

Abstract
The myeloproliferative neoplasms (MPNs) are characterized by hematopoietic stem/progenitor cell (HSPC) expansion and overproduction of blood cells. The acquired mutation JAK2V617F plays a central role in these disorders. Mechanisms responsible for MPN HSPC expansion is not fully understood, limiting the effectiveness of current treatments. Endothelial cells (ECs) carrying the JAK2V617F mutation can be detected in patients with MPNs, suggesting that ECs are involved in the pathogenesis of MPNs. Here we report that JAK2V617F-bearing primary murine ECs have increased cell proliferation and angiogenesis in vitro compared to JAK2WT ECs. While there was no difference between JAK2V617F and JAK2WT HSPC proliferation when co-cultured with JAK2WT EC, the JAK2V617F HSPC displayed a relative growth advantage over the JAK2WT HSPC when co-cultured on JAK2V617F EC. In addition, the thrombopoietin (TPO) receptor MPL is up regulated in JAK2V617F ECs and contributes to the maintenance/expansion of the JAK2V617F clone over JAK2WT clone in vitro. Considering that ECs are an essential component of the hematopoietic niche and most HSPCs reside in the perivascular niche, our studies suggest that the JAK2V617F-bearing ECs form an important component of the MPN vascular niche and contribute to mutant stem/progenitor cell expansion, likely through a critical role of the TPO/MPL signaling axis.
Keywords: myeloproliferative neoplasms, hematopoietic stem/progenitor cells, microenvironment, endothelial cells, JAK2V617F, MPL
Introduction
The marrow consists of the hematopoietic cells and non-hematopoietic stromal cells, including fibroblasts, reticular cells, endothelial cells (ECs), macrophages, adipocytes and osteoblasts. In addition to its role in normal HSPC biology, an altered microenvironment is an important contributor to the development of hematologic malignancies.1–3 In a reciprocal fashion, myeloid malignancies also affect the function of the marrow microenvironment to impair normal hematopoiesis while favoring malignant stem cell expansion.4,5
The chronic Philadelphia chromosome (Ph1) negative myeloproliferative neoplasms (MPNs), including polycythemia vera (PV), essential thrombocythemia (ET) and primary myelofibrosis (PMF), are clonal stem cell disorders characterized by HSPC expansion, overproduction of mature blood cells, a tendency to extramedullary hematopoiesis, and transformation to acute leukemia or myelofibrosis at variable rates. The acquired signaling kinase mutation JAK2V617F plays a central role in the pathogenesis of MPN, but mechanism(s) responsible for MPN HSPC expansion is not fully understood, limiting the effectiveness of current treatments. Although the etiology of dysregulated hematopoiesis has been mainly attributed to the molecular alterations within the hematopoietic stem/progenitor cells, abnormalities of the marrow microenvironment are beginning to be recognized as an important factor in MPN development.1,5–7 The diseased niche could impair normal hematopoiesis and favor the competing malignant stem cells, which could contribute to the poor engraftment and treatment-related mortality following allogeneic stem cell transplantation, the only curative treatment for patients with MPNs.1–3,5,8,9
ECs are an essential component of the hematopoietic niche and most hematopoietic stem/progenitor cells (HSPCs) reside close to a marrow sinusoid (the “perivascular niche”).10–13 In addition, ECs are an important niche component of the extramedullary (splenic) hematopoiesis, which is almost always present in patients with MPNs and is associated with MPN disease progression.13,14 Although the existence and cell of origin of endothelial progenitors is still a matter of debate, JAK2V617F mutation can be detected in endothelial progenitors derived from the hematopoietic lineage (the so-called endothelial cell colony-forming units; CFU-ECs) and, in some reports, in the true endothelial colony-forming cells (ECFC) based on in vitro assays.15–19 JAK2V617F mutation is also present in ECs isolated by microdissection from liver and spleen samples of patients with MPNs.17,20 In addition, we and others have shown that JAK2V617F ECs are critical in the development of the bleeding abnormalities in a murine model of JAK2V617F-positive MPNs in which JAK2V617F is expressed in all hematopoietic cells and endothelial cells.21 All of these observations suggest that ECs are involved in the pathogenesis of MPNs.
Previously, we and others have shown that the thrombopoietin (TPO) receptor MPL is essential for the development of HSPC expansion in MPNs.22,23 MPL is expressed in long-term HSPCs and is associated with both HSPC repopulating activity and HSPC quiescence.24,25 MPL is also expressed on several types of endothelium.26–28 Whether EC MPL receptor affects the vascular niche function and contributes to MPN development is not known. In this study, we examined the roles of JAK2V617F-bearing ECs in MPN hematopoiesis. We found that the MPN vascular niche contributes to the growth advantage of JAK2V617F HSPC over the JAK2WT HSPC. We also found that the EC MPL receptor is important for the JAK2V617F clonal expansion in MPNs.
Materials and Methods
Experimental mice
JAK2V617F Flip-Flop (FF1) mice29 were kindly provided by Radek Skoda (University Hospital, Basal, Switzerland), Tie2-Cre mice30 by Mark Ginsberg (University of California, San Diego), and MPL knockout mice (MPL−/−)31 by Warren Alexander (Melbourne, Australia). FF1 mice were crossed with Tie2-Cre to express JAK2V617F specifically in hematopoietic cells and ECs (Tie2/FF1 mice) as we previously did.21 All mice used were on a C57BL/6 background and were bred in a pathogen-free mouse facility at Stony Brook University. CD45.1+ congenic mice (SJL) were purchased from Taconic Inc. (Albany, NY). Animal experiments were performed in accordance with the guidelines provided by the Institutional Animal Care and Use Committee at Stony Brook University.
Isolation of murine hematopoietic stem/progenitor cells (HSPCs)
14–18-week old mice were euthanized and the femurs and tibias removed. A 25-gauge needle was used to flush the marrow with PBS + 2% FBS. Cells were triturated and filtered through 70μM nylon mesh (BD Biosciences, San Jose, CA) to obtain a single cell suspension. For depletion of mature hematopoietic cells, the Lineage Cell Depletion Kit (Miltenyi Biotec, Cat. 130-090-858, San Diego, CA) was used. The lineage (CD5, CD45R, CD11b, Ter119, and GR-1) negative cells were collected and then positively selected for CD117+ (cKit+) cells using CD117 microbead (Miltenyi Biotec, Cat. 130-091-224) to yield LineagenegcKit+ (Lin−cKit+) HSPCs.
