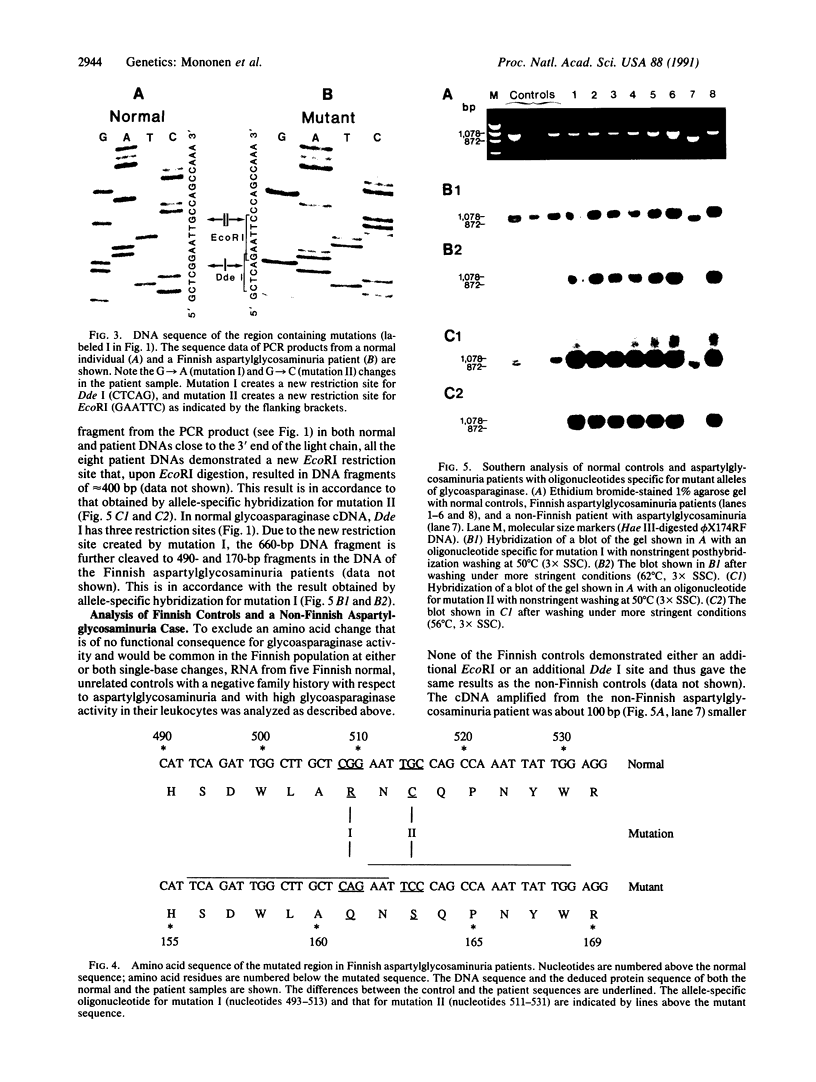
Aspartylglycosaminuria is an inherited lysosomal storage disease caused by deficiency of glycoasparaginase (EC 3.5.1.26) and occurs with higher frequency among Finns than other populations. We have purified human glycoasparaginase and determined about 90% of the amino acid sequence of its light subunit and greater than 70% of that of its heavy subunit by Edman degradation and mass spectrometry. Additional sequence data were obtained from the cloning and subsequent nucleotide analysis of a cDNA corresponding to the normal human glycoasparaginase gene. The enzyme is encoded by a single mRNA as a single polypeptide that is posttranslationally processed to generate the subunits and is glycosylated. After preparing first-strand cDNA from leukocyte and fibroblast total RNA, we used the polymerase chain reaction to amplify the glycoasparaginase cDNA of eight Finnish aspartylglycosaminuria patients. We demonstrate that the Finnish patients' mRNA sequence differed from the normal sequence by two single-base changes six nucleotides apart from one another in the heavy chain of glycoasparaginase. The first change resulted in the replacement of arginine by glutamine (R161Q), whereas the second change resulted in a cysteine to serine substitution (C163S). Both mutations resulted in novel restriction endonuclease sites and were present in all eight Finnish aspartylglycosaminuria patients originating from different pedigrees, but they were absent from Finnish and non-Finnish controls and a non-Finnish case of aspartylglycosaminuria. These results indicate molecular homogeneity in aspartylglycosaminuria alleles in the Finnish population.
Full text
PDF




Images in this article

Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Erickson A. K., Payne D. M., Martino P. A., Rossomando A. J., Shabanowitz J., Weber M. J., Hunt D. F., Sturgill T. W. Identification by mass spectrometry of threonine 97 in bovine myelin basic protein as a specific phosphorylation site for mitogen-activated protein kinase. J Biol Chem. 1990 Nov 15;265(32):19728–19735. [PubMed] [Google Scholar]
- Fisher K. J., Tollersrud O. K., Aronson N. N., Jr Cloning and sequence analysis of a cDNA for human glycosylasparaginase. A single gene encodes the subunits of this lysosomal amidase. FEBS Lett. 1990 Sep 3;269(2):440–444. doi: 10.1016/0014-5793(90)81211-6. [DOI] [PubMed] [Google Scholar]
- Heisterkamp N., Groffen J., Stephenson J. R. The human v-abl cellular homologue. J Mol Appl Genet. 1983;2(1):57–68. [PubMed] [Google Scholar]
- Hunt D. F., Yates J. R., 3rd, Shabanowitz J., Winston S., Hauer C. R. Protein sequencing by tandem mass spectrometry. Proc Natl Acad Sci U S A. 1986 Sep;83(17):6233–6237. doi: 10.1073/pnas.83.17.6233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaartinen V., Mononen I. Assay of aspartylglycosylaminase by high-performance liquid chromatography. Anal Biochem. 1990 Oct;190(1):98–101. doi: 10.1016/0003-2697(90)90140-5. [DOI] [PubMed] [Google Scholar]
- Kawasaki E. S., Clark S. S., Coyne M. Y., Smith S. D., Champlin R., Witte O. N., McCormick F. P. Diagnosis of chronic myeloid and acute lymphocytic leukemias by detection of leukemia-specific mRNA sequences amplified in vitro. Proc Natl Acad Sci U S A. 1988 Aug;85(15):5698–5702. doi: 10.1073/pnas.85.15.5698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell G. A., Brody L. C., Sipila I., Looney J. E., Wong C., Engelhardt J. F., Patel A. S., Steel G., Obie C., Kaiser-Kupfer M. At least two mutant alleles of ornithine delta-aminotransferase cause gyrate atrophy of the choroid and retina in Finns. Proc Natl Acad Sci U S A. 1989 Jan;86(1):197–201. doi: 10.1073/pnas.86.1.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mononen I., Kaartinen V., Mononen T. Laboratory detection of aspartylglycosaminuria. Scand J Clin Lab Invest Suppl. 1988;191:7–11. [PubMed] [Google Scholar]
- Mononen T., Parviainen M., Penttilä I., Mononen I. Liquid-chromatographic detection of aspartylglycosaminuria. Clin Chem. 1986 Mar;32(3):501–502. [PubMed] [Google Scholar]
- Norrander J., Kempe T., Messing J. Construction of improved M13 vectors using oligodeoxynucleotide-directed mutagenesis. Gene. 1983 Dec;26(1):101–106. doi: 10.1016/0378-1119(83)90040-9. [DOI] [PubMed] [Google Scholar]
- Sanger F., Nicklen S., Coulson A. R. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A. 1977 Dec;74(12):5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenzuela D. M., Groffen J. Four human carcinoma cell lines with novel mutations in position 12 of c-K-ras oncogene. Nucleic Acids Res. 1986 Jan 24;14(2):843–852. doi: 10.1093/nar/14.2.843. [DOI] [PMC free article] [PubMed] [Google Scholar]