

Abstract
Dendritic cells (DCs), have been implicated as important regulators of innate and adaptive inflammation in many diseases including atherosclerosis. However, the molecular mechanisms by which DCs mitigate or promote inflammatory pathogenesis are only partially understood. Previous studies have shown an important anti-inflammatory role for the transcription factor KLF2 in regulating activation of various cell types that participate in atherosclerotic lesion development, including endothelial cells, macrophages, and T cells. We used a pan-DC, CD11c-specific cre-lox gene knockout mouse model to assess the role of KLF2 in DC activation, function, and control of inflammation in the context of hypercholesterolemia and atherosclerosis. We found that KLF2 deficiency enhanced surface expression of costimulatory molecules CD40 and CD86 in DCs and promoted increased T cell proliferation and apoptosis. Transplant of bone marrow from mice with KLF2-deficient DCs into Ldlr−/− mice aggravated atherosclerosis compared to control mice, most likely due to heightened vascular inflammation evidenced by increased DC presence within lesions, enhanced T cell activation and cytokine production, and increased cell death in atherosclerotic lesions. Together these data indicate that KLF2 governs the degree of DC activation and hence the intensity of pro-atherogenic T cell responses.
Introduction
Atherosclerosis is a chronic inflammatory disease involving both the innate and adaptive arms of the immune response that is characterized by the development of lipid laden plaques in the arterial wall (1–4). Dendritic cells (DCs), a class of innate immune cells that function at the junction of innate and adaptive immunity, have been shown to play a central role in the initiation of atherosclerosis. During early lesion development, DCs as well as macrophages accumulate and transform into the early foam cells of the so-called “fatty streak” (5). DCs are also responsible for priming pro-atherogenic T cell responses against modified lipid and other athero-related antigens (6, 7). However, DCs have also been shown to play a protective role in atherosclerosis. DC-mediated nasal immunization with heat shock protein −65 ameliorated atherosclerosis in hypercholesterolemic mice (8), as did the adoptive transfer of tolerogenic DCs loaded with Apo B-100 (9). Other studies have also shown that the maintenance of atherosclerosis-suppressing Treg responses requires MyD88-dependent signaling in classical, Flt3-Flt3L-dependent DCs, the loss of which aggravates atherosclerosis (10, 11). Thus, DCs are involved in the complex interplay of proinflammatory and regulatory mechanisms that impact atherosclerosis.
While the generation of DCs with pro-inflammatory or tolerogenic T cell priming capabilities in vitro has been well established (9, 12–14), the molecular mechanisms by which DCs mature and function in vivo to drive either pro- or anti-atherogenic T cell responses in the context of hypercholesterolemia are poorly understood. Krüppel-like factor 2 (KLF2) is a transcription factor with well-established regulatory functions, including maintenance of quiescence in numerous cell types important in atherosclerosis, such as endothelial cells, macrophages, and T cells (15–21). Recent research has shown that KLF2 modulates development and inflammatory activity in macrophages and neutrophils (17, 22, 23). KLF2 hemizygous mice showed increased inflammatory Ly-6Chi monocytes in the circulation and increased recruitment of Ly-6Chi macrophages to the peritoneum (23). Importantly, pan-myeloid deletion of KLF2 in Lyz2cre Klf2fl/fl mice led to spontaneous macrophage activation and a fatal sepsis-like innate immune response against bacterial infection (17).
With regard to T cell responses, studies in our lab have shown that statin-induced expression of KLF2 negatively regulates inflammatory functions of T cells (18). DCs, the principal antigen presenting cells for naïve T cells, express relatively low levels of KLF2 mRNA, and the biological significance of DC-KLF2 is not clear. Therefore we examined the effects of Klf2 deletion in CD11C-expressing cells on DC phenotype and function, and on T cell priming and activation in vitro and in vivo. We also determined the effects of DC-KLF2 deficiency on atherosclerosis in mice by transplanting bone marrow from mice with KLF2-deficient DCs into LDLR KO mice and characterizing lesion development after feeding them a high-fat/cholesterol diet for 10 weeks. We found that loss of DC-KLF2 exacerbated atherosclerosis, while paradoxically inducing profound leukopenia, primarily via loss of T cells. KLF2-deficient DCs expressed higher levels of costimulatory molecules such as CD40 and CD86 following LPS-induced maturation, and were more efficient at stimulating both CD4+ and CD8+ T cell proliferation, cytokine production, and apoptosis.
Materials and Methods
Mice
All animals used in this study were bred and housed in the pathogen-free facility at the Warren Alpert Building (Harvard Medical School, Boston, MA) in accordance with IACUC guidelines. LDL receptor knockout mice (B6.129S7-Ldlrtm1Her) and CD11c-cre mice (C57BL/6J-Tg (Itgax-cre,-EGFP) 4097Ach/J) were purchased from Jackson Laboratories (Bar Harbor, ME). Klf2fl/fl mice were a kind gift from Mark L. Kahn (24). Klf2fl/fl mice were bred with Itgaxcre-cre mice to yield Itgaxcre -Klf2fl/fl mice which have conditional deficiency of KLF2 expression in CD11c-expressing cells, including most dendritic cells. Klf2fl/fl littermates lacking cre expression were used as control animals.
Cells and treatment
Bone marrow-derived dendritic cells (BMDC) or macrophages (BM-DM) were generated as previously described (25, 26). In short, bone marrow cells were cultured in complete RPMI-1640 medium supplemented with L-glutamine, sodium pyruvate, MEM-NEAA, and either 20 ng/ml GM-CSF (BMDCs; Peprotech) or 10 ng/ml M-CSF (BM-DMs; Peprotech) for 5–7 days and BMDCs/BM-DMs harvested from cultures. For in vitro experiments, BMDCs were matured by treatment with LPS (1 μg/ml) (Sigma-Aldrich) for 24h before use. For measurements of DC Klf2 expression, some BMDC preparations were pre-treated for 24h with low-dose simvastatin (0.5 μM) and rapamycin (1 nM) prior to LPS activation.
Bone Marrow Transplant
Male and female 8-week-old Ldlr−/− mice were subjected to 950 rad of total body irradiation delivered 4 hours apart in 2 doses (450 rad and 500 rad) and reconstituted with 2 × 106 bone marrow cells from Itgaxcre -Klf2fl/fl or Klf2fl/fl mice via tail vein injection. Bone marrow recipients received Sulfatrim (Sulfamethoxazole/Trimethoprim) treatment administered in drinking water for 1 week prior to and 4 weeks following BMT. All animals were allowed to recover on a chow diet for 6 weeks after BMT and then fed an atherogenic high-fat diet (HFD) containing 1.25% cholesterol (Cat. No. D1218C, Research Diets, Inc.) (27) for 10 weeks.
Histological analysis and morphometric analysis of aortic atherosclerosis
After sacrifice following 10 weeks of atherogenic diet feeding, aortic roots were dissected, embedded in OCT, and serial frozen section sections prepared. Analysis of atherosclerotic lesion size was performed on 5 Oil-Red-O stained cryosections (10 μm each) spanning 160 μm of the three valve area of the aortic root, as described (28, 29).
Immunization
For ovalbumin (Ova) immunization/T cell restimulation studies, Itgaxcre -Klf2fl/fl or Klf2fl/fl control mice were immunized by injecting 20 μl of 1 mg/ml Ova mixed 1:1 with Complete Freund’s Adjuvant (CFA; Sigma-Aldrich) into the hock with a 27-gauge needle as previously described (30). Draining lymph node cells were harvested from inguinal and popliteal lymph nodes 10 days after immunization. Lymph node cells were cultured in 96-well flat-bottom plates at 2.5×105 cells/well in complete DMEM supplemented with L-glutamine, sodium pyruvate, MEM-NEAA, and 2-mercaptoethanol and re-challenged with 100 μg/ml Ova or left untreated (control) for either 2 or 4 days. For cell proliferation experiments, lymph node cells were loaded with carboxyfluorescein diacetate succinimidyl ester (CFDA-SE) as previously described (31) prior to antigen re-challenge.
Serum Lipid Analysis
Mouse serum cholesterol and triglycerides were quantified using the c501 module of the Cobas 6000 analyzer (Roche Diagnostics, Indianapolis, IN). More detailed lipoprotein analyses were performed using high-performance liquid chromatography (HPLC; Liposearch, Tokyo, Japan).
