

Summary
An Escherichia coli strain that cannot scavenge hydrogen peroxide has been used to identify the cell processes that are most sensitive to this oxidant. Low micromolar concentrations of H2O2 completely blocked the biosynthesis of leucine. The defect was tracked to the inactivation of isopropylmalate isomerase. This enzyme belongs to a family of [4Fe-4S] dehydratases that are notoriously sensitive to univalent oxidation, and experiments confirmed that other members were also inactivated. In vitro and in vivo analyses showed that H2O2 directly oxidizes their solvent-exposed clusters in a Fenton-like reaction. The oxidized cluster then degrades to a catalytically inactive [3Fe-4S] form. Experiments indicate that H2O2 accepts two consecutive electrons during the oxidation event; as a consequence, hydroxyl radicals are not released, the polypeptide undamaged, and the enzyme is competent for reactivation by repair processes. Strikingly, in scavenger-deficient mutants the H2O2 that was generated as an adventitious by-product of metabolism (< 1 μM) was sufficient to damage these [4Fe-4S] enzymes. This result demonstrates that aerobic organisms must synthesize H2O2 scavengers to avoid poisoning their own pathways. The extreme vulnerability of these enzymes may explain why many organisms, including mammals, deploy H2O2 to suppress microbial growth.
Introduction
Virtually all organisms express peroxidases and catalases to protect themselves from hydrogen peroxide. H2O2 is continuously formed by the autoxidation of redox enzymes (reviewed in [1]), and scavenging enzymes may have originally evolved to protect cells against these internal sources of H2O2. The peroxidases and catalases are sufficiently abundant and active that they probably drive the steady-state level of intracellular H2O2 into the low nanomolar range (2). Nevertheless, it is widely suspected that even this dose of H2O2 may comprise a chronic, low-level stress that gradually debilitates cells and, in higher organisms, drives the deterioration of tissue function as part of the aging process.
Exogenous H2O2 rapidly diffuses across cell membranes [Seaver and Imlay, J.Bact. 183: 7182-7189, 2001] and can impose a much more acute stress on cells; accordingly, it is often used as a biological weapon. For example, H2O2 is formed by phagocytes and may accumulate to 10−4 M inside phagosomes that have engulfed invading bacteria. Lactic acid bacteria suppress the growth of competitors by releasing H2O2 as a primary metabolic product, achieving millimolar concentrations in lab cultures. And redox-cycling antibiotics, which are produced as microbicides both by plants and bacteria, suffuse target organisms with a continuous stream of H2O2.
If we wish to understand the severity and nature of the stress that H2O2 imposes upon cells, we must identify the biomolecules with which it primarily reacts. This problem has not been easy to solve. In vitro studies have shown that H2O2 can oxidize methionine (3) and cysteine (4) residues, but the rates with which it does so suggest that these types of damage will be scant at physiological doses of H2O2, unless the surrounding polypeptide context somehow strongly activates the residues. Reactions between H2O2 and loosely bound iron generate hydroxyl radicals and are suspected of being involved in protein carbonylation, lipid peroxidation, and DNA oxidation (5):
| (Eq. 1) |
Early measurements indicated that this reaction (the Fenton reaction) is relatively slow as well (6), prompting some workers to question its significance in real-world scenarios (discussed in [5]); however, subsequent work revealed that anionic ligands activate ferrous iron to the point that it reacts quickly with micromolar H2O2 (7, 8).
An alternative approach to pinpointing the important targets of H2O2 is to expose cells to increasing doses in a way that identifies the first cell processes to fail. E. coli and other organisms have calibrated their defensive systems to detect submicromolar levels of H2O2 (2,9), so we anticipate that these low concentrations are sufficient to threaten the most sensitive biomolecules. Unfortunately, if scavenging enzymes are active, it is difficult to impose such a low dose of H2O2 over an extended period of time, since the enzymes will degrade the H2O2 and end the stress. Therefore, these experiments are most easily conducted with scavenger-deficient mutants. We have constructed E. coli strains that lack peroxidase and catalase activities (10). These mutants grow at wild-type rates in anaerobic environments, but when they are exposed to oxygen they grow at reduced rates in complex medium, and they fail to grow at all in a minimal medium. The former defect is due, at least in part, to Fenton-mediated DNA damage (8). The second defect stems from problems with biosynthetic pathways. In this study we identify the mechanism by which micromolar H2O2 blocks leucine biosynthesis, and we find that this class of injury affects multiple pathways in the cell.
Experimental procedures
Strains and culture conditions
Strains and plasmids used in this study are listed in Table 1. Anaerobic cultures were grown in an anaerobic chamber (Coy Laboratory Products, Inc.), and aerobic cultures were grown with vigorous shaking in a water bath at 37°C. Standard minimal medium contained minimal A salts (11), 0.2% glucose, 1 mM MgCl2, 5 mg/liter thiamine, and 0.5 mM each of histidine, phenylalanine, tyrosine, and tryptophan. Histidine was always added to the media because the parent strain, MG1655, is a histidine auxotroph anaerobically; to minimize the difference between anaerobic and aerobic cultures, histidine was also added to aerobic cultures. Where indicated, lactose (0.2%), gluconate (0.2%), or malate (40 mM) was substituted for glucose. Where indicated, other amino acids, α-ketoisovalerate (TCI America), or α-ketoisocaproate (Sigma) were present at 0.5 mM.