Isolation of murine lung endothelial cells
Primary murine lung EC isolation was performed as we previously did.32 Briefly, 14–18wk old mice were euthanized using 100% CO2 inhalation followed by cervical dislocation. The chest was immediately opened through a midline sternotomy. The left ventricle was identified and the ventricular cavity was entered through the apex with a 27-gauge needle. The right ventricle was identified and an incision was made in the free wall to exsanguinate the animal and to allow the excess perfusate to exit the vascular space. The animal was perfused with 30 ml of cold PBS. The lung tissue was collected and minced finely with scissors. The tissue fragments were digested in DMEM medium containing 1 mg/mL Collagenase D (Roche, Switzerland), 1 mg/mL Collagenase/Dispase (Roche) and 25 U/mL DNase (Sigma, St. Louis, MO) at 37°C for 2hr with shaking, after which the suspension was homogenized by triturating. The homogenate was filtered through a 70μm nylon mesh (BD Biosciences, San Jose, CA) and pelleted by centrifugation (400g for 5 min). Cells were first depleted for CD45+ cells (Miltenyi Biotec) and then positively selected for CD31+ cells (Miltenyi Biotec) using magnetically labeled microbeads according to the manufacturer’s protocol. Isolated ECs (CD45−CD31+) were cultured in EC culture medium with no medium change for the first 72hrs to allow EC attachment followed by medium change every 2–3 days. Cells were re-selected for CD31+ cells when they reach >70–80% confluence (usually after 3–4 days of culture).
Flow cytometry
CD45.1 (A20) antibody or isotype control was used for chimerism studies. All staining steps were performed in ice-cold PBS containing 2% fetal bovine serum. All samples were analyzed by flow cytometry using a FACSCalibur flow cytometer (Becton Dickinson, Franklin Lakes, NJ). Postacquisition data analysis was performed with FlowJo software V9.2.3 (Treestar, CA).
Polymerase Chain Reaction
Human JAK2 cDNA-specific primers (5′-GAAGAACTTCAGCAGTCTTAAAGATC-3′ and 5′-CCATGCCAACTGTTTAGCAACTTC-3′) were used to detect the expression of human JAK2V617F in ECs from Tie2/FF1 mice using reverse transcription polymerase chain reaction (RT-PCR). The primers amplify a 573bp fragment that would be detected on 2% agarose gel.
The TaqMan® Gene Expression Assay (Applied Biosystems) was used for real-time quantitative polymerase chain reaction (qPCR) to verify differential expression of MPL (Mm00440306_g1) on an ABI ViiA™ 7 Real-Time PCR machine (Applied Biosystems). The gene expression levels were normalized to Actin beta (Actb) expression and relative fold changes was calculated by the 2ΔΔCT method. All assays were performed in triplicate.
Assays to examine endothelial cell in vitro angiogenesis
EC tube formation assay was performed as a measure of angiogenesis in vitro.32 Matrigel® matrix (10mg/ml, Corning Inc., Corning, NY) were thaw overnight at 4°C and kept on ice until use. 150ul Matrigel per well was added to pre-chilled 48-well culture plate. After gelation at 37°C for 30minutes, gels were overlaid with 6 × 104 JAK2V617F ECs (from Tie2/FF1 mice) or JAK2WT ECs (from control mice) (passage 3–4) in 300ul of complete EC medium. Tube formation was inspected after a period of 2, 4, 6, and 8hrs and images were captured with a phase-contrast microscope (AMEX-1200, AMG, Bothell, WA). The quantification of the capillary tube formation was performed using the ImageJ® software (National Institute of Health, Bethesda, MD) by counting the number of nodes (or branch points), loops, and tubes in 4 non-overlapping areas at ×40 magnification in two duplicate wells.
In vitro cell culture
Lin−cKit+ HSPCs were cultured in StemSpan® serum-free expansion medium (SFEM) containing 100 ng/mL recombinant mouse SCF, 6 ng/mL recombinant mouse IL3 and 10 ng/mL recombinant human IL-6 (all from Stem Cell Technologies, Vancouver, BC)
ECs were cultured on 1% gelatin coated plates in complete EC medium which is consisted of advanced DMEM/F12 (ThermoFisher, Waltham, MA) medium containing 20% fetal bovine serum, 50μg/ml endothelial cell growth supplement (Alfa Aesar, Ward Hill, MA), 1% Antibiotic-antimyotic solution (Cat. 15240-062, ThermoFisher), 10mM HEPES buffer (ThermoFisher), 5μM SB431542 small molecule (R&D, Minneapolis, MN), 50μg/ml Heparin (Sigma), 1% Glutamax 100× solution (ThermoFisher), 1% non-essential amino acid (ThermoFisher), recombinant mouse VEGF 10ng/ml (PeproTech, Rocky Hill, NJ; add fresh when changing medium) and recombinant human FGF2 20ng/ml (PeproTech; add fresh when changing medium). Cells were cultured at 37°C in a humidified 5% CO2 atmosphere with medium change every 2–3 days until they reach 70–80% confluence. 0.05 % trypsin-EDTA solution was used for cell passaging. EC-conditioned media (ECCM) was collected when cell density reach 50–60% confluency.
Co-culture of Lin-cKit+ cells with EC
Three days prior to co-culture with HSPCs, 2 × 104 primary murine lung ECs (passage 1–3) were seeded into 1% gelatin coated 24-well plate in complete EC medium. On Day 0, 104 Lin-cKit+ cells were seeded onto the EC monolayer in 0.3ml SFEM containing 100 ng/mL recombinant mouse SCF, 6 ng/mL recombinant mouse IL3 and 10 ng/mL recombinant human IL-6. 0.3ml fresh SFEM with cytokines was added on Day 3 and cells were counted on Day 5.
In vitro competitive growth assay
On Day 0, 103 WT Lin-cKit+ HSPCs (CD45.1) and 103 Tie2/FF1 Lin-cKit+ HSPCs (CD45.2) were mixed and culture together in 24-well plate with 0.2ml SFEM, or SFEM + WT ECCM (1:1 vol:vol), or SFEM + MPL−/− ECCM (1:1 vol:vol) containing 100 ng/mL recombinant mouse SCF, 6 ng/mL recombinant mouse IL3 and 10 ng/mL recombinant human IL-6. 0.2ml fresh medium with cytokines was added on Day 3. After 6 days of culture, cells were stained with CD45.1 antibody for flow cytometry analysis.