Flow Cytometry
BMDCs were stained using the following antibodies (Biolegend): CD11c (N418), I-Ab (AF6-120.1), CD40 (3/23), and CD86 (GL-1). BMDC viability was assessed using Zombie Aqua fixable viability dye (Biolegend). Staining for monocyte and DC populations in peripheral blood leukocyte preparations was done using antibodies against CD11c (N418), CD11b (M1/70), Ly-6C (HK1.4), I-A/I-E (M5/114.15.2), Flt3 (A2F10), CD90.2 (53-2.1), B220 (RA3-6B2), Ly-6G (1AB), NK1.1 (PK136), and CD49b (DX5; Suppl. Figure 1). Splenocytes and peritoneal macrophages were stained for macrophage markers CD11b (M1/70), F4/80 (BM8), and CD11c (N418). T cell staining of splenocytes was done using the following antibodies: CD3 (145-2C11), CD4 (RM4-5), CD8 (53-6.7), and B220 (RA3-6B2). Splenocytes were separately stained for activated (CD25+) CD4+ T cells and Tregs (CD25+FoxP3+) using the following antibodies in addition to Zombie Aqua fixable viability dye (Biolegend): CD4 (RM4-5), CD25 (PC61), and FoxP3 (FJK-16S). Permeabilization of cells for FoxP3 staining was done using a FoxP3 Staining Buffer Set (eBioscience). Measurements of T cell apoptosis in lymph node cells from Ova-immunized mice were done by staining with CD90.2 (53-2.1), CD4 (RM4-5), and CD8 (53-6.7) monoclonal antibodies (Biolegend) and PE Annexin V Apoptosis Detection Kit I (BD Biosciences). Cell viability was assessed using 7-Aminoactinomycin D (7-AAD) viability dye (BD Biosciences). Apoptotic cell percentages were calculated by adding early (Annexin V+) and late (Annexin V+7-AAD+) events. For proliferation studies, lymph node cells were loaded with CFDA-SE prior to culture as previously noted and stained with CD90.2 (53-2.1) and CD4 (RM4-5) monoclonal antibodies and 7-AAD viability dye (BD Biosciences). All samples were acquired on a DxP11 flow cytometer (Cytek). Flow cytometry data were analyzed using FlowJo V10 analysis software (Treestar, Inc.).
Immunohistochemistry and immunofluorescence staining of aortic lesions
Frozen sections (7 μm) of aortic sinus lesions were stained with antibodies specific for CD4 (RM4-5, BD Pharmingen) macrophage marker Mac-3 (M3/84, BD Pharmingen), and neutrophil marker recognizing Ly6C and Ly6G (NIMP-R14, Abcam) and visualized with appropriate secondary antibodies. Neutral lipid accumulation in lesion sections was measured by Oil Red O staining. Nuclei for all immunohistochemistry stains were counterstained using Gil’s Hematoxylin. Immunohistochemistry stains were visualized with a Nikon Microphot-Fxa microscope (Nikon) equipped with an FX-35-DX digital camera (Nikon). Images were captured using either 4× / 0.13, 10× / 0.45, or 40× / 0.70 objective lens as indicated using the ACT-2U imaging software (Nikon). For immunofluorescence stains, sections were stained with biotinylated anti-CD11c (HL3) mAb, and visualized with streptavidin-conjugated Dylight 549 (Jackson Immunoresearch, West Grove, PA). Sections were mounted in Prolong Gold Antifade mounting medium with 4′,6-diamidino-2-phenylindole (DAPI; ThermoFisher Scientific) and visualized with an Olympus Fluoview FV1000 microscope (Olympus) and Olympus DP72 digital camera (Olympus). Images were captured using either a 10× / 0.30 or 20× / 0.75 objective lens and the FV10-ASW imaging software (Olympus). All images were modified post-capture to enhance brightness and contrast independently for each color channel using the level function in Adobe Photoshop (Adobe Systems, Inc.)
Quantitative RT-PCR (qRT-PCR) analyses
Gene expression levels in RNA samples were determined using a StepOnePlus Real Time PCR System (Applied Biosystems). RNA was extracted using an RNEasy Mini Kit (Qiagen) per the manufacturer instructions, and cDNA made using ThermoScript RT-PCR Kit (ThermoFisher Scientific). qRT-PCR was performed with forward and reverse primer sets as follows: Beta-actin F 5′-TCC TTC GTT GCC GGT CCA-3′, R 5′-ACC AGC GCA GCG ATA TCG TC-3′ Klf2 F 5′-ACA GAC TGC TAT TTA TTG GAC CTT AG-3′, R5′-CAG AAC TGG TGG CAG AGT CAT TT-3′; Ifng F5′-AAC GCT ACA CAC TGC ATC TTG G-3′, R 5′-GCC GTG GCA GTA ACA GCC-3′; Il17a F 5′-GCT CCA GAA GGC CCT CAG A-3′, R 5′-AGC TTT CCC TCC GCA TTG A-3′; Il4 F 5′-ACA GGA GAA GGG ACG CCA T-3′, R 5′-GAA GCC CTA CAG ACG AGC TCA-3′; Tbx21 F 5′-CAA CAA CCC CTT TGC CAA AG-3′. R 5′-TCC CCC AAG CAG TTG ACA GT-3′; Rorc F 5′-GGA GCC AAG TTC TCA GTC ATG AGA ACA CA-3′, R 5′-GCC CTT GCA CCC CTC ACA GGT-3′; Gata3 F 5′-AGA ACC GGC CCC TTA TCA A-3′, R 5′-AGT TCG CGC AGG ATG TCC-3′; Foxp3 F 5′-GGC CCT TCT CCA GGA CAG A-3′, R 5′-GCT GAT CAT GGC TGG GTT GT-3′. Levels of specific gene expression were normalized to endogenous levels of Actb gene expression and normalized against control groups or expressed as % Actb.
Acetylated-LDL Uptake Assay
BMDCs were generated by culture of bone marrow with GM-CSF (20 ng/ml) for seven days. Cells were washed and treated with 1 μg/ml or 10 μg/ml human Dil-Acetylated-LDL (Thermo Fisher) for 4 hours. Cells were stained with antibodies and Dil-AcLDL uptake was quantified by flow cytometry.
Multiplexed Cytokine Assays
CD4+ T cells were purified from splenocyte preparations by MACS cell separation using anti-CD4 (L3T4) microbeads (Miltenyi Biotec) as per manufacturer instructions and stimulated in vitro overnight (18h) with plate-bound anti-CD3 mAb (145-2C11, Biolegend). Supernatant cytokine levels were analyzed by proprietary multiplex analysis using the Mouse TH17 Array 25-plex Discovery Array (Cat #: MTH17-25-106; Eve Biotechnology, Calgary, Alberta, Canada). Supernatants from BMDCs treated overnight with LPS (Sigma-Aldrich) were also analyzed using the Mouse Cytokine Array/Chemokine Array 31-plex Discovery Assay (Cat #: MD31).
Statistical analyses
All statistical analyses were performed using GraphPad Prism software (GraphPad Software, Inc.). Values are expressed as mean ± SEM unless otherwise noted. Pairwise comparisons were performed using Student t tests or Mann-Whitney U tests (non-parametric data). Multiple comparisons of means for matched subjects were performed using 2-way ANOVA followed by Sidak’s multiple comparison tests. Differences between groups were considered significant at probability values below 0.05.
Results
CD11c-specific cre-lox deletion of KLF2 enhances DC co-stimulatory molecule expression
In order to determine the phenotype of KLF2 deletion in dendritic cells, we generated Itgaxcre -Klf2fl/fl mice that lack KLF2 in CD11c-expressinf cells, and compared them to Klf2fl/fl mice without a Cre transgene. KLF2 mRNA expression in CD11c+MHC-II+ bone marrow-derived DCs (BMDCs) from Itgaxcre -Klf2fl/fl mice was reduced more than 90% compared to expression in control mice. Furthermore, Itgaxcre -Klf2fl/fl BMDCs were unresponsive to KLF2-enhancing statin/rapamycin treatment (Fig. 1A). Naïve CD4+ T cells, which express high levels of KLF2, but not CD11c, showed no difference in KLF2 expression between Itgaxcre -Klf2fl/fl and Klf2fl/fl control mice (Fig. 1B). An expected loss of KLF2 expression, however, was seen in peritoneal macrophages (Fig. 1C), as many of these cells also express CD11c (Suppl. Fig. 2). Unlike BMDCs, Itgaxcre -Klf2fl/fl peritoneal macrophages maintained KLF2-responsiveness upon statin/rapamycin treatment (Fig. 1D), consistent with the interpretation that many of these cells are CD11c negative macrophages, which retain functional Klf2 genes.