Table 1.
strains and plasmids
| Strain or plasmid | Genotype or characteristics | Source or ref. |
|---|---|---|
| MG1655 | Wild type E. coli | (43) |
| LC106 | Δ(katG17::Tn10)1 Δ(ahpC-ahpF') kan::'ahpF Δ(katE12::Tn10)1 | (10) |
| JH400 | Δ(dgs-fumCA-manA)1 Δ(mel-fumB) zdg232::Tn10 metB1 spoT1 relA | John Guest |
| OD570 | ΔfumC::cm | (19) |
| OD571 | ΔfumCA::cm | (19) |
| SJ19 | ΔfumC::cm in LC106 | This study |
| SJ20 | ΔfumAC::cm in LC106 | This study |
| SJ34 | ΔfumB::cm in BW25113 | This study |
| SJ37 | As LC106 plus ΔfumC ΔfumB::cm | This study |
| SJ38 | As LC106 plus ΔfumAC ΔfumB::cm | This study |
| BW25113 | lacIq rrnBT14 ΔacZWJ16 hsdR514 ΔaraBADAH33 ΔrhaBADLD78 | (12) |
| pWKS30 | Plac polylinker Amr | (18) |
| pLEUCD2 | pWKS30 containing leuCD insert | This study |
| pCKR101 | Plac–lacIq Ptac polylinker Amr | Jeff Gardner |
| pLEUCD3 | pCKR101 containing leuCD insert | This study |
| pFUMA | pCKR101 containing fumA insert | This study |
| pCP20 | temperature-sensitive FLP expression plasmid | (44) |
| pKD3 | FRT-flanked cat | (12) |
| pKD46 | λ Red recombinase expression plasmid | (12) |
Mutations were introduced into new strains by P1 transduction (11). To create a null mutation of fumB using the Red recombinase method (12), the forward primer 5’-TAACAAATACAGAGTTACAGGCTGGAAGCTGTAGGCTGGAGCTGC and the reverse primer 5’-AGCATGCTGCCAGGCGCTGGGCCGAAGAGGATATGAATATCCTCC were used. Mutations were confirmed by PCR analysis and enzyme assays.
Aerobic cell growth
To ensure that cells were growing exponentially before they were exposed to oxygen, anaerobic overnight cultures of Hpx− cells were diluted to an OD600 of 0.005 in fresh anaerobic minimal glucose medium. Cells were then grown anaerobically to an OD600 of approximately 0.1 prior to dilution into aerobic medium.
Enzyme assays
Cell extracts were prepared by suspending and sonicating cells in anaerobic buffers inside an anaerobic chamber. Isopropylmalate isomerase (IPMI) activity was measured by monitoring the decrease in the absorbance (235 nm) of citraconate (Sigma) (13), an analogue of isopropylmalate; reactions contained 100 mM Tris-Cl (pH 7.6) and 0.4 mM citraconate. Fumarase activity was determined from the appearance of fumarate (250 nm) in a reaction containing 50 mM sodium phosphate (pH 7.4) and 30 mM malate (Sigma) (14). To assay 6-phosphogluconate dehydratase, lysates were prepared from cultures grown in a minimal medium containing 0.2% gluconate. Turnover of 6-phosphogluconate dehydratase produces pyruvate; its formation (in 50 mM Tris-Cl, pH 7.65) was determined in a second reaction catalyzed by lactate dehydrogenase (Sigma) (15). To assay NADH dehydrogenase I (Ndh1), inverted membrane vesicles were isolated from extracts after sonication in 50 mM MES, pH 6.0 (16). NdhI oxidizes deaminoNADH (340 nm) in 50 mM potassium phosphate buffer (pH 7.0), whereas NADH dehydrogenase II does not. Aconitase was assayed by the conversion of isocitrate to aconitate (17).
Plasmid construction
The leuCD ORF was PCR-amplified from E. coli MG1655 by using the forward primer 5′-ATATCGAATTCTTAACGATATCATTGCCCGCTATATAGCAG-3′ and the reverse primer 5’-CTGGATCTAGATTAATTCATAAACGCAGGTTG-3’. To construct the pLEUCD2 plasmid, the PCR products were digested with XbaI and EcoRI and cloned into pWKS30 (18) vector behind the lac promoter. The plasmid was confirmed by restriction analysis and by the isopropylmalate isomerase assay. Hpx− cells containing pLEUCD2 or pWKS30 were cultured in lactose minimal medium with histidine, aromatic amino acids, and 50 μg/ml ampicillin. This plasmid was used in complementation experiments; the leuCD genes were induced three-fold above wild-type levels by 1 mM IPTG, which was added to both anaerobic precultures and final aerobic cultures.
For EPR studies and purification purposes, IPMI and fumarase A were strongly overproduced by expression from a tac promoter. The leuCD genes were excised from pLEUCD2 with XbaI and EcoRI and cloned into pCKR101 vector to construct pLEUCD3. The fumA ORF was PCR-amplified from E. coli MG1655 by using the forward primer 5′-ATATCGAATTCTTAACATAACCAAACCAGGCAGTAAGTG-3′ and the reverse primer 5’-CATGGATCTAGATTATTTCACACAGCGGGTGCATTG-3. PCR products were digested with XbaI and EcoRI and cloned into the pCRK101 vector to construct pFUMA. Cells containing the plasmids were cultured in glucose minimal medium with histidine, aromatic amino acids, and 50 μg/ml of ampicillin. These plasmids overproduced IPMI and fumarase A more than 60-fold above wild-type levels.
Inactivation of enzymes
Hpx− cells were grown anaerobically to 0.2 (OD600). H2O2 was added to the cultures when they were aerated. At time points, aliquots were removed, catalase was added to 200 U/ml, and cells were returned to the anaerobic chamber for lysis and assay.