Statistical Analysis
Statistical analyses were performed using Student’s unpaired, 2-tailed t tests using Excel software (Microsoft). A p value of less than 0.05 was considered significant. For all bar graphs, data are presented as mean ± standard error of the mean (SEM).
Results
JAK2V617F mutant ECs show increased cell proliferation and angiogenesis in vitro compared to JAK2WT ECs
FF1 mice were crossed with Tie2-Cre mice to generate mice that express JAK2V617F specifically in hematopoietic cells and ECs (Tie2/FF1 mice) as was previously described.21 Primary lung ECs were isolated from Tie2/FF1 mice (JAK2V617F ECs) and age-matched littermate control mice (JAK2WT EC). The expression of human JAK2V617F mutation in Tie2/FF1 ECs was confirmed by RT-PCR. (Figure 1A) Consistent with a previous report on lentiviral-transduced JAK2V617F-mutant human umbilical vein endothelial cells (HUVECs),33 primary murine lung ECs carrying the JAK2V617F mutation proliferated to a greater extent than JAK2WT ECs (2.0-fold, p = 0.031). (Figure 1B) To study the effects of JAK2V617F mutation on EC function, tube formation assay was performed on JAK2WT and JAK2V617F ECs as a measure of their capacity for in vitro angiogenesis. JAK2V617F ECs had significantly increased angiogenesis in Matrigel™ compared to JAK2WT EC. (Figure 1C) In addition, the tubular structures formed by the JAK2V617F ECs in vitro were more stable than those from JAK2WT ECs. (Figure 1D) This result suggests that the JAK2V617F mutation in EC might be responsible for the increased microvascular density in the marrow of patients with MPNs.34–36 Since ECs are an essential component of the hematopoietic niche, and most HSPCs reside in the perivascular niche,10–13 these results prompted us to study how JAK2V617F-bearing ECs affect hematopoiesis in MPNs.
Figure 1. JAK2V617F-mutant ECs have increased cell proliferation and angiogenesis in vitro compared to JAK2WT ECs.
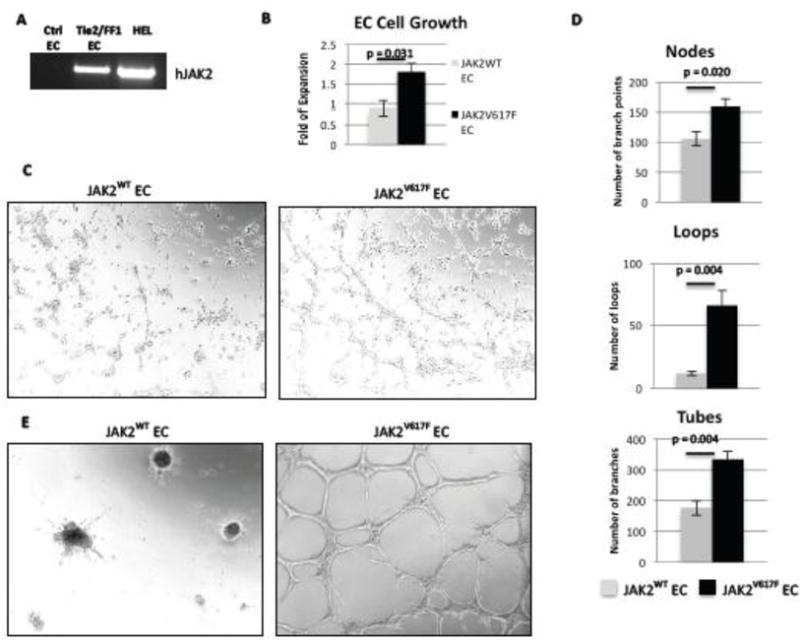
(A) As determined by RT-PCR, human JAK2V617F was expressed in ECs from Tie2/FF1 mice, but not in ECs from control mice. Human erythroleukemia (HEL) cells were used as the positive control. (B) JAK2V617F ECs proliferated more than JAK2WT EC (2.0-fold, p = 0.031). Results of 2 independent experiments (with triplicate in each experiments) are shown here. (C) JAK2WT and JAK2V617F ECs (6×104) were seeded in Matrigel matrix. EC tube formation was observed after an 8-hour incubation. A representative picture is shown. Magnification: 100×. (D) Quantification of tube formation was performed on images taken at 40× magnification by counting the number of nodes (or branch points), loops, and tubes in 4 non-overlapping fields. Results are expressed as the mean ± SEM (n=4). Data are from one of two independent experiments that gave similar results. (E) The tubular structures formed by the JAK2V617F ECs in vitro were more stable than the JAK2WT ECs after 24-hour incubation.
The JAK2V617F vascular niche contributes to the maintenance/expansion of JAK2V617F HSPC in preference to JAK2WT HSPCs
Lin-cKit+ HSPCs were isolated from Tie2/FF1 and age-matched littermate control mice. To examine the role of JAK2WT and JAK2V617F ECs in MPN hematopoiesis, we used an ex vivo co-culture system in which Lin−cKit+ HSPCs were cultured on a feeder layer of JAK2WT or JAK2V617F ECs under serum-free conditions. We compared JAK2WT and JAK2V617F HSPC proliferation on JAK2WT or JAK2V617F ECs respectively. Consistent with previous reports that JAK2V617F does not confer a significant growth advantage to HSPCs,37–42 we did not observe any significant difference between JAK2WT and JAK2V617F HSPC proliferation in vitro in SFEM. (Figure 2A) While there was no difference between JAK2V617F and JAK2WT HSPC proliferation when co-cultured with JAK2WT EC, the JAK2V617F HSPC displayed a relative growth advantage over the JAK2WT HSPC (1.24-fold, p = 0.041) when co-cultured on JAK2V617F EC. (Figure 2B–C) These results suggest that the MPN vascular niche (i.e. JAK2V617F EC) contributes to the maintenance/expansion of the JAK2V617F HSPCs in preference to JAK2WT HSPCs. This is consistent with other reports that the marrow microenvironment of myeloid malignancies are altered to impair normal hematopoiesis while favoring malignant stem cell expansion.4,5
Figure 2. JAK2V617F vascular niche contributes to the growth advantage of JAK2V617F HSPC over JAK2WT HSPC.
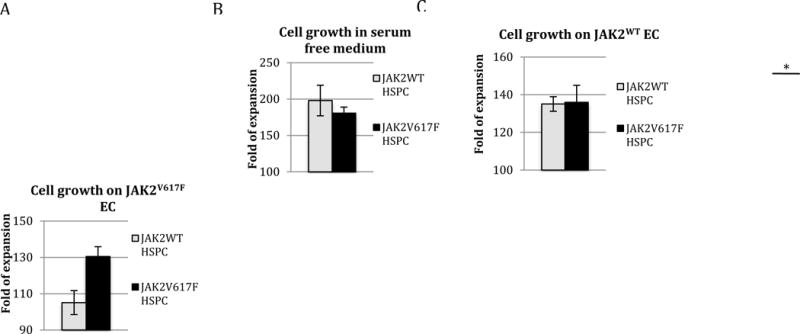
JAK2WT (from control mice) and JAK2V617F (from Tie2/FF1 mice) Lin−cKit+ HSPCs were cultured on a feeder layer of JAK2WT or JAK2V617F ECs under serum-free conditions. (A) There was no significant difference between JAK2WT and JAK2V617F HSPC proliferation in vitro in SFEM. (B) There was no significant difference between JAK2WT and JAK2V617F HSPC proliferation when co-cultured with JAK2WT EC. (C) JAK2V617F HSPC displayed a relative growth advantage over the JAK2WT HSPC when co-cultured with JAK2V617F EC. Cell proliferation was shown as fold of expansion which is the ratio of the final cell count to starting cell count. The results were expressed as mean ±s.e.m. (n=3) Data are from one of two independent experiments (with triplicates in each experiment) performed by two investigators (C.L. and H.Z.) that gave similar results. * p < 0.05
The EC MPL receptor contributes to the maintenance/expansion of the JAK2V617F HSPC over JAK2WT HSPCs
Thrombopoietin (TPO) and its receptor, the proto-oncogene MPL, are key regulators of HSPC activity.24,25,43–46 MPL is expressed in long-term HSPCs and is associated with both HSPC repopulating activity and HSPC quiescence.24,25 Previously, we and others have shown that MPL is essential for the development of an increased neoplastic stem cell pool in MPNs.22,23 Specifically, reducing MPL expression attenuated MPN severity and stem cell numbers, suggesting a gene dosage effect of receptor expression levels on the disease process. MPL is also expressed on endothelial cells (ECs) and TPO can stimulate EC growth and angiogenesis.26–28 Both TPO knockout (TPO−/−) and MPL knockout (MPL−/−) mice have significantly decreased HSPC populations and impaired angiogenesis.46–48 We checked MPL expression levels in JAK2WT and JAK2V617F ECs by qPCR. We found that MPL expression was increased by 46% in the JAK2V617F ECs compared to JAK2WT ECs (p = 0.003). (Figure 3A) Therefore, we hypothesized that MPN JAK2V617F-mutant clone expansion might depend on the endothelial microenvironment, as mediated by the endothelial MPL receptor.
Figure 3. MPL receptor on vascular ECs contributes to JAK2V617F HSPC maintenance/expansion in MPN.
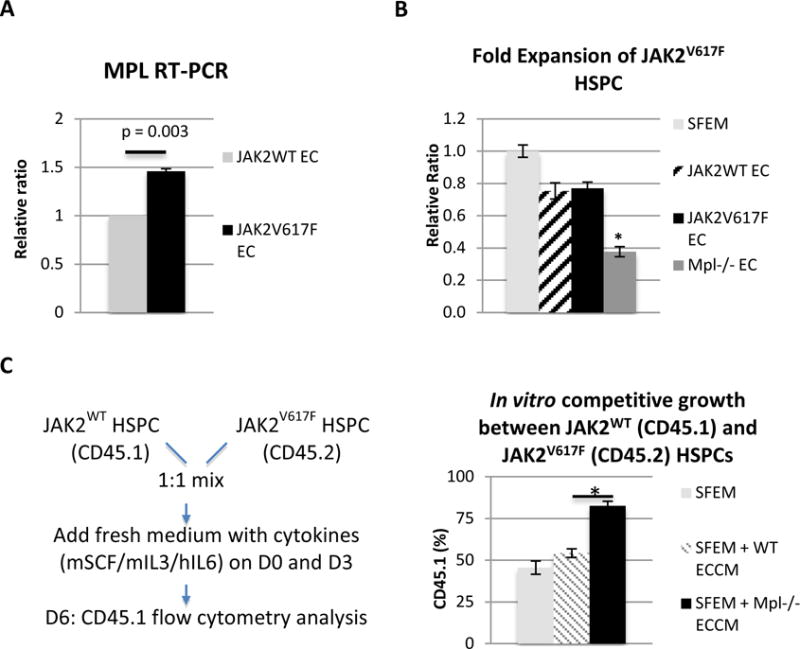
(A) MPL expression was increased by 46% in the JAK2V617F ECs compared to JAK2WT ECs (p = 0.003). MPL expression in JAK2V617F ECs was shown as the relative ratio compared to its expression in JAK2WT ECs which was set as “1”. (B) JAK2V617F HSPC (Lin-cKit+) cell proliferation was significantly decreased when co-cultured with MPL−/− EC, compared to when co-cultured with JAK2WT ECs or JAK2V617F ECs. Cell proliferation in EC co-culture was shown as the relative ratio compared to cell proliferation in SFEM which was set as “1”. (C) Left: experimental design of an in vitro competitive growth assay where JAK2WT HSPCs (CD45.1) and JAK2V617F HSPCs (CD45.2) were cultured together (1:1 mix) in the presence of ECCM collected from either WT EC or MPL−/− EC. Right: while there were equal numbers of JAK2WT HSPCs (CD45.1) and JAK2V617F HSPCs (CD45.2) in the presence of WT ECCM, there were significantly more JAK2WT HSPCs than JAK2V617F HSPCs in the presence of MPL−/− ECCM. The results were expressed as mean±s.e.m. (n=3). Data were from one of two independent experiments (with triplicates in each experiment) performed by two investigators (C.L. and H.Z.) that gave similar results.
To test this hypothesis, we obtained primary murine lung ECs isolated from the MPL knockout mice (MPL−/− EC)31 and performed ex vivo co-culture with JAK2V617F Lin-cKit+ HSPCs. We found that JAK2V617F HSPC (Lin-cKit+) cell proliferation was significantly decreased when co-cultured with MPL−/− EC, compared to when co-cultured with JAK2WT ECs or JAK2V617F ECs. (Figure 3B) This suggests that the EC MPL receptor is important for the JAK2V617F HSPC expansion in the MPN vascular niche.
Next, since both JAK2WT clones and JAK2V617F mutant clones coexist in most patients with MPNs, we asked whether the EC MPL receptor contributes to the JAK2V617F mutant clonal expansion over JAK2WT clones. We designed a in vitro competitive growth experiment where both JAK2WT HSPCs (CD45.1) and JAK2V617F HSPCs (CD45.2) were cultured together (1:1 mix) in the presence of EC conditioned medium (ECCM) collected from either WT EC or MPL−/− EC. At the end of the 6-day culture, there were equal numbers of JAK2WT HSPCs (CD45.1 48.9±2.6%) and JAK2V617F HSPCs in the presence of WT ECCM, which was what we expected since there was no significant difference between JAK2V617F and JAK2WT HSPC proliferation when co-cultured with JAK2WT EC (Figure 2B). However, there were significantly more JAK2WT HSPCs (CD45.1 77.3±2.6%) than JAK2V617F HSPCs in the presence of MPL−/− ECCM compared to in WT ECCM (p = 0.002), suggesting that EC MPL receptor contributes to the maintenance/expansion of the JAK2V617F clone over the JAK2WT clone in MPNs. (Figure 3C)
Discussion
ECs carrying the JAK2V617F mutation can be detected in patients with MPNs.16,17,20 In addition, ECs are an important niche component of the extramedullary (splenic) hematopoiesis, which is almost always present in patients with MPNs and is associated with MPN disease progression.13,14 All of these observations suggest that ECs are involved in the malignant process leading to MPNs. Previously, we and others have shown that JAK2V617F ECs are critical in the development of the bleeding abnormalities in a murine model of JAK2V617F-positive MPNs in which JAK2V617F was expressed in all hematopoietic cells and endothelial cells (Tie2-Cre/FF1).21,29,30 In this study, using primary ECs isolated from wild-type control (JAK2WT EC), Tie2/FF1 (JAK2V617F EC), and MPL−/− mice (MPL−/− EC), we have found that JAK2V617F-bearing ECs contribute to the competitive advantage of JAK2V617F HSPC over the JAK2WT HSPC, and the MPL receptor on vascular ECs is important for JAK2V617F HSPC maintenance/expansion in vitro.
Consistent with the enhanced angiogenesis and increased microvascular density in the marrow of patients with MPNs compared to normal marrow,34–36 JAK2V617F-bearing ECs proliferated to a greater extent than JAK2WT EC (2.0-fold, p = 0.031) (Figure 1B) and displayed increased angiogenesis in vitro, as measured by the tube formation assay. (Figure 1C) In addition, we found that JAK2V617F HSPC gained a competitive advantage compared to JAK2WT HSPC when co-cultured on JAK2V617F EC, while there was no difference between JAK2V617F and JAK2WT HSPC proliferation when co-cultured on JAK2WT EC. Both JAK2WT and JAK2V617F-mutant HSPC cell proliferation was decreased when co-cultured on ECs compared to cultured alone in SFEM, suggesting that our EC co-culture system inhibits HSPC proliferation or promotes HSPC quiescence by the elaboration of secreted factors and/or by cell-cell contacts. We hypothesize that the JAK2V617F mutation alters both HSPC and EC function to provide the relative growth advantage of the JAK2V617F-bearing HSPCs over JAK2WT HSPCs when co-cultured on JAK2V617F EC. As no significant difference was detected between JAK2V617F and JAK2WT HSPC proliferation when co-cultured with JAK2WT EC, our study suggests that both mutant HSPCs and mutant vascular niche are required for the development of MPNs. The stem cell compartment in MPN is heterogeneous with the presence of both JAK2WT clones and JAK2V617F mutant clones in most patients with MPNs. The mechanism(s) that contribute to the JAK2V617F-positive HSPC proliferation and expansion is poorly understood, limiting the effectiveness of current treatments. Since ECs are an essential component of the hematopoietic niche, and most HSPCs reside in the perivascular niche,10–13 our results suggest that the JAK2V617F-bearing vascular niche contributes to the JAK2V617F- mutant clonal expansion in MPNs. Therefore, targeting the MPN vascular niche could provide more effective adjuvant therapies to treatments directly focusing on the MPN neoplastic cells.
The TPO receptor, MPL, is a key regulator of HSPC activity.24,25,44–46,49 MPL is expressed in long-term HSPCs and is associated with both HSPC repopulating activity and HSPC quiescence.24,25 MPL is also expressed on several types of endothelium.26–28 Our previous study showed that the EC MPL receptor does not contribute significantly to the regulation of TPO levels or to steady-state platelet counts.50 Whether the EC MPL receptor could affect vascular niche function, and contribute to the critical role of TPO/MPL signaling in HSPC maintenance, is not known. In this study, we found that MPL expression was increased in the JAK2V617F ECs compared to JAK2WT ECs. (Figure 3A) Ablating the MPL receptor in ECs (i.e. MPL−/− ECs) significantly decreased cell proliferation of the co-cultured JAK2V617F HSPCs compared to JAK2WT ECs or JAK2V617F ECs did. (Figure 3B) In addition, an in vitro competitive growth experiment showed that the EC MPL receptor contributes to the maintenance/expansion of the JAK2V617F clone over the JAK2WT clone when cultured together. (Figure 3C) Together with our previous report,22 we have shown that the MPL receptor on HSPCs and ECs is important for JAK2V617F HSPC expansion in MPNs. Since MPL relies on JAK2 protein for its downstream signaling pathways, and JAK2 can affect MPL receptor stability and cell surface expression, we hypothesize that the JAK2V617F mutation could alter the vascular niche function and HSPC-EC interactions through altered TPO/MPL signaling, which in turn could contribute to altered hematopoietic factor and adhesion molecule production/expression in the MPN vascular niche. Considering that most HSPCs reside adjacent to a marrow sinusoid (the “perivascular niche”),10–13 our results suggest that JAK2V617F-bearing endothelial niche could maintain/expand the JAK2V617F HSPC clone via altered TPO/MPL signaling.