Figure 1. Cre-lox deletion of KLF2 selectively depletes Klf2 expression from CD11c expressing cells, and enhances co-stimulatory molecule expression in DCs following activation.
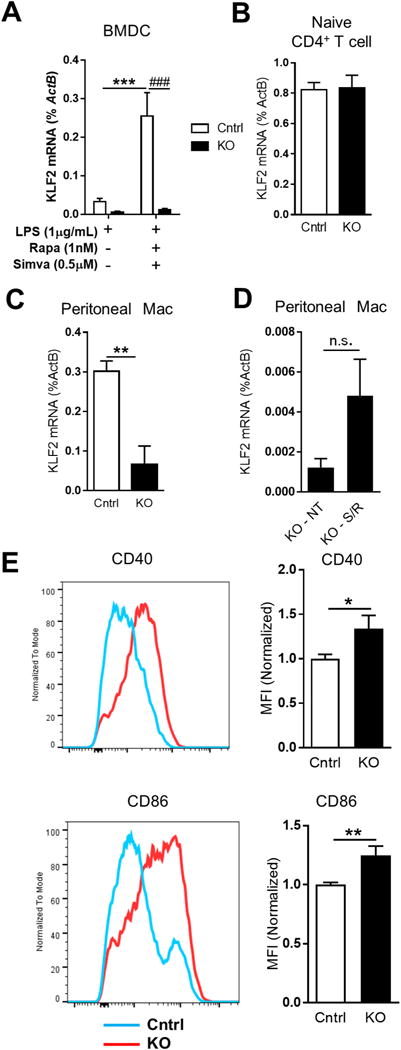
A, BMDCs were generated from Klf2fl/fl (Cntrl, white bars) or Itgaxcre-Klf2fl/fl (KO, black bars) bone marrow, matured with LPS alone or LPS plus simvastatin/rapamycin, and Klf2 mRNA were measured by qRT-PCR. Klf2 mRNA was measured in purified splenic naïve CD4+ T cells (B), peritoneal macrophages (C), and untreated (NT) or simvastatin/rapamycin-treated (S/R) peritoneal macrophages (D). E, CD40 and CD86 expression on LPS-matured CD11c+MHC-II+ BMDCs was measured by flow cytometry. Histograms show representative staining from 4 independent experiments. Bar graphs quantify CD40 and CD86 mean fluorescence intensity (MFI) normalized to control. A, n=10; B-D, n= 3; E-F, n=6. *, p<0.05; **, p<0.01; ***, p<0.001
Since we found that rapamycin enhances KLF2 expression in DCs, and others have shown that rapamycin induces a tolerogenic DC phenotype that expresses lower levels of co-stimulatory markers CD40 and CD80/CD86 (32), we conducted in vitro studies to determine if loss of DC-KLF2 enhances co-stimulatory molecule expression in mature DCs. Itgaxcre -Klf2fl/fl BMDCs showed increased surface expression of both CD40 and CD86 following LPS activation (Fig. 1E), consistent with a more activated, pro-inflammatory phenotype. Analyses of supernatants from these BMDC cultures showed no difference in DC secretion of IL-1, TNFα, IL-12, or IL-10 (data not shown).
CD11C-KLF2 depletion induces leukopenia that is exacerbated by hypercholesterolemia
Previous research has shown that LysM-Cre-specific loss of KLF2 in macrophages and neutrophils triggers spontaneous basal macrophage activation, enhances microbe-induced macrophage activation, and aggravates atherosclerosis (17, 22). We hypothesized that DC-KLF2 may function to suppress pro-atherogenic T cell responses, and therefore examined the effect of CD11C-KLF2 deficiency on atherosclerosis in Ldlr−/− mice. For this purpose, we transplanted bone marrow from Itgaxcre -Klf2fl/fl and Klf2fl/fl control mice into Ldlr−/− mice and placed them on a high fat and cholesterol diet for 10 weeks. Transplant of Itgaxcre -Klf2fl/fl bone marrow into Ldlr−/− mice successfully ablated KLF2 expression in both BMDCs and bone marrow-derived macrophages (Fig. 2A–B). Loss of DC-KLF2 had no effect on the percentage of circulating CD11c+MHC-II+ DCs but there was a trend towards increased CD11c+Flt3+ pre-cDCs (Fig. 2C–D), suggesting that KLF2-deficiency does not drastically disrupt overall DC development and hematopoiesis. Interestingly, Itgaxcre -Klf2fl/fl mice showed an unexpected and profound reduction in the total numbers of circulating leukocytes (Suppl. Fig. 3A–B). This was characterized by an almost 2-fold reduction in the number of lymphocytes, monocytes, and eosinophils. A reduction in blood leukocytes, neutrophils, lymphocytes, and monocytes was also seen in native LDLR-expressing Cd11c-Cre+ Klf2fl/fl mice compared with Cd11c-Cre− Klf2fl/fl control animals, albeit not as profound as seen in the bone marrow chimeras (Suppl. Fig. 3A–B). Hypercholesterolemia enhanced the reduction in total leukocyte counts, although there was HFD-induced monocytosis and eosinophilia in Itgaxcre -Klf2fl/fl mice as well as in controls. Cell numbers in both spleen and heart lymph node were also reduced in hypercholesterolemic Itgaxcre-Klf2fl/fl mice compared to controls (Fig. 3C–D), indicating a global KLF2-dependent leukopenia.
Figure 2. Loss of KLF2 in DCs induces leukopenia and promotes a more pro-atherogenic inflammatory monocyte phenotype under hypercholesterolemic conditions.
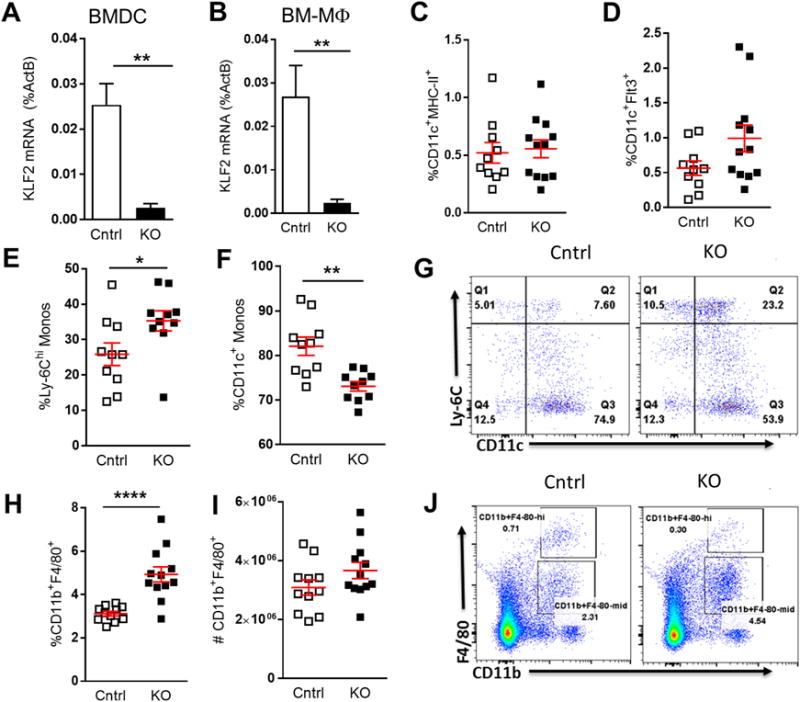
Itgaxcre-Klf2fl/fl (KO) or Klf2fl/fl (Cntrl) bone marrow was transplanted into Ldlr−/− recipients and fed a high-fat diet for 10 weeks. A–B, qRT-PCR for Klf2 mRNA was performed on BMDC and macrophages (BM-Mɸ) generated from Cntrl and KO bone marrow recipients following sacrifice. C–D, Flow cytometry was performed on blood to determine circulating levels of mature DCs (C), and DC precursors (D). E–G, Circulating monocytes were stained for Ly-6C and CD11c expression. E and F show percentage of Ly-6Chi and CD11c+ monocytes respectively. G, shows a representative flow cytometry plot for Ly-6C and CD11c staining. H–J, splenocytes were stained for macrophage markers CD11b and F4/80. H, shows macrophages as percent of total splenocytes and I shows splenic macrophage numbers calculated from total splenocyte counts. J, shows a representative splenic F4/80, CD11b flow cytometry plot. A, n=11; B, n=6; C–J n=10–12. *, p<0.05; **, p<0.01; ****, p<0.0001
Figure 3. Loss of CD11C-KLF2 aggravates atherosclerosis.