In vivo inactivation of IPMI by endogenous H2O2 was initiated by aerating heretofore anaerobic cultures without any addition of exogenous H2O2. Under these conditions the cells steadily generate H2O2. The H2O2 equilibrates so quickly across membranes that in Hpx− cultures the intracellular H2O2 concentration is essentially equivalent to the extracellular concentration (Seaver and Imlay, J. Bacteriol. 183: 7182-7189, 2001). The extracellular H2O2 was measured directly by the amplex red/horseradish peroxidase method (2). At the same time, extracts were prepared and assayed in the anaerobic chamber.
The inactivation of enzymes in vitro was accomplished by the addition of H2O2 to lysates or to purified enzyme in anaerobic buffer. The H2O2 was subsequently removed by catalase prior to anaerobic assay. In some cases, damaged iron-sulfur clusters were chemically rebuilt by incubation with 50 μM Fe(NH4)2(SO4)2 (Sigma) and 2.5 mM dithiothreitol (17) (Sigma) at room temperature.
EPR analysis
In vivo EPR samples were prepared with Hpx− cells that overproduce IPMI or fumarase A. To overexpress the structural genes, 1 mM IPTG was added when cells reached an OD600 of 0.2. After another 2 hours of incubation, the cells were harvested by centrifugation, and the cell pellets were resuspended in 1/500th of original culture volume in 10 % glycerol. The resuspended cells were incubated with H2O2 for 1 min at 37°C. The cell suspension (250 μl) then was transferred into an EPR tube and frozen in dry ice.
EPR spectra of [3Fe-4S]+ clusters (19) were obtained with the following settings: microwave power, 1 milliwatt; microwave frequency, 9.05 GHz; modulation amplitude, 8 Gauss at 100 KHz; time constant, 0.032.
Purification of enzymes
Four-liter cultures of the Hpx− mutant that overproduces fumarase A were grown anaerobically to an OD600 of 0.2 at 37°C. Expression of fumA was induced by incubation with 1 mM IPTG for 2 hours at 37°C. The cultures were harvested by centrifugation, the cell pellets were resuspended in 15 ml of anaerobic 50 mM Tris-Cl/10 mM Mg2+ (pH 8) and disrupted by sonication, and cell debris was removed by centrifugation. The purification procedure was derived by the protocol described by Flint (20). All steps of the purification were conducted in an anaerobic chamber at room temperature, and all buffers were anaerobic. Protamine sulfate (1 %) was added to the supernatant to remove nucleic acids. The supernatant was then loaded onto a DEAE-Sepharose column (16 mm by 140 mm) and eluted by a 0–1 M gradient of KCl in 50 mM Tris-Cl/10 mM Mg2+. The fractions containing the highest activity were pooled. Ammonium sulfate (1.5 M) was added, precipitate was removed by centrifugation, and the supernatant was loaded onto a phenyl sepharose column (16 mm by 110 mm). The fractions were eluted by a 1.5 – 0 M gradient of ammonium sulfate in 50 mM Tris-Cl/10 mM Mg2+. The fractions containing the highest activity were pooled and concentrated (Amicon). The enzyme was loaded onto a Superdex column (16 mm by 50 mm). The fractions were eluted with 500 mM KCl in 50 mM Tris-Cl/10 mM Mg2+. The three fractions containing the highest activity were frozen in dry containing ethanol. Purified fumarase A was more than 90% pure based on SDS-PAGE. The enzyme was quantified by dye-binding assay using bovine serum albumin as a standard and the conversion factor determined by Flint (20).
Aconitase A was overproduced and purified as described (17).
Detection of iron released upon cluster oxidation
EPR spectroscopy was used to quantitatively correlate the creation of [3Fe-4S]+ clusters with the release of iron when FumA was damaged in vitro. Desferrioxamine (1 mM, Sigma) was added to purified fumarase A (18 μM) immediately prior to the addition of H2O2. The sample was then transferred into an EPR tube and frozen. The [3Fe-4S]+ signal was detected as described above, while EPR spectra of free iron were obtained with the following settings: microwave power, 10 milliwatt; microwave frequency, 9.05 GHz; modulation amplitude, 12.5 Gauss at 100 KHz; time constant, 0.032. Iron concentrations were quantified using standard solutions of FeCl3 (Sigma) in 50 mM Tris-Cl containing 10 mM Mg2+ and 1 mM desferrioxamine.
Desferrioxamine binds both ferric and ferrous iron and triggers the oxidation of the latter species. Thus the EPR method did not distinguish whether the released iron was in the ferric or ferrous form.
Ferene is commonly used to detect ferrous iron that is released by metalloenzymes, but we found that ferene itself directly inactivated fumarase A. Dipyridyl did not. Dipyridyl binds ferrous iron in a complex that exhibits an absorbance maximum at 522 nm (21); we determined that this complex cannot be oxidized by H2O2. To detect Fe2+ that is released during cluster decomposition, 1 mM dipyridyl (Sigma) was added to 18 μM of fumarase A prior to H2O2 addition. Control experiments confirmed that this concentration of dipyridyl captured ferrous iron before the H2O2 could oxidize it: when Fe(NH4)2(SO4)2 was added to the H2O2-containing reaction mixture, we were able to quantitatively recover it as a dipyridyl chelate, and we could accurately detect as little as 2 μM.
Protein mass spectroscopy
Fumarase A (3 μM) was treated with 5μM H2O2 for 2 min at room temperature
The reaction was terminated by the addition of 0.1 U/μl catalase. Assays showed that >98 % of the enzyme had been inactivated. For mass spectroscopic analysis, 2 μg of sample was desalted using the Genotech (St. Louis) Perfect Focus 2-D sample Clean up kit. The desalted sample was suspended in 25 mM ammonium bicarbonate containing 12.5 ug/ml trypsin and was incubated for 12 hours at 37 °C. The sample was then dried, suspended in 5% acetonitrile/0.1% formic acid, and injected into a quadrupole time-of-flight mass spectrometer (Waters Q-ToF) via an HPLC interface. Peptide masses were detected using a data-dependent method and were subjected to MS/MS for de novo sequencing and characterization of post-translational modifications. Analysis was done using Waters Protein Lynx Global Server 2.1, MASCOT (Matrix Sciences), and PEAKS (Bioinformatics Solutions, Inc.)
Results
Hydroperoxidase mutants that cannot scavenge H2O2 (katG katE ahp, here denoted Hpx−) grow well in anaerobic glucose minimal medium but stop growing when they are aerated. The cells resume growth when aromatic amino acids are supplied, indicating that endogenously formed H2O2 poisons some step in the aromatic biosynthetic pathway. The mechanism is unknown and is the subject of a separate investigation. However, aromatic supplements do not fully restore these mutants to a wild-type growth rate, and the addition of small amounts of exogenous H2O2 exacerbates the residual defect. Eight micromolar H2O2 competely blocked growth in this medium (Fig. 1A). Supplementation with casamino acids restored growth, implying that stasis was a result of a second amino-acid biosynthetic defect. Experiments using different amino acids determined that leucine was the critical one.
Figure 1. Low concentrations of H2O2 block leucine biosynthesis.

(A) Hpx− cells were cultured in aerobic glucose medium. Where indicated, 8 μM H2O2, branched-chain amino acids (BCAA: Leu, Ile, and Val), and/or leucine were added. (B) Hpx− cells containing either pLEUCD2 (overproducing isopropylmalate isomerase) or pWKS30 (an empty vector) were cultured in lactose minimal medium with or without leucine supplementation. All media included histidine and aromatic amino acids. The pLEUCD2 plasmid boosted the isopropylmalate isomerase activity of anaerobic cultures about three-fold.
Hpx− growth in the presence of H2O2 was improved by supplementation with exogenous α-ketoisocaproate, the final intermediate in the leucine pathway, but not by α-ketoisovalerate, the first one (data not shown). There are five reactions between the two intermediates, catalyzed by four enzymes. We anticipated that H2O2 might inhibit or inactivate one of them. Indeed, the growth defect was partially relieved by a plasmid that overproduces isopropylmalate isomerase three-fold (Fig. 1B), suggesting that this is the rate-limiting enzyme in the pathway during H2O2 stress.
Enzyme assays confirmed that isopropylmalate isomerase (IPMI) lost activity rapidly when 8 μM H2O2 was added to cultures (Fig. 2A). Furthermore, significant enzyme inactivation occurred upon aeration even without the addition of exogenous H2O2 (Fig. 2B), indicating that < 0.4 μM intracellular H2O2 was sufficient to poison a substantial fraction of IPMI. Thus this enzyme is exquisitely sensitive, and scavenging enzymes are needed to protect it from endogenously formed H2O2.
Figure 2. H2O2 rapidly inactivates isopropylmalate isomerase in vivo.

(A) Hpx− cells were anaerobically cultured to an OD600 of 0.2, and 8 μM H2O2 was then added to the cultures. At intervals catalase was added and residual enzyme activity was determined. (B) Hpx− cells were cultured in anaerobic medium and then aerated starting at time zero. Isopropylmalate isomerase activity (squares) and H2O2 concentrations (circles) were monitored.
The nature of IPMI damage
IPMI activity was lost when H2O2 was added to anaerobically prepared Hpx− extracts (Fig. 3A), indicating that inactivation occurs by direct action of H2O2 upon the enzyme. Catalase protected completely (data not shown).
Figure 3. H2O2 inactivates isopropylmalate isomerase by converting its [4Fe-4S]2+ cluster to a [3Fe-4S]+ cluster in vitro.

(A) An extract of anaerobically grown Hpx− cells was exposed to the indicated concentrations of H2O2 for 2 min at room temperature. Catalase then was added to remove H2O2. The experiment was conducted anaerobically. (B) Isopropylmalate isomerase from anaerobically cultured Hpx− cells was inactivated with 2 μM H2O2 for 5 min. Catalase was added to remove H2O2, and then 50 μM Fe(NH4)2(SO4)2 and 2.5 mM dithiothreitol were added. The activity was measured after 3 min incubation. (C) Hpx− cells that overproduce IPMI were harvested when the cells were OD600 of 0.2. The cell pellets were resuspended in 1/500 of original culture volume of 10% glycerol. The resuspended cells were incubated with 100 μM H2O2 for 1 min at 37°C. The cell suspension (250 μl) then was transferred into an EPR tube and frozen.
IPMI is a dehydratase, and its protein sequence suggests that it belongs to family of enzymes that employ [4Fe-4S] clusters as active-site catalysts (22). The solvent-exposed clusters of these enzymes both coordinate substrate and act as Lewis acids in abstracting the hydroxide anion from the bound substrate (23). These enzymes are typified by aconitase, and they are notoriously sensitive to inactivation by univalent oxidants such as superoxide and ferricyanide (15, 24-27). These agents abstract a single electron from the cluster, converting it to a [4Fe-4S]3+ form. The cluster is unstable in that valence and releases the substrate-binding iron atom as Fe2+, so that the residual cluster is left in a [3Fe-4S]+ form that lacks the key iron atom and is catalytically inactive.
Consistent with this model, the addition of citraconate, a pseudosubstrate of IPMI, protected the enzyme from H2O2 in vitro, suggesting that H2O2 must directly contact the cluster in order to inactivate the enzyme (data not shown). Damaged clusters can often be reassembled chemically by treatment with dithiothreitol and ferrous iron; when damaged IPMI was subjected to this protocol, 60% of the activity was regained within three minutes (Fig. 3B).
IPMI was overexpressed inside Hpx− cells, and cells were then exposed to H2O2. A strong [3Fe-4S]+ signal appeared (Fig. 3C) which was absent from non-overproducing controls. This result confirmed both that IPMI has a cluster and that it is destroyed by H2O2. In fact, ca. 85% of the IPMI activity was recovered when the extracts from H2O2-exposed cells were treated with dithiothreitol and ferrous iron.
Other [4Fe-4S] dehydratases are similarly sensitive to H2O2
Other cluster-containing dehydratases—6-phosphogluconate dehydratase and fumarases A and B--also lost activity when they were exposed to low concentrations of H2O2 in vitro (Fig. 4A, B, and C). In each case full activity could be restored by subsequent treatment with dithiothreitol and ferrous iron (data not shown). Further, a strong [3Fe-4S]+ signal was detected when H2O2 was added to Hpx− cells that overproduced fumarase A (Fig. 5). Thus it appears that H2O2 efficiently damages the clusters of all members of this enzyme family.
Figure 4. H2O2 inactivates other [4Fe-4S] dehydratases.

(A) 6-phosphogluconate dehydratase. (B) Fumarase A. (C) Fumarase B. Lysates were prepared from anaerobic cultures of LC106, SJ37, and SJ20, respectively, and the indicated concentrations of H2O2 were added for 5 min. Catalase was added to terminate the stress, and residual activity was determined.
Figure 5. H2O2 inactivates fumarase A by converting its [4Fe-4S]2+ cluster to a [3Fe-4S]+ cluster in vivo.

SJ37 cells that overproduce fumarase A were harvested at an OD600 of 0.2, washed, and resuspended at 1/500 of original culture volume in 10 % glycerol. The resuspended cells were incubated with 200 or 20 μM H2O2 for 1 min at 37°C. The cell suspension (250 μl) then was transferred into an EPR tube and frozen.
Glucose medium, which we employed for our initial growth studies, does not demand that E. coli process substrate through its TCA cycle in order to generate ATP; therefore, growth would not have been affected by inactivation of aconitase or fumarase. However, the Hpx− strain exhibited a severe growth defect when it was cultured in malate medium (Fig. 6). This result indicates that the submicromolar H2O2 that is generated by endogenous processes is sufficient to debilitate this pathway.
Figure 6. The Hpx− mutant is defective at using malate as the sole carbon source.

Wild-type (stars) and Hpx− (open circles) cells were cultured in aerobic glucose medium containing to 0.15 – 0.25 (OD600). At the arrow aliquots were removed, washed, and resuspended in aerobic malate medium. All media included histidine and aromatic amino acids.
Iron-sulfur clusters are also used by respiratory enzymes to transfer electrons between active sites. The NADH dehydrogenase I complex is characteristic of these enzymes, as it contains at least 9 Fe-S clusters (28). However, even 5 mM H2O2 was unable to diminish its activity (data not shown). Because these clusters are buried within polypeptide, the implication is that H2O2 can only oxidize those clusters which it can contact directly.
The mechanism of cluster inactivation
IPMI is a two-subunit enzyme that dissociates during purification. Therefore, fumarase A and aconitase A were chosen for purification and further examination. The isolated enzymes were acutely sensitive to H2O2, exhibiting inactivation rate constants of 4 × 103 M−1 s−1 and 3 × 102 M−1 s−1, respectively, at 0° C. The fumarase inactivation constant was 0.5-1 × 105 M−1 s−1 at 25° C, and inactivation at 37° C was too fast for us to measure.
Strikingly, the fumarase rate constants were orders of magnitude higher than the apparent constant that we calculated in vivo using Hpx− mutants. We suspected that substrates protect the enzyme inside the cell. Indeed, both malate and fumarate fully protected fumarase when they were added in saturating concentrations (Fig. 7). The doses needed for half-maximal protection (0.4 mM for malate and 0.3 mM for fumarate, measured at 0° C [Ref: Flint, D.H. Initial kinetic and mechanistic characterization of Escherichia coli fumarase A. Arch. Biochem. Biophys. 311: 509-516, 1994]) were in reasonable agreement with the Km's of the enzyme for those substrates (0.7 and 0.6 mM respectively, at 37° C). Thus it is likely that the inactivation of the enzyme in vivo is tempered by the consequent accumulation of substrate. These data also support the suspicion that H2O2 must intimately contact the cluster in order to inactivate it.
Figure 7. Substrates protect fumarase against H2O2.
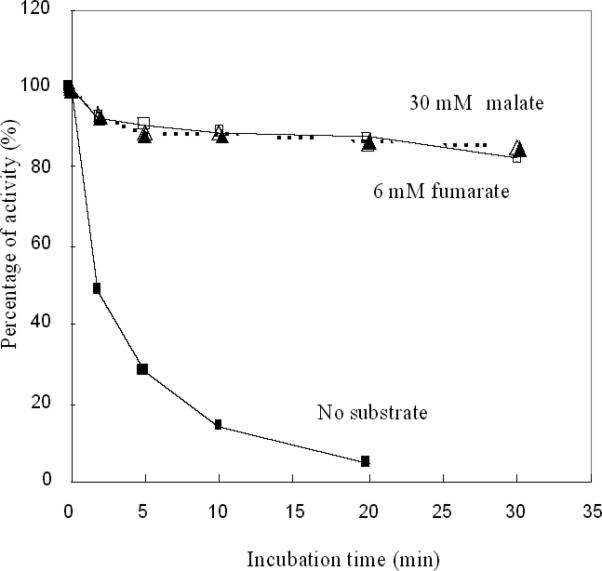
Purified fumarase A (5 nM) was exposed to 1 μM H2O2 at 0 °C. Where indicated 30 mM malate or 6 mM fumarate were included in the buffer. At time points, aliquots were removed, catalase was added, and residual activity was measured.
The standard model of univalent cluster oxidation posits that a [3Fe-4S]+ form is initially generated with release of one ferrous ion (27). However, if the oxidant is H2O2, then a hydroxyl radical should also be formed.
| (Eq. 2) |
| (Eq. 3) |
Hydroxyl radicals react with most organic biomolecules, including amino acids, at nearly diffusion-limited rates; therefore, if one were formed within the active site of a dehydratase, the likely consequence would be the direct oxidation of the protein. However, we were able to quantitatively reactivate H2O2-treated fumarase (data not shown), which seemed inconsistent with the oxidation of active-site residues. SDS-PAGE analysis of the protein showed that the polypeptide chain was not cleaved. Mass spectroscopy failed to detect any oxidation of histidyl, lysyl, prolyl, or methionyl residues (data not shown).
An alternative model is that a ferryl-like species is generated by the initial electron transfer from the cluster to liganded H2O2, and that the ferryl radical then abstracts a second electron from the cluster rather than release a hydroxyl radical:
| (Eq. 4) |
| (Eq. 5) |
To test this possibility, we used desferrioxamine and dipyridyl to quantify the release of total iron and ferrous iron, respectively. Ferrous iron is generated by the first mechanistic scheme (eq. 3) but not by the second (eq. 6). While we detected stoichiometric release of one iron atom per cluster (Fig. 8), none of this iron was in the ferrous form (< 0.1 atom/cluster) (data not shown). From this result, and from the absence of polypeptide oxidation, wee infer that a free hydroxyl radical is not generated.
Figure 8. H2O2 degrades the [4Fe-4S]2+ cluster of purified fumarase A to a [3Fe-4S]+ cluster with loss of one iron atom.

Purified fumarase A (18 μM) was inactivated by 100 μM H2O2 in 50 mM Tris-Cl/10 mM Mg2+ containing 1 mM desferrioxamine. At time points, aliquots were removed, catalase was added, and the reaction mixture was frozen. The residual cluster (panel A) and released ferric iron (panel B) were analyzed by EPR spectroscopy.
Previous experiments, which were conducted with millimolar doses of oxidant, indicated that with time the cluster disintegration might continue past the [3Fe-4S]+ state (19). However, the ease with which we were able to chemically reactivate damaged dehydratases was inconsistent with the formation of apoprotein, which is only slowly reactivated. In fact, EPR analysis showed that the [3Fe-4S]+ fumarase cluster was unaffected by a ten-minute exposure to 100 μM H2O2 (Fig. 8A). We conclude that over this period physiological doses of H2O2 do not directly degrade clusters beyond the easily repaired [3Fe-4S]+ state.
Discussion
The experiments reported here reveal that sub-micromolar concentrations of H2O2 are sufficient to destroy enzymic iron-sulfur clusters. Multiple catabolic and biosynthetic pathways are thereby disrupted. Earlier work with the same strain demonstrated that the endogenous H2O2 also reacts with unincorporated iron to produce high levels of DNA damage (8). The two phenotypes are of a piece in that both result from Fenton-type reactions.
The sensitivity of Fe-S clusters to univalent oxidants has been appreciated for some time; in fact, much of the toxicity of superoxide is due to inactivation of the same enzymes that were identified in this study. Damage by millimolar doses of H2O2 has been reported (17,29). However, while the chemistry is not unexpected, the surprise is that these reactions occur so rapidly. The rate constant of the Fenton reaction was measured to be 76 M−1 s−1 at pH 3, and this number has been cited widely (6). It carried with it the implication that reactions between iron and H2O2 are too slow to occur at an important rate in most biological scenarios, where H2O2 concentrations are micromolar or lower. However, more-recent measurements have shown that at physiological pH the rate constant for hexaqueous iron is actually orders of magnitude higher, presumably because at neutral pH hydroxide anion coordinates iron and lowers its reduction potential. Extrapolating from measurements made at lower temperatures (8), we estimate that the rate may be 20,000-30,000 M−1 s−1 at 37° C. The same enhancement evidently pertains to the substrate-binding iron atom within a dehydratase iron-sulfur cluster.
The biological consequence of this high reactivity is that far less H2O2 is needed to create toxicity than had been suspected. The data directly show that E. coli must synthesize scavenging enzymes to avoid being poisoned by the high-nanomolar H2O2 that it makes through the adventitious oxidation of its own redox enzymes. Basal levels of the NADH peroxidase solve that problem. However, this basic vulnerability is still exploited by phagocytes and plants, both of which suppress the growth of invading bacteria by generating far higher levels of H2O2, either directly or through the synthesis of redox-active antibiotics (30-33).
These deliberate oxidative assaults, as well as the H2O2 that accumulates in the environment through chemical oxidation processes, are a true threat to bacteria. Because H2O2 influx across membranes can outstrip the action of scavenging enzymes, extracellular concentrations of 5 μM H2O2 are sufficient to raise the intracellular H2O2 in wild-type (scavenger-proficient) cells to about 1 μM (2). Therefore, as little as 5 μM of environmental H2O2 is sufficient to enzyme and pathway defects that werereported in this study.
Interestingly, lactic acid bacteria employ a similar strategem to inhibit competing organisms, elaborating lactate and pyruvate oxidases which can drive H2O2 levels in their immediate environment up to millimolar levels (34,35). How do the lactic acid bacteria themselves tolerate the stress that they are creating? Lactic-acid bacteria do not rely upon pathways that contain Fe-S dehydratases. Instead, they obtainfrom their environment the amino acids that dehydratase-dependent biosynthetic pathways would normally produce, and they ferment sugars to lactatic acid rather than using the TCA cycle.
To defend themselves against such H2O2-mediated assaults, microbes throughout the biotic world have evolved H2O2-inducible defenses, most of which are either homologues or analogues of the OxyR system of E. coli. The front-line defense of these systems is the strong induction of scavenging enzymes, but the repair of damaged clusters is also an important feature. Ongoing cluster repair may contribute to the disparity between the rates of enzyme damage that we measured in vitro and the apparent rates that we determined in vivo. In previous studies we had observed that cells quantitatively repair H2O2-damaged dehydratases (19), but we were perplexed as to why the hydroxyl radical, which presumably had been formed upon cluster oxidation, did not irreversibly damage active-site amino-acid residues. This puzzle is resolved by the observation that ferric (rather than ferrous) iron is released during cluster oxidation. Therefore a ferryl radical--or a non-diffusing hydroxyl radical--evidently pulls a second electron from the residual cluster rather than allowing an unexpended hydroxyl radical to be released into the active-site bulk solution. Interestingly, an analogous phenomenon was noted for PerR protein of Bacillus subtilis, in that a Fenton reaction between its prosthetic ferrous iron atom and H2O2 caused immediate oxidation of the iron ligands, rather than the more widely distributed oxidation of the polypeptide as might have been expected from a diffusible hydroxyl radical.
In the dehydratase case we do not stipulate whether the second electron transfer would generate an intermediate all-ferric [4Fe-4S]4+ cluster or if it would create a disulfide bond concomitant with ferric iron release. However, in either case polypeptide oxidation would be avoided. The physiological significance is that the enzyme can be restored to its native state by cellular processes that reduce and remetallate the [3Fe-4S]+ cluster. The components that catalyze that repair process have not yet been identified. However, the OxyR system responds to H2O2 by inducing the Suf proteins (36), which comprise a backup system for cluster assembly.
A secondary consequence of cluster damage is that iron is released in an uncontrolled manner into the cytosol. Loose iron can bind to DNA and catalyze the formation of damaging hydroxyl radicals; therefore, this iron leak has the effect of accelerating the rate at which H2O2 produces DNA damage (37-39). The OxyR system addresses this threat by inducing the synthesis of Dps, a ferritin-like storage protein that scavenges loose iron and sequesters it into an unreactive ferric hydroxide core (8,40-42). Thus it has become clear that several aspects of the inducible defense against H2O2 are well-matched to the threat that H2O2 poses.
Acknowledgements
We thank Mark Nilges of the Illinois EPR Research Center for assistance with EPR experiments, Peter Yau of the University of Illinois Biotechnology Center for assistance with protein mass spectrometry, and John Guest for generously providing strains that were used in this study. This work was supported by grant GM49640 from the National Institutes of Health.
References
- 1.Imlay JA. Ann. Rev. Microbiol. 2003;57:395–418. doi: 10.1146/annurev.micro.57.030502.090938. [DOI] [PubMed] [Google Scholar]
- 2.Seaver LC, Imlay JA. J. Bacteriol. 2001;183:7182–7189. doi: 10.1128/JB.183.24.7182-7189.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Griffiths SW, Cooney CL. Biochemistry. 2002;41:6245–6252. doi: 10.1021/bi025599p. [DOI] [PubMed] [Google Scholar]
- 4.Winterbourn CC, Metodiewa D. Free Rad. Biol. Med. 1999;27:322–328. doi: 10.1016/s0891-5849(99)00051-9. [DOI] [PubMed] [Google Scholar]
- 5.Halliwell B, Gutteridge JMC. Meth. Enzymol. 1990;186:1–85. doi: 10.1016/0076-6879(90)86093-b. [DOI] [PubMed] [Google Scholar]
- 6.Walling C. Accounts of Chemical Research. 1975;8:125–131. [Google Scholar]
- 7.Rush JD, Maskos Z, Koppenol WH. FEBS Letters. 1990;261:121–123. [Google Scholar]
- 8.Park S, You X, Imlay JA. Proc. Natl. Acad. Sci. USA. 2005;102:9317–9322. doi: 10.1073/pnas.0502051102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aslund F, Zheng M, Beckwith J, Storz G. Proc. Natl. Acad. Sci. USA. 1999;96:6161–6165. doi: 10.1073/pnas.96.11.6161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seaver LC, Imlay JA. J. Bacteriol. 2001;183:7173–7181. doi: 10.1128/JB.183.24.7173-7181.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller JH. Experiments in Molecular Genetics. Cold Spring Harbor Laboratory; Cold Spring Harbor, N.Y.: 1972. [Google Scholar]
- 12.Datsenko KA, Wanner BL. Proc. Natl. Acad. Sci. USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kohlhaw GB. Meth. Enzymol. 1988;166:423–429. doi: 10.1016/s0076-6879(88)66055-1. [DOI] [PubMed] [Google Scholar]
- 14.Massey V. Meth. Enzymol. 1955;1:729–735. [Google Scholar]
- 15.Gardner PR, Fridovich I. J. Biol. Chem. 1991;266:1478–1483. [PubMed] [Google Scholar]
- 16.Matsushita K, Ohnishi T, Kaback HR. Biochemistry. 1987;26:7732–7737. doi: 10.1021/bi00398a029. [DOI] [PubMed] [Google Scholar]
- 17.Varghese SM, Tang Y, Imlay JA. J. Bacteriol. 2003;185:221–230. doi: 10.1128/JB.185.1.221-230.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang RF, Kushner SR. Gene. 1991;100:195–199. [PubMed] [Google Scholar]
- 19.Djaman O, Outten FW, Imlay JA. J. Biol. Chem. 2004;279:44590–44599. doi: 10.1074/jbc.M406487200. [DOI] [PubMed] [Google Scholar]
- 20.Flint DH, Emptage MH, Guest JR. Biochemistry. 1992;31:10331–10337. doi: 10.1021/bi00157a022. [DOI] [PubMed] [Google Scholar]
- 21.Mason EC, Adarraga-Elizaran A. J. Clin. Pathol. 1963;16:604–606. doi: 10.1136/jcp.16.6.604-b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hentze MW, Argos P. Nucl. Acids Res. 1991;19:1739–1740. doi: 10.1093/nar/19.8.1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lauble H, Kennedy MC, Beinert H, Stout CD. Biochemistry. 1992;31:2735–2748. doi: 10.1021/bi00125a014. [DOI] [PubMed] [Google Scholar]
- 24.Gardner PR, Fridovich I. J. Biol. Chem. 1991;266:19328–19333. [PubMed] [Google Scholar]
- 25.Kuo CF, Mashino T, Fridovich I. J. Biol. Chem. 1987;262:4724–4727. [PubMed] [Google Scholar]
- 26.Liochev SI, Fridovich I. Proc. Natl. Acad. Sci. USA. 1992;89:5892–5896. doi: 10.1073/pnas.89.13.5892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Flint DH, Tuminello JF, Emptage MH. J. Biol. Chem. 1993;268:22369–22376. [PubMed] [Google Scholar]
- 28.Ohnishi T. Biochim. Biophys. Acta. 1998;1364:186–206. doi: 10.1016/s0005-2728(98)00027-9. [DOI] [PubMed] [Google Scholar]
- 29.Brown NM, Kennedy MC, Antholine WE, Eisenstein RS, Walden WE. J. Biol. Chem. 2002;277:7246–7254. doi: 10.1074/jbc.M110282200. [DOI] [PubMed] [Google Scholar]
- 30.Klebanoff SJ, Locksley RM, Jong EC, Rosen H. Ciba Found. Symp. 1983;99:92–112. doi: 10.1002/9780470720806.ch6. [DOI] [PubMed] [Google Scholar]
- 31.Wojtaszek P. Biochem. J. 1997;15:681–692. doi: 10.1042/bj3220681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Inbaraj JJ, Chignell CF. Chem. Res. Toxicol. 2004;17:55–62. doi: 10.1021/tx034132s. [DOI] [PubMed] [Google Scholar]
- 33.Ran H, Hassett DJ, Lau GW. Proc. Natl. Acad. Sci. USA. 2003;100:14315–14320. doi: 10.1073/pnas.2332354100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ito A, Sato Y, Kudo S, Sato S, Nakajima H, Toba T. Curr. Microbiol. 2003;47:231–236. doi: 10.1007/s00284-002-3993-1. [DOI] [PubMed] [Google Scholar]
- 35.Pericone CD, Park S, Imlay JA, Weiser JN. J Bacteriol. 2003;185:6815–6825. doi: 10.1128/JB.185.23.6815-6825.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zheng M, Wang X, Templeton LJ, Smulski DR, LaRossa RA, Storz G. J. Bacteriol. 2001;183:4562–4570. doi: 10.1128/JB.183.15.4562-4570.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Touati D, Jacques M, Tardat B, Bouchard L, Despied S. JBact. 1995;177:2305–2314. doi: 10.1128/jb.177.9.2305-2314.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liochev SI, Fridovich I. Free Rad Biol Med. 1994;16:29–33. doi: 10.1016/0891-5849(94)90239-9. [DOI] [PubMed] [Google Scholar]
- 39.Keyer K, Imlay JA. PNAS. 1996;93:13635–13640. doi: 10.1073/pnas.93.24.13635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Almiron M, Link AJ, Furlong D, Kolter R. Genes & Development. 1992;6:2646–2654. doi: 10.1101/gad.6.12b.2646. [DOI] [PubMed] [Google Scholar]
- 41.Altuvia S, Almiron M, Huisman G, Kolter R, Storz G. Mol. Microbiol. 1994;13:265–272. doi: 10.1111/j.1365-2958.1994.tb00421.x. [DOI] [PubMed] [Google Scholar]
- 42.Zhao G, Ceci P, Ilari A, Giangiacomo L, Laue TM, Chiancone E, Chasteen ND. J. Biol. Chem. 2002;277:27689–27696. doi: 10.1074/jbc.M202094200. [DOI] [PubMed] [Google Scholar]
- 43.Neidhardt FC, R Curtiss I, Ingraham JL, Lin ECC, Low KB, Magasanik B, Reznikoff WS, Riley M, Schaechter M, Umbarger HE. In: Escherichia coli and Salmonella. Neidhardt FC, editor. ASM Press; Washington, D.C.: 1996. [Google Scholar]
- 44.Cherepanov PP, Wackernagel W. Gene. 1995;158:9–14. doi: 10.1016/0378-1119(95)00193-a. [DOI] [PubMed] [Google Scholar]