In this study, primary lung ECs were used to model the hematopoietic vascular niche in vitro. Since lung ECs might have different structural, phenotypic, or functional attributes from the marrow or spleen ECs,51 we have also used primary murine splenic ECs from wild-type control (JAK2WT EC), Tie2/FF1 (JAK2V617F EC), and MPL−/− mice (MPL−/− EC) and verified selected findings in splenic ECs (data not shown). In contrary to previous reports that ECs (e.g. HUVECs, human brain ECs, transfected/immortalized HUVECs, or primary murine ECs) support hematopoietic stem/progenitor cell (HSPC) expansion in vitro, our study found decreased Lin-cKit+ HSPC cell expansion when cultured on lung ECs.52–57 The different results could be due to differing HSPC populations, differences in the type of ECs employed, as well as culture medium and growth factors used in these studies. Further investigation will be required to explore whether the MPN vascular niche (with the JAK2V617F-bearing ECs) contributes to the JAK2V617F HSPC clonal expansion in vivo and whether this process also depends on the EC MPL receptor. Based on results reported here (i.e. JAK2WT HSPC did not display any growth advantage on JAK2V617F EC compared to on JAK2WT EC), competitive marrow transplantation experiments are likely required to model the human diseases and quantify JAK2WT and JAK2V617F HSPC numbers and functions in the JAK2V617F-bearing vascular niche.
In summary, our studies have shown that JAK2V617F-bearing ECs form an important part of the MPN HSPC niche and contribute to JAK2V617F clonal expansion in vitro. Further, we have demonstrated that the EC MPL receptor is important for the maintenance/expansion of the JAK2V617F HSPC over JAK2WT HSPCs, providing a potential mechanism through which the absence of MPL prevents MPN development.22,23 Considering that both JAK2WT clones and JAK2V617F mutant clones coexist in most patients with MPNs, our findings provide a possible new mechanism for the mutant clone expansion seen over time in patients with MPNs, and suggests that the MPN vascular niche and the EC MPL receptor may be excellent therapeutic targets to eradicate the malignant clone in patients with MPNs.
Acknowledgments
The authors thank Dr. Yupo Ma (Stony Brook University, NY) for his scientific consultation on endothelial cell and hematopoietic stem/progenitor cell co-culture experiments. This research was supported by the Veterans Affairs Career Development Award CDA210959632 (H.Z.) and National Institute of Diabetes and Digestive and Kidney Diseases grant R01DK049855 (K.K.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contribution
C.L. performed experiments, analyzed data, and reviewed the manuscript; K.K. analyzed data and wrote the manuscript; and H.Z. designed and performed experiments, analyzed data, and wrote the manuscript. All authors approved the submitted version of the manuscript.
References
- 1.Walkley CR, Olsen GH, Dworkin S, et al. A microenvironment-induced myeloproliferative syndrome caused by retinoic acid receptor gamma deficiency. Cell. 2007;129:1097–110. doi: 10.1016/j.cell.2007.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Raaijmakers MH, Mukherjee S, Guo S, et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature. 2010;464:852–7. doi: 10.1038/nature08851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Geyh S, Oz S, Cadeddu RP, et al. Insufficient stromal support in MDS results from molecular and functional deficits of mesenchymal stromal cells. Leukemia. 2013;27:1841–51. doi: 10.1038/leu.2013.193. [DOI] [PubMed] [Google Scholar]
- 4.Zhang B, Ho YW, Huang Q, et al. Altered microenvironmental regulation of leukemic and normal stem cells in chronic myelogenous leukemia. Cancer cell. 2012;21:577–92. doi: 10.1016/j.ccr.2012.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schepers K, Pietras EM, Reynaud D, et al. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell stem cell. 2013;13:285–99. doi: 10.1016/j.stem.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arranz L, Sanchez-Aguilera A, Martin-Perez D, et al. Neuropathy of haematopoietic stem cell niche is essential for myeloproliferative neoplasms. Nature. 2014;512:78–81. doi: 10.1038/nature13383. [DOI] [PubMed] [Google Scholar]
- 7.Mager LF, Riether C, Schurch CM, et al. IL-33 signaling contributes to the pathogenesis of myeloproliferative neoplasms. The Journal of clinical investigation. 2015;125:2579–91. doi: 10.1172/JCI77347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Colmone A, Amorim M, Pontier AL, Wang S, Jablonski E, Sipkins DA. Leukemic cells create bone marrow niches that disrupt the behavior of normal hematopoietic progenitor cells. Science. 2008;322:1861–5. doi: 10.1126/science.1164390. [DOI] [PubMed] [Google Scholar]
- 9.Rondelli D, Goldberg JD, Isola L, et al. MPD-RC 101 prospective study of reduced-intensity allogeneic hematopoietic stem cell transplantation in patients with myelofibrosis. Blood. 2014;124:1183–91. doi: 10.1182/blood-2014-04-572545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121:1109–21. doi: 10.1016/j.cell.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 11.Sipkins DA, Wei X, Wu JW, et al. In vivo imaging of specialized bone marrow endothelial microdomains for tumour engraftment. Nature. 2005;435:969–73. doi: 10.1038/nature03703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Acar M, Kocherlakota KS, Murphy MM, et al. Deep imaging of bone marrow shows non-dividing stem cells are mainly perisinusoidal. Nature. 2015;526:126–30. doi: 10.1038/nature15250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Inra CN, Zhou BO, Acar M, et al. A perisinusoidal niche for extramedullary haematopoiesis in the spleen. Nature. 2015;527:466–71. doi: 10.1038/nature15530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang B, Lewis SM. The splenomegaly of myeloproliferative and lymphoproliferative disorders: splenic cellularity and vascularity. European journal of haematology. 1989;43:63–6. doi: 10.1111/j.1600-0609.1989.tb01253.x. [DOI] [PubMed] [Google Scholar]
- 15.Yoder MC, Mead LE, Prater D, et al. Redefining endothelial progenitor cells via clonal analysis and hematopoietic stem/progenitor cell principals. Blood. 2007;109:1801–9. doi: 10.1182/blood-2006-08-043471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Teofili L, Martini M, Iachininoto MG, et al. Endothelial progenitor cells are clonal and exhibit the JAK2(V617F) mutation in a subset of thrombotic patients with Ph-negative myeloproliferative neoplasms. Blood. 2011;117:2700–7. doi: 10.1182/blood-2010-07-297598. [DOI] [PubMed] [Google Scholar]
- 17.Rosti V, Villani L, Riboni R, et al. Spleen endothelial cells from patients with myelofibrosis harbor the JAK2V617F mutation. Blood. 2013;121:360–8. doi: 10.1182/blood-2012-01-404889. [DOI] [PubMed] [Google Scholar]
- 18.Sozer S, Ishii T, Fiel MI, et al. Human CD34+ cells are capable of generating normal and JAK2V617F positive endothelial like cells in vivo. Blood Cells Mol Dis. 2009;43:304–12. doi: 10.1016/j.bcmd.2009.08.005. [DOI] [PubMed] [Google Scholar]
- 19.Piaggio G, Rosti V, Corselli M, et al. Endothelial colony-forming cells from patients with chronic myeloproliferative disorders lack the disease-specific molecular clonality marker. Blood. 2009;114:3127–30. doi: 10.1182/blood-2008-12-190991. [DOI] [PubMed] [Google Scholar]
- 20.Sozer S, Fiel MI, Schiano T, Xu M, Mascarenhas J, Hoffman R. The presence of JAK2V617F mutation in the liver endothelial cells of patients with Budd-Chiari syndrome. Blood. 2009;113:5246–9. doi: 10.1182/blood-2008-11-191544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Etheridge SL, Roh ME, Cosgrove ME, et al. JAK2V617F-positive endothelial cells contribute to clotting abnormalities in myeloproliferative neoplasms. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:2295–300. doi: 10.1073/pnas.1312148111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sangkhae V, Etheridge SL, Kaushansky K, Hitchcock IS. The thrombopoietin receptor, MPL, is critical for development of a JAK2V617F-induced myeloproliferative neoplasm. Blood. 2014;124:3956–63. doi: 10.1182/blood-2014-07-587238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marty C, Pecquet C, Nivarthi H, et al. Calreticulin mutants in mice induce an MPL-dependent thrombocytosis with frequent progression to myelofibrosis. Blood. 2016;127:1317–24. doi: 10.1182/blood-2015-11-679571. [DOI] [PubMed] [Google Scholar]
- 24.Solar GP, Kerr WG, Zeigler FC, et al. Role of c-mpl in early hematopoiesis. Blood. 1998;92:4–10. [PubMed] [Google Scholar]
- 25.Yoshihara H, Arai F, Hosokawa K, et al. Thrombopoietin/MPL signaling regulates hematopoietic stem cell quiescence and interaction with the osteoblastic niche. Cell stem cell. 2007;1:685–97. doi: 10.1016/j.stem.2007.10.020. [DOI] [PubMed] [Google Scholar]
- 26.Cardier JE, Dempsey J. Thrombopoietin and its receptor, c-mpl, are constitutively expressed by mouse liver endothelial cells: evidence of thrombopoietin as a growth factor for liver endothelial cells. Blood. 1998;91:923–9. [PubMed] [Google Scholar]
- 27.Brizzi MF, Battaglia E, Montrucchio G, et al. Thrombopoietin stimulates endothelial cell motility and neoangiogenesis by a platelet-activating factor-dependent mechanism. Circulation research. 1999;84:785–96. doi: 10.1161/01.res.84.7.785. [DOI] [PubMed] [Google Scholar]
- 28.Eguchi M, Masuda H, Kwon S, et al. Lesion-targeted thrombopoietin potentiates vasculogenesis by enhancing motility and enlivenment of transplanted endothelial progenitor cells via activation of Akt/mTOR/p70S6kinase signaling pathway. Journal of molecular and cellular cardiology. 2008;45:661–9. doi: 10.1016/j.yjmcc.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 29.Tiedt R, Hao-Shen H, Sobas MA, et al. Ratio of mutant JAK2-V617F to wild-type Jak2 determines the MPD phenotypes in transgenic mice. Blood. 2008;111:3931–40. doi: 10.1182/blood-2007-08-107748. [DOI] [PubMed] [Google Scholar]
- 30.Constien R, Forde A, Liliensiek B, et al. Characterization of a novel EGFP reporter mouse to monitor Cre recombination as demonstrated by a Tie2 Cre mouse line. Genesis. 2001;30:36–44. doi: 10.1002/gene.1030. [DOI] [PubMed] [Google Scholar]
- 31.Alexander WS, Roberts AW, Nicola NA, Li R, Metcalf D. Deficiencies in progenitor cells of multiple hematopoietic lineages and defective megakaryocytopoiesis in mice lacking the thrombopoietic receptor c-Mpl. Blood. 1996;87:2162–70. [PubMed] [Google Scholar]
- 32.Zhan H, Ma Y, Lin CH, Kaushansky K. JAK2V617F-mutant megakaryocytes contribute to hematopoietic stem/progenitor cell expansion in a model of murine myeloproliferation. Leukemia. 2016 doi: 10.1038/leu.2016.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Badr Kilani, J VD, Virginie Gourdou-Latyszenok, Eric Lippert, Raj Sewduth, Cécile Duplaa, Jean-Luc Villeval, Thierry Couffinhal, Chloe James. Consequences of the Presence of the JAK2V617F Mutation in Endothelial Cells: Towards a Better Understanding of the Increased Angiogenesis in Myeloproliferative Neoplasms. American Society of Hematology Annual Meeting (abstract) Blood. 2014;124 [Google Scholar]
- 34.Medinger M, Skoda R, Gratwohl A, et al. Angiogenesis and vascular endothelial growth factor-/receptor expression in myeloproliferative neoplasms: correlation with clinical parameters and JAK2-V617F mutational status. British journal of haematology. 2009;146:150–7. doi: 10.1111/j.1365-2141.2009.07726.x. [DOI] [PubMed] [Google Scholar]
- 35.Boveri E, Passamonti F, Rumi E, et al. Bone marrow microvessel density in chronic myeloproliferative disorders: a study of 115 patients with clinicopathological and molecular correlations. British journal of haematology. 2008;140:162–8. doi: 10.1111/j.1365-2141.2007.06885.x. [DOI] [PubMed] [Google Scholar]
- 36.Gianelli U, Vener C, Raviele PR, et al. VEGF expression correlates with microvessel density in Philadelphia chromosome-negative chronic myeloproliferative disorders. American journal of clinical pathology. 2007;128:966–73. doi: 10.1309/FP0N3LC8MBJUFFA6. [DOI] [PubMed] [Google Scholar]
- 37.Mullally A, Lane SW, Ball B, et al. Physiological Jak2V617F expression causes a lethal myeloproliferative neoplasm with differential effects on hematopoietic stem and progenitor cells. Cancer cell. 2010;17:584–96. doi: 10.1016/j.ccr.2010.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kent DG, Li J, Tanna H, et al. Self-renewal of single mouse hematopoietic stem cells is reduced by JAK2V617F without compromising progenitor cell expansion. PLoS biology. 2013;11:e1001576. doi: 10.1371/journal.pbio.1001576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li J, Kent DG, Godfrey AL, et al. JAK2V617F homozygosity drives a phenotypic switch in myeloproliferative neoplasms, but is insufficient to sustain disease. Blood. 2014;123:3139–51. doi: 10.1182/blood-2013-06-510222. [DOI] [PubMed] [Google Scholar]
- 40.James C, Mazurier F, Dupont S, et al. The hematopoietic stem cell compartment of JAK2V617F-positive myeloproliferative disorders is a reflection of disease heterogeneity. Blood. 2008;112:2429–38. doi: 10.1182/blood-2008-02-137877. [DOI] [PubMed] [Google Scholar]
- 41.Li J, Spensberger D, Ahn JS, et al. JAK2 V617F impairs hematopoietic stem cell function in a conditional knock-in mouse model of JAK2 V617F-positive essential thrombocythemia. Blood. 2010;116:1528–38. doi: 10.1182/blood-2009-12-259747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Godfrey AL, Chen E, Pagano F, et al. JAK2V617F homozygosity arises commonly and recurrently in PV and ET, but PV is characterized by expansion of a dominant homozygous subclone. Blood. 2012;120:2704–7. doi: 10.1182/blood-2012-05-431791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fox N, Priestley G, Papayannopoulou T, Kaushansky K. Thrombopoietin expands hematopoietic stem cells after transplantation. The Journal of clinical investigation. 2002;110:389–94. doi: 10.1172/JCI15430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sitnicka E, Lin N, Priestley GV, et al. The effect of thrombopoietin on the proliferation and differentiation of murine hematopoietic stem cells. Blood. 1996;87:4998–5005. [PubMed] [Google Scholar]
- 45.Kimura S, Roberts AW, Metcalf D, Alexander WS. Hematopoietic stem cell deficiencies in mice lacking c-Mpl, the receptor for thrombopoietin. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:1195–200. doi: 10.1073/pnas.95.3.1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Qian H, Buza-Vidas N, Hyland CD, et al. Critical role of thrombopoietin in maintaining adult quiescent hematopoietic stem cells. Cell stem cell. 2007;1:671–84. doi: 10.1016/j.stem.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 47.Amano H, Hackett NR, Rafii S, Crystal RG. Thrombopoietin gene transfer-mediated enhancement of angiogenic responses to acute ischemia. Circulation research. 2005;97:337–45. doi: 10.1161/01.RES.0000179534.17668.f8. [DOI] [PubMed] [Google Scholar]
- 48.Jin DK, Shido K, Kopp HG, et al. Cytokine-mediated deployment of SDF-1 induces revascularization through recruitment of CXCR4+ hemangiocytes. Nature medicine. 2006;12:557–67. doi: 10.1038/nm1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kaushansky K, Lok S, Holly RD, et al. Promotion of megakaryocyte progenitor expansion and differentiation by the c-Mpl ligand thrombopoietin. Nature. 1994;369:568–71. doi: 10.1038/369568a0. [DOI] [PubMed] [Google Scholar]
- 50.Geddis AE, Fox NE, Kaushansky K. The Mpl receptor expressed on endothelial cells does not contribute significantly to the regulation of circulating thrombopoietin levels. Experimental hematology. 2006;34:82–6. doi: 10.1016/j.exphem.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 51.Nolan DJ, Ginsberg M, Israely E, et al. Molecular signatures of tissue-specific microvascular endothelial cell heterogeneity in organ maintenance and regeneration. Dev Cell. 2013;26:204–19. doi: 10.1016/j.devcel.2013.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Butler JM, Nolan DJ, Vertes EL, et al. Endothelial cells are essential for the self-renewal and repopulation of Notch-dependent hematopoietic stem cells. Cell stem cell. 2010;6:251–64. doi: 10.1016/j.stem.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li N, Eljaafari A, Bensoussan D, et al. Human umbilical vein endothelial cells increase ex vivo expansion of human CD34(+) PBPC through IL-6 secretion. Cytotherapy. 2006;8:335–42. doi: 10.1080/14653240600845062. [DOI] [PubMed] [Google Scholar]
- 54.Yildirim S, Boehmler AM, Kanz L, Mohle R. Expansion of cord blood CD34+ hematopoietic progenitor cells in coculture with autologous umbilical vein endothelial cells (HUVEC) is superior to cytokine-supplemented liquid culture. Bone Marrow Transplant. 2005;36:71–9. doi: 10.1038/sj.bmt.1705001. [DOI] [PubMed] [Google Scholar]
- 55.Chute JP, Saini AA, Chute DJ, et al. Ex vivo culture with human brain endothelial cells increases the SCID-repopulating capacity of adult human bone marrow. Blood. 2002;100:4433–9. doi: 10.1182/blood-2002-04-1238. [DOI] [PubMed] [Google Scholar]
- 56.Chute JP, Muramoto GG, Fung J, Oxford C. Soluble factors elaborated by human brain endothelial cells induce the concomitant expansion of purified human BM CD34+CD38- cells and SCID-repopulating cells. Blood. 2005;105:576–83. doi: 10.1182/blood-2004-04-1467. [DOI] [PubMed] [Google Scholar]
- 57.Li W, Johnson SA, Shelley WC, Yoder MC. Hematopoietic stem cell repopulating ability can be maintained in vitro by some primary endothelial cells. Exp Hematol. 2004;32:1226–37. doi: 10.1016/j.exphem.2004.09.001. [DOI] [PubMed] [Google Scholar]