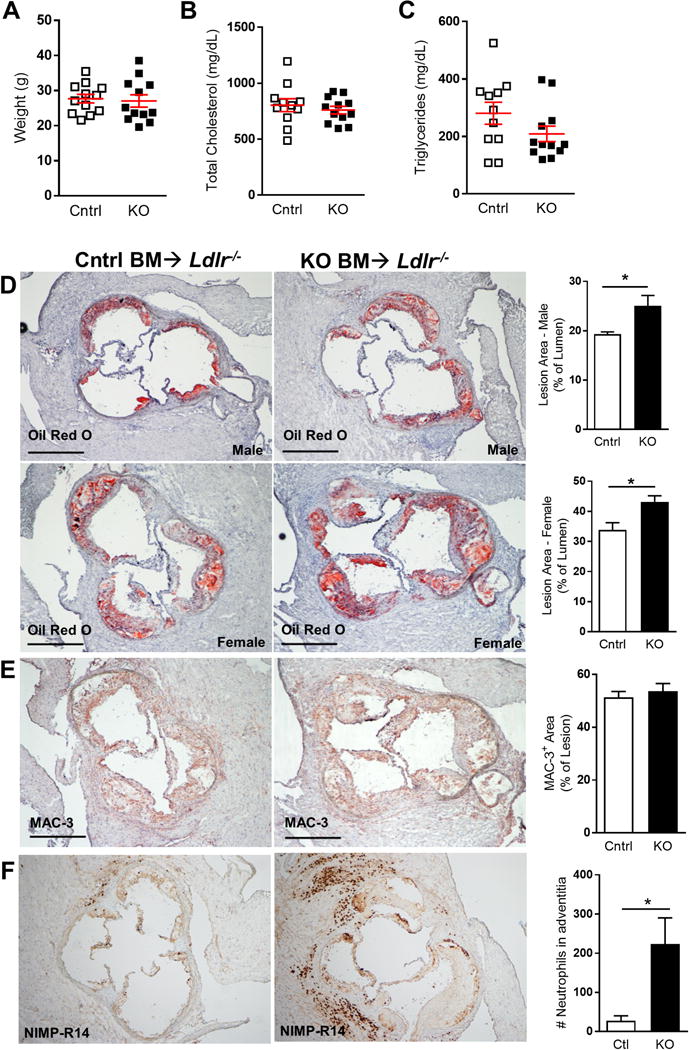
Klf2fl/flCd11c-Cre−/− (Cntrl) or Klf2fl/flCd11c-Cre+/− (KO) bone marrow was transplanted into 12 male and 12 female Ldlr−/− recipients and fed a high-fat diet for 10 weeks. A, Shows animal weight at sacrifice. B–C, Blood serum was taken at sacrifice and lipid levels measured to determine total cholesterol and triglyceride levels. D–F, Frozen sections from aortic sinus were obtained and atherosclerotic lesions from the 3-valve region were stained with Oil Red O for neutron lipid (D), or by immunohistochemistry with antibodies specific for macrophage marker MAC-3 (E), anti-neutrophil antibody (NIMP-R14) (F). Nuclei were counterstained with hematoxylin. Isotype controls for MAC-3 and NIMP-R14 staining was negative (not shown). Bar graphs indicate lesion area as percent of lumen for male or female mice. Scale bars = 500 μm. A–C, n=12; D, n=20; E, n=12; F, n=6. *, p<0.05
Loss of KLF2 in CD11c-expressing cells promotes a more pro-atherogenic inflammatory monocyte phenotype under hypercholesterolemic conditions
While reduced lymphocyte numbers were noted previously in Lysmcre-Klf2fl/fl mice (22), hypercholesterolemia-induced monocytosis has been well established as an underlying factor in murine models of atherosclerosis (33, 34). We therefore asked if KLF2-deficiency influenced the inflammatory/non-classical monocyte balance. While total monocyte levels were reduced in Itgaxcre-Klf2fl/fl mice, Ly-6C staining showed an increased percentage of pro-atherogenic Ly-6Chi, inflammatory monocytes (Fig. 2E). This was perhaps related to a reduced percentage of CD11c+ monocytes which make up the vast majority of Ly-6Cmid/lo non-classical monocytes (Fig. 2F–G). In contrast to monocytes, splenic macrophages accounted for a higher fraction of total spleen cells in Itgaxcre-Klf2fl/fl mice versus control mice (Fig. 2H), while total splenic macrophage numbers did not significantly differ (Fig. 2I–J). Together, these data show that KLF2 deletion in our model is restricted to CD11c-expressing myeloid cells, and includes DCs as well as a significant percentage of monocytes and macrophages that normally express CD11c. Furthermore, we observed both leukopenia and an inflammatory Ly-6Chi skewed monocyte fraction in the CD11C-KLF2−/−, which based on previous studies, should have opposing effects on atherosclerosis.
Loss of DC-KLF2 exacerbates atherosclerosis development
Low density lipoprotein receptor deficient (Ldlr−/−)− mice develop hypercholesterolemia and atherosclerosis when fed a high fat/cholesterol containing diet, even after they are irradiated and reconstituted with Ldlr+/+ bone marrow, due to a deficiency in hepatic lipoprotein uptake, and these mice have been used extensively to study the impact of immune inflammatory processes in atherosclerosis (35). Ldlr−/− recipients of Itgaxcre-Klf2fl/fl bone marrow showed no difference in weight, total serum cholesterol, or serum triglycerides after 10 weeks of pro-atherogenic diet, compared with recipients of control bone marrow (Fig. 3A–C). However, oil red O analysis of aortic sinus lesions showed enhanced atherosclerosis in the Itgaxcre-Klf2fl/fl group in both male and female mice (Fig. 3D). The percent macrophage content of lesions as determined by MAC-3 staining did not differ between groups (Fig. 3E), suggesting that KLF2 deficiency in CD11c+ monocyte/macrophages does not influence plaque macrophage numbers. There was a significant increase in aortic root adventitial neutrophils (clone: NIMP-R14) (Fig. 3F). Anti-neutrophil stained cells co-localized with myeloperoxidase staining, suggesting that the stained cells were neutrophils rather than monocytes/macrophages (data not shown). The pro-atherogenic effect observed in our model is seen despite the fact that the development of atherosclerosis is usually accompanied by a marked monocytosis (34, 36). Instead, our hypercholesterolemic Itgaxcre-Klf2fl/fl recipients showed a comparatively blunted monocytosis and deepened lymphopenia compared to hypercholesterolemic Klf2fl/fl control mice. Thus, it appears that the pro-inflammatory phenotype of KLF2-deficient DCs has a dominant pro-atherosclerotic effect over the net reduction in circulating monocytes and other leukocytes that contributes to lesion growth.
Increased intimal DC accumulation corresponds with reduced intimal CD4+ T cells and increased apoptosis within lesions
In order to better understand how KLF2-deficient DCs could enhance lesion inflammation and growth, we analyzed the numbers and localization of lesional DCs and T cells. CD11c immunofluorescence staining of aortic sinus lesions showed that intimal DCs accumulate in significantly greater numbers in Itgaxcre-Klf2fl/fl lesions (Fig. 4A). Intimal DCs from Itgaxcre-Klf2fl/fl lesions also showed a different pattern of localization compared to control lesions, with more DCs residing deep within the lesion, beyond the sub-endothelial space where intimal DCs tend to reside. Interestingly, acetylated LDL uptake was not different between Itgaxcre-Klf2fl/fl vs. control DCs (Supplemental Fig. 4), consistent with the interpretation that the impact of DC KLF2 deficiency on enhanced lesion development is indirectly related to effects on other cells. Immunostaining of CD4 showed a significant reduction in intimal, but not adventitial CD4+ T cells in Itgaxcre-Klf2fl/fl lesions (Fig. 4B). The reduced lesional T cell numbers are consistent with the global lymphopenia observed in these mice. This is in contrast with previous studies that have shown that increased T cell numbers are associated with more lesion development (37). Surprisingly, we noted the loss of intimal, but not adventitial T cells, which corresponded roughly with increased intimal DC accumulation, suggesting that interactions between KLF2-deficient DCs and T cells may be a root cause of reduced T cell numbers. In support of this interpretation, TUNEL staining showed enhanced intimal apoptosis within Itgaxcre-Klf2fl/fl mouse lesions (Fig. 4C), suggesting that the loss of DC-KLF2 results in enhanced inflammation and cell death within lesions. Thus, even though pro-atherogenic T cell accumulation may be reduced in lesions, the inflammatory consequences of their early activation, and perhaps other inflammatory results of KLF2-deficient DCs results in more lesion development.
Figure 4. KLF2-deficient DCs accumulate within intimal lesions corresponding with intimal CD4+ T cell loss.
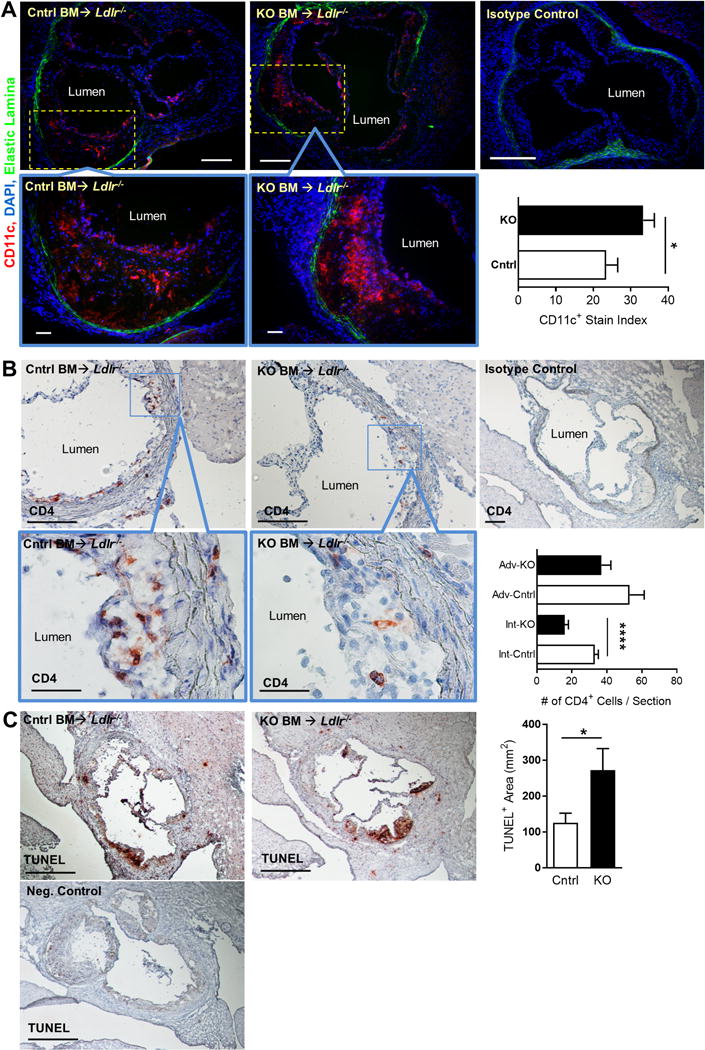
Frozen sections of aortic sinus were obtained from Ldlr−/− recipients of Klf2fl/fl (Cntrl) or Itgaxcre -Klf2fl/fl (KO) bone marrow following 10 weeks of high-fat diet and stained for CD11c (A) or CD4 (B). A, Micrographs show CD11c staining (red), nuclear staining by 4′,6-diamidino-2-phenylindole (DAPI) (blue), and elastic laminae autofluorescence (green). Bars, 200 μm; inset bars 50 μm. Bar graph shows CD11c stain index (# of red pixels/# of blue pixels) for Cntrl (white bars) or KO (black bars) bone marrow recipients. B, Micrographs show CD4 staining (red) or isotype control. Nuclei are counterstained with hematoxylin. Bars, 200 μm; inset bars 50 μm. Bar graph shows number of intimal (Int) and adventitial (Adv) CD4 T cells per section indicate lesion area as percent of lumen for male or female mice. C, Micrographs show TUNEL staining or negative control. Nuclei are counterstained with hematoxylin. A–C, n=10–12. *, p<0.05; ****, p<0.0001
DC-KLF2 modulates T cell activation in hypercholesterolemic mice
In order to explain how atherosclerosis can increase in the context of lymphopenia and reduced monocyte numbers, we interrogated splenocytes from the hypercholesterolemic Ldlr−/− mice transplanted with either Itgaxcre-Klf2fl/fl or Klf2fl/fl control bone marrow. Flow cytometry analyses of spleen show that, as seen in circulating leukocytes, hypercholesterolemic Itgaxcre-Klf2fl/fl bone marrow recipients had markedly reduced numbers of T cells, but not B-cells (Fig. 5A–C). This reduction in T cell numbers was seen in both CD4+ and CD8+ T cell compartments (Fig. 5D–F), suggesting that some aspect of T cell development, activation, or maintenance is disrupted in the absence of DC-KLF2. We next sought to determine whether or not loss of DC-KLF2 affects Treg development or T cell activation. CD25 and FoxP3 staining of splenic CD4+ T cells showed that Itgaxcre-Klf2fl/fl bone marrow recipients were as capable of maintaining splenic Tregs as controls (Fig. 6G), but had more activated CD25+FoxP3− T cells (Fig. 5H–I).
Figure 5. Pan-CD11c KLF2-deficiency enhances T cell activation while reducing overall T cell numbers in hypercholesterolemic mice.
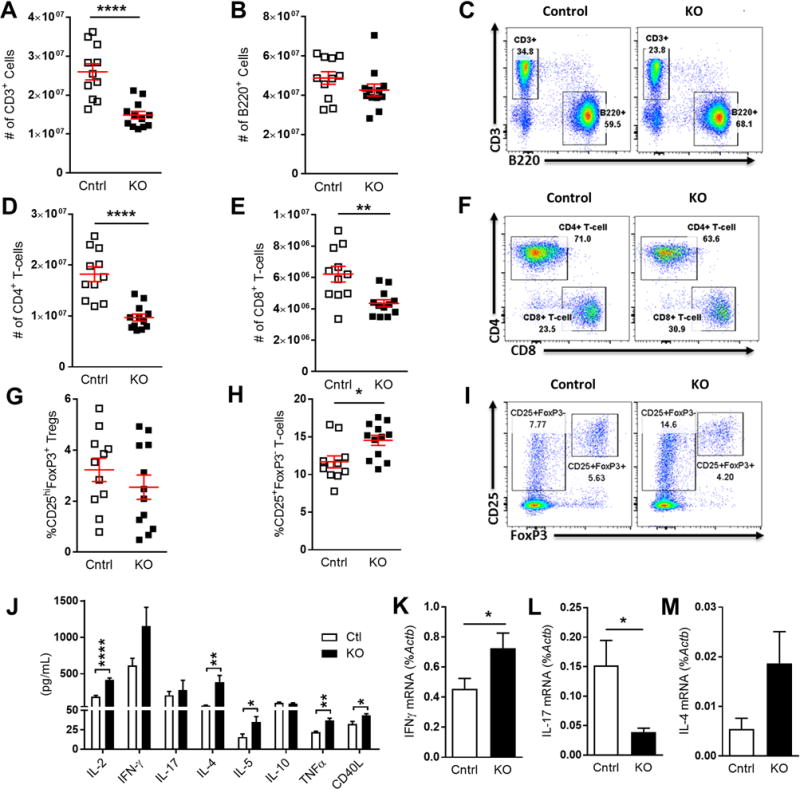
Splenocytes from Klf2fl/fl(Cntrl) or Itgaxcre -Klf2fl/fl (KO) bone marrow recipients were stained for CD3, B220, CD4, and CD8 and analyzed by flow cytometry. A and B, Total numbers of CD3+ T cells and B220+ B-cells per spleen. C, Representative FACS plots show CD3 and B220 staining of splenocytes. D and E, Total numbers of CD4+ and CD8+ T cells per spleen. F, Representative FACS plots of CD4 and CD8 staining gated on CD3+ splenocytes. G–I, Splenocytes were also stained for CD4, CD25, and FoxP3. G, percent of CD4+ T cells that are CD25hiFoxP3+ Tregs. H, Percentage of activated CD4+ T cells (CD4+CD25+FoxP3−). I, Representative staining and gating of CD4+ T cell data shown in G and H. J, CD4+ splenocytes were isolated by MACS and stimulated overnight with αCD3 Ab. Graph shows supernatant cytokine levels determined by multiplex cytokine array. K–M, RNA was also taken from αCD3-stimulated cultures and qRT-PCR run on Ifng (K), Il17a (L), and Il4 (M). A–H, n=12; J, n=6; K–M, n=10. *, p<0.05; **, p<0.01; ****, p<0.0001
Figure 6. KLF2-deficient DCs promote enhanced T cell proliferation and activation-induced cell death (apoptosis).
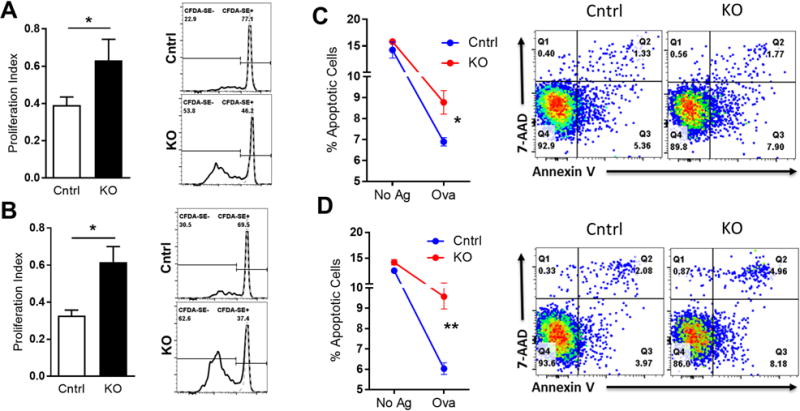
Klf2fl/f (Cntrl) or Itgaxcre -Klf2fl/fl(KO) mice were immunized in the hock with whole ovalbumin (Ova) and complete Freund’s adjuvant (CFA) and draining lymph node cells were harvested 10 days later. A–B, Draining lymph node cells from Ova-immunized mice were loaded with carboxyfluorescein diacetate succinimidyl ester (CFDA-SE) and re-challenged with Ova or left unstimulated. After 4 days, cultures were harvested and stained with Abs against CD90.2 and CD4, as well as the viability dye 7-AAD. A and B, proliferation index for CD4+ and CD8+ T cells respectively measured by CFDA-SE dilution in response to Ova (black lines in histograms) over unstimulated baseline (dashed grey lines). C–D, Draining lymph node cells from Ova-immunized mice were re-challenged with Ova (Ova) or left unstimulated (No Ag) and stained with CD90.2, CD4, CD8, Annexin V, and 7-AAD after 2 days. Shown are the sum early (Q3) and late (Q2) apoptotic CD4+ (C) and CD8+ (D) T cells. Representative FACS plots are shown on the right for 2 independent experiments. A–B, n=8; C–D, n=6. *, p<0.05; **, p<0.01.
To further explore whether DC-KLF2 influences T cell activation or Th-subset lineage commitment, splenic CD4+ T cells from the Itgaxcre-Klf2fl/fl or Klf2fl/fl control bone marrow recipients were re-stimulated with αCD3 antibodies. RT-qPCR of Th-lineage-specific mRNA from these experiments showed no difference in Th1-restricted Tbx21, Th17-restricted Rorc, or Treg-restricted Foxp3 expression, while expression of Th2-restricted Gata3, which is usually associated with reduced atherosclerosis, was increased in the Itgaxcre-Klf2fl/fl group (data not shown). Stimulated CD4+ T cell culture supernatants showed a 2-fold increase in IL-2 production in Itgaxcre-Klf2fl/fl bone marrow recipient mice, as well as significantly increased IL-4, IL-5, TNFα, and soluble CD40L production (Fig. 5J). The increased IL-2 production is consistent with the enhanced activation-induced cell death we observed, since this cytokine is known to enhance Fas-Fas-ligand induced death (38). IFN-γ secretion was also enhanced in Itgaxcre-Klf2fl/fl cells, although this trend was not significant, while Th17-produced IL-17 and Treg-produced IL-10 showed no difference. RT-qPCR data support the cytokine data, showing increased expression of pro-inflammatory, pro-atherogenic Th1 cytokine Ifng in CD4+ T cell cultures from Itgaxcre-Klf2fl/fl mice while Il17a mRNA expression was reduced (Fig. 5K–L). Il4 mRNA showed a non-significant trend towards increased expression in Itgaxcre-Klf2fl/fl samples in line with the increased Gata3 expression and IL-4 and IL-5 secretion (Fig. 5M). Together, these data show a highly activated CD4+ T cell phenotype in the absence of DC-KLF2, which surprisingly favors both Th1 and Th2 T cell activity, but not Th17 cells. However, the lack of a compensatory increase in Treg IL-10 production or Treg numbers implies that Treg responses may not keep pace with enhanced T cell activation in response to hypercholesterolemia, perhaps explaining how plaque burden increased in Itgaxcre-Klf2fl/fl mice despite reduced monocyte and T cell numbers.
Loss of DC-KLF2 enhances T cell proliferation and apoptosis
We next examined if T cells from mice with KLF2-deficient DCs showed enhanced activation in vitro and whether or not KLF2-deficient DCs induce higher rates of apoptosis in activated T cells. Itgaxcre-Klf2fl/fl and control mice were immunized in the hock with ovalbumin (Ova) protein in complete Freund’s adjuvant (CFA). Draining lymph nodes were harvested 10 days later and restimulated in vitro with ovalbumin. Ova-specific T cell proliferation was tested by CFDA-SE dilution in 4 day cultures. Both CD4+ and CD8+ T cells from Itgaxcre-Klf2fl/fl mice showed significantly enhanced antigen-induced proliferation compared to controls, consistent with enhanced in vivo naïve T cell priming by KLF2-deficient DCs (Fig. 6A–B). Apoptosis was measured in 2 day cultures via Annexin V staining, which showed that both CD4+ (Fig. 6C) and CD8+ (Fig. 6D) T cells from Itgaxcre-Klf2fl/fl mice had increased rates of apoptosis compared with T cells from control mice following in vitro antigen re-challenge. However, there was no significant difference in cell death between the two groups in the absence of Ova. These data show that while DC-KLF2 is not required for Treg development or maintenance, it does play a role in modulating effector T cell activation, expansion, and activation-induced cell death. The enhanced T cell death is consistent with less T cells in the blood, spleen, and lesions of Itgaxcre-Klf2fl/fl mice despite increased ability of KLF2−/− DCs to activate T cells.
Discussion
An important role for DCs in the modulation of inflammation has been established for atherosclerosis (39). Evidence has been found showing that DCs can both promote and protect against atherosclerosis, depending either on lineage or cytokine milieu. Thus, a better understanding of the molecular mechanisms that influence DC control of pro- and anti-atherogenic T cell responses is needed in order to identify future molecular targets of drug or gene therapies. However the role of KLF2 in modulating how DCs influence atherosclerosis has not been previously explored.
We first looked at how KLF2 influences DCs by performing phenotypic and functional studies in Itgaxcre-Klf2fl/fl and control BMDCs. Our data show that lack of DC-KLF2 enhanced DC expression of costimulatory molecules CD40 and CD86 (Fig. 1E). This is consistent with a report that silencing KLF2 expression in human monocyte derived DCs enhanced of CD80 (40). A previous report describing Lyz2cre-specific deletion of KLF2 in myeloid cells showed spontaneous myeloid cell activation in the absence of myeloid KLF2 (17). However, Itgaxcre-Klf2fl/fl deletion did not influence cytokine production in BMDCs prior to and following LPS-induced activation (data not shown). Thus, while KLF2 has been shown to function as a critical factor in maintaining cellular quiescence in other cell types (15, 17, 18, 41–43), our cytokine data suggest that in DCs, KLF2 plays a more subtle role, controlling instead the “volume” of context-dependent DC activation rather than suppressing spontaneous DC activation. This interpretation is further supported by the absence of a pronounced autoimmune phenotype in our Itgaxcre-Klf2fl/fl mice, as we did not observe spontaneous tissue specific or systemic inflammation, nor increased mortality. CD40 signaling between DCs and T cells has been shown to enhance several aspects important to T cell stimulation, including upregulation of intercellular adhesion molecule-1 (ICAM-1) that serves to stabilize the DC/T cell interaction, increased expression of CD80 and CD86 that provides co-stimulatory signals needed for T cell activation (44), increased expression and stability of MHCII-peptide complexes (45, 46), prolonged DC survival (47, 48), and prolonged antigen persistence following LPS-induced maturation (46, 49–51). Together, these findings indicate that DC-KLF2 deficiency may promote a more pro-inflammatory DC phenotype by enhancing CD40-CD40L signaling between DCs and T cells, independent of DC cytokine production. CD40-CD40L signaling, in turn, likely enhances DC survival, helper (CD4+) T cell differentiation, and priming cytotoxic CD8+ T cell responses. This establishes our Itgaxcre-Klf2fl/fl model as phenotypically distinct from previous myeloid lineage tissue-specific KLF2 knockout models.
KLF2 expression was also reduced in CD11c-expressing monocytes and macrophages (Fig. 1C–D). A monocyte/macrophage phenotype was also seen in our atherosclerosis model where Ldlr−/− Itgaxcre-Klf2fl/fl bone marrow recipients showed reduced KLF2 expression in BMDCs and BMDMs as well (Fig. 2A–B). Hypercholesterolemic Itgaxcre-Klf2fl/fl recipients showed a significantly increased skewing of the monocyte compartment towards an inflammatory phenotype compared with controls, which can be explained by the fact that a majority of Ly-6Clo/mid non-classical monocytes express CD11c (Fig. 2E–G). Prior studies have shown that inflammatory macrophages express less KLF2 than non-classical macrophages (52, 53). From our data, it is possible that many CD11c-expressing Ly-6Clo/mid non-classical monocytes lose KLF2 and upregulate Ly-6C, becoming inflammatory Ly-6Chi monocytes as Itgaxcre-Klf2fl/fl recipients had increased numbers of CD11c+Ly-6Chi, but not CD11c−Ly-6Chi compared with controls (Fig. 2G). This pro-inflammatory skewing of the monocyte compartment in Itgaxcre-Klf2fl/fl mice was accompanied by a paradoxical reduction in circulating monocyte numbers (Fig. 4A–B), which corresponds with findings in previous studies of myeloid-specific KLF2 knockout mice (17, 22). This monocyte loss was accompanied by an even greater reduction in lymphocytes in both hypercholesterolemic Itgaxcre-Klf2fl/fl bone marrow recipients and steady state Itgaxcre-Klf2fl/fl mice maintained on chow diet (Supplemental Fig. 3A–B). Reduced leukocyte numbers were also found in spleen and heart draining lymph node of hypercholesterolemic mice (Supplemental Fig. 3C–D), suggesting that this cell loss occurs in multiple compartments. These findings are distinct from those previously reported in myeloid-restricted KLF2-KO models and highlight the Itgaxcre-Klf2fl/fl phenotype as a unique indicator of KLF2 function in DCs.
The major finding in this study is that, despite significantly reduced monocyte and lymphocyte numbers, loss of CD11C-KLF2 enhanced atherosclerotic plaque burden in hypercholesterolemic mice without enhancing body weight or blood cholesterol levels beyond that of control mice (Fig. 4). Myeloid KLF2 depletion in LysM-Cre mice was previously shown to enhance atherosclerosis in Ldlr−/− mice (22). However, that study found a mechanism of action involving enhanced neutrophil and macrophage adhesion and homing to atherosclerotic plaques via enhanced expression of CD11b. In our model, Itgaxcre-Klf2fl/fl plaques had equal amounts of macrophages as shown by MAC-3 staining as control plaques (Fig. 3E), despite CD11c+ macrophage populations in spleen and peritoneum expressing significantly more CD11b as well (data not shown). Thus, it is likely that reduced monocyte numbers may in part be due to enhanced lymphoid and peripheral tissue recruitment of monocytes and a higher rate of tissue macrophage activation and death (Fig. 2H, Fig. 4).
Immunostaining of lesions showed increased numbers of intimal CD11c+ cells present in Itgaxcre-Klf2fl/fl plaques (Fig. 5). This is perhaps explained by our finding of increased circulating pre-cDCs which serve as precursors for tissue resident DCs (54) (Fig. 2D). It is possible that some of the CD11c staining is on macrophages, which we cannot distinguish from CD11c+ DCs in the lesions, but both cell types should be competent at activating T cells within the lesion, and both would be susceptible to CD11c-Cre driven Klf2fl/fl deletion. Detailed electron scanning microscopy studies of human atherosclerotic lesions have shown immature vascular DCs lay in contact with the basal side of the endothelial layer before activating and joining a webbed network of sub-endothelial leukocytes. These activated DCs then continue to migrate deeper into the growing lesion and show morphological evidence of greater inflammatory activity as disease progresses (55–57). As shown in Fig. 4A, increased numbers of CD11c+ staining cells appear deeper within intimal lesions, which points to a more activated, inflammatory DC phenotype, consistent with our in vitro data showing enhanced KLF2−/− DC maturation and increased expression of co-stimulatory molecules. Reduced intimal CD4+ T cell numbers coupled with increased TUNEL staining within Itgaxcre-Klf2fl/fl lesions also supports the theory of enhanced DC-mediated T cell activation in vivo as lesional T cell activation is a well-known contributor to cell death and necrotic core formation (2, 3).
It is paradoxical, then, that atherosclerosis was enhanced in Itgaxcre-Klf2fl/fl bone marrow recipients in the context of reduced circulating and splenic T cells (Fig. 5). While prior studies of LysM-Cre KLF2−/− mice reported pro-inflammatory changes in mainly monocyte/macrophage and neutrophil populations (17, 22), our studies of hypercholesterolemic Itgaxcre-Klf2fl/fl mice showed profound changes in T cell responses. Most notably, there were more activated CD25+ splenic CD4+ T cells in Itgaxcre-Klf2fl/fl bone marrow recipients, while Treg responses remained unchanged compared to control (Fig. 5G–I). Purified splenic CD4+ T cells from Itgaxcre-Klf2fl/fl mice produced increased levels of both Th1 and Th2 cytokines upon restimulation with αCD3 mAb, including IL-2, IFN-γ, TNFα, IL-4, and IL-5 (Fig. 5J–M). Meanwhile, Th17 and Treg responses were either reduced or unchanged. One possible explanation for increased Th1 and Th2 responses in the absence of a correspondingly increased Treg response comes from prior work in a Leishmania infection model, which shows that CD40lo DCs are more effective at inducing Tregs, while CD40hi DCs primarily induce effector T cell responses (58). These findings indicate that KLF2-deficient DCs enhance a broad spectrum of T cell activation.
We confirmed there was enhanced CD4+ and CD8+ T cell activation by KLF2-deficient DCs in in independent experiments by immunizing Itgaxcre-Klf2fl/fl or control mice with Ova and measuring Ova-induced proliferation of primed cells ex vivo (Fig. 6A–B). T cell apoptosis following antigen re-challenge was also increased in Itgaxcre-Klf2fl/fl mice (Fig. 6C–D). Together with increased T cell activation and IL-2 production, enhanced T cell apoptosis indicates that the reduced global T cell numbers seen in hypercholesterolemic Itgaxcre-Klf2fl/fl bone marrow recipients likely occurs at least in part due to activation induced cell death (AICD). AICD has long been known to be driven by IL-2 binding to its high-affinity receptor, CD25, which was also increased on the surface Itgaxcre-Klf2fl/fl T cells (59–61).
Taken together our findings highlight the previously unknown importance of KLF2 in regulating T cell inflammatory responses. DCs lacking KLF2 express higher levels of co-stimulatory molecules like CD40 and CD86 upon activation and “over” activate T cells, leading to increased inflammation and AICD. This aggravated atherosclerosis development despite reducing T cell and monocyte numbers in the circulation.
Supplementary Material
Acknowledgments
None.
References
- 1.Ross R. Atherosclerosis–an inflammatory disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 2.Andersson J, Libby P, Hansson GK. Adaptive immunity and atherosclerosis. Clin Immunol. 2010;134:33–46. doi: 10.1016/j.clim.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 3.Hansson GK, Libby P. The immune response in atherosclerosis: a double-edged sword. Nat Rev Immunol. 2006;6:508–519. doi: 10.1038/nri1882. [DOI] [PubMed] [Google Scholar]
- 4.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 5.Paulson KE, Zhu SN, Chen M, Nurmohamed S, Jongstra-Bilen J, Cybulsky MI. Resident intimal dendritic cells accumulate lipid and contribute to the initiation of atherosclerosis. Circ Res. 2010;106:383–390. doi: 10.1161/CIRCRESAHA.109.210781. [DOI] [PubMed] [Google Scholar]
- 6.Erkkila L, Laitinen K, Haasio K, Tiirola T, Jauhiainen M, Lehr HA, Aalto-Setala K, Saikku P, Leinonen M. Heat shock protein 60 autoimmunity and early lipid lesions in cholesterol-fed C57BL/6JBom mice during Chlamydia pneumoniae infection. Atherosclerosis. 2004;177:321–328. doi: 10.1016/j.atherosclerosis.2004.08.021. [DOI] [PubMed] [Google Scholar]
- 7.Hjerpe C, Johansson D, Hermansson A, Hansson GK, Zhou X. Dendritic cells pulsed with malondialdehyde modified low density lipoprotein aggravate atherosclerosis in Apoe(−/−) mice. Atherosclerosis. 2010;209:436–441. doi: 10.1016/j.atherosclerosis.2009.10.003. [DOI] [PubMed] [Google Scholar]
- 8.Xiong Q, Li J, Jin L, Liu J, Li T. Nasal immunization with heat shock protein 65 attenuates atherosclerosis and reduces serum lipids in cholesterol-fed wild-type rabbits probably through different mechanisms. Immunol Lett. 2009;125:40–45. doi: 10.1016/j.imlet.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 9.Hermansson A, Johansson DK, Ketelhuth DF, Andersson J, Zhou X, Hansson GK. Immunotherapy with tolerogenic apolipoprotein B-100-loaded dendritic cells attenuates atherosclerosis in hypercholesterolemic mice. Circulation. 2011;123:1083–1091. doi: 10.1161/CIRCULATIONAHA.110.973222. [DOI] [PubMed] [Google Scholar]
- 10.Subramanian M, Thorp E, Hansson GK, Tabas I. Treg-mediated suppression of atherosclerosis requires MYD88 signaling in DCs. J Clin Invest. 2013;123:179–188. doi: 10.1172/JCI64617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi JH, Cheong C, Dandamudi DB, Park CG, Rodriguez A, Mehandru S, Velinzon K, Jung IH, Yoo JY, Oh GT, Steinman RM. Flt3 signaling-dependent dendritic cells protect against atherosclerosis. Immunity. 2011;35:819–831. doi: 10.1016/j.immuni.2011.09.014. [DOI] [PubMed] [Google Scholar]
- 12.Raich-Regue D, Glancy M, Thomson AW. Regulatory dendritic cell therapy: from rodents to clinical application. Immunol Lett. 2014;161:216–221. doi: 10.1016/j.imlet.2013.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Habets KL, van Puijvelde GH, van Duivenvoorde LM, van Wanrooij EJ, de Vos P, Tervaert JW, van Berkel TJ, Toes RE, Kuiper J. Vaccination using oxidized low-density lipoprotein-pulsed dendritic cells reduces atherosclerosis in LDL receptor-deficient mice. Cardiovasc Res. 2010;85:622–630. doi: 10.1093/cvr/cvp338. [DOI] [PubMed] [Google Scholar]
- 14.Van Brussel I, Lee WP, Rombouts M, Nuyts AH, Heylen M, De Winter BY, Cools N, Schrijvers DM. Tolerogenic dendritic cell vaccines to treat autoimmune diseases: Can the unattainable dream turn into reality? Autoimm Rev. 2014;13:138–150. doi: 10.1016/j.autrev.2013.09.008. [DOI] [PubMed] [Google Scholar]
- 15.SenBanerjee S, Lin Z, Atkins GB, Greif DM, Rao RM, Kumar A, Feinberg MW, Chen Z, Simon DI, Luscinskas FW, Michel TM, Gimbrone MA, Jr, Garcia-Cardena G, Jain MK. KLF2 Is a novel transcriptional regulator of endothelial proinflammatory activation. J Exp Med. 2004;199:1305–1315. doi: 10.1084/jem.20031132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sharma N, Lu Y, Zhou G, Liao X, Kapil P, Anand P, Mahabeleshwar GH, Stamler JS, Jain MK. Myeloid Kruppel-like factor 4 deficiency augments atherogenesis in ApoE−/− mice–brief report. Arterio Thromb, Vasc Bio. 2012;32:2836–2838. doi: 10.1161/ATVBAHA.112.300471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mahabeleshwar GH, Kawanami D, Sharma N, Takami Y, Zhou G, Shi H, Nayak L, Jeyaraj D, Grealy R, White M, McManus R, Ryan T, Leahy P, Lin Z, Haldar SM, Atkins GB, Wong HR, Lingrel JB, Jain MK. The myeloid transcription factor KLF2 regulates the host response to polymicrobial infection and endotoxic shock. Immunity. 2011;34:715–728. doi: 10.1016/j.immuni.2011.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bu DX, Tarrio M, Grabie N, Zhang Y, Yamazaki H, Stavrakis G, Maganto-Garcia E, Pepper-Cunningham Z, Jarolim P, Aikawa M, Garcia-Cardena G, Lichtman AH. Statin-induced Kruppel-like factor 2 expression in human and mouse T cells reduces inflammatory and pathogenic responses. J Clin Invest. 2010;120:1961–1970. doi: 10.1172/JCI41384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hart GT, Wang X, Hogquist KA, Jameson SC. Kruppel-like factor 2 (KLF2) regulates B-cell reactivity, subset differentiation, and trafficking molecule expression. Proc Nat Acad Sci USA. 2011;108:716–721. doi: 10.1073/pnas.1013168108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Batal I, De Serres SA, Safa K, Bijol V, Ueno T, Onozato ML, Iafrate AJ, Herter JM, Lichtman AH, Mayadas TN, Guleria I, Rennke HG, Najafian N, Chandraker A. Dendritic Cells in Kidney Transplant Biopsy Samples Are Associated with T Cell Infiltration and Poor Allograft Survival. J Am Soc Neph JASN. 2015;26:3102–3113. doi: 10.1681/ASN.2014080804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee JS, Yu Q, Shin JT, Sebzda E, Bertozzi C, Chen M, Mericko P, Stadtfeld M, Zhou D, Cheng L, Graf T, MacRae CA, Lepore JJ, Lo CW, Kahn ML. Klf2 is an essential regulator of vascular hemodynamic forces in vivo. Develop Cell. 2006;11:845–857. doi: 10.1016/j.devcel.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 22.Lingrel JB, Pilcher-Roberts R, Basford JE, Manoharan P, Neumann J, Konaniah ES, Srinivasan R, Bogdanov VY, Hui DY. Myeloid-specific Kruppel-like factor 2 inactivation increases macrophage and neutrophil adhesion and promotes atherosclerosis. Circ Res. 2012;110:1294–1302. doi: 10.1161/CIRCRESAHA.112.267310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Das M, Lu J, Joseph M, Aggarwal R, Kanji S, McMichael BK, Lee BS, Agarwal S, Ray-Chaudhury A, Iwenofu OH, Kuppusamy P, Pompili VJ, Jain MK, Das H. Kruppel-like factor 2 (KLF2) regulates monocyte differentiation and functions in mBSA and IL-1beta-induced arthritis. Curr Mol Med. 2012;12:113–125. doi: 10.2174/156652412798889090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sebzda E, Zou Z, Lee JS, Wang T, Kahn ML. Transcription factor KLF2 regulates the migration of naive T cells by restricting chemokine receptor expression patterns. Nat Immunol. 2008;9:292–300. doi: 10.1038/ni1565. [DOI] [PubMed] [Google Scholar]
- 25.Son YI, Egawa S, Tatsumi T, Redlinger RE, Jr, Kalinski P, Kanto T. A novel bulk-culture method for generating mature dendritic cells from mouse bone marrow cells. J Immunol Meth. 2002;262:145–157. doi: 10.1016/s0022-1759(02)00013-3. [DOI] [PubMed] [Google Scholar]
- 26.Fortier AH, Falk LA. Isolation of murine macrophages. Current protocols in immunology / edited by John E Coligan .. [et al.] 2001 doi: 10.1002/0471142735.im1401s11. Chapter 14: Unit 14 11. [DOI] [PubMed] [Google Scholar]
- 27.Lichtman AH, Clinton SK, Iiyama K, Connelly PW, Libby P, Cybulsky MI. Hyperlipidemia and atherosclerotic lesion development in LDL receptor-deficient mice fed defined semipurified diets with and without cholate. Arterioscl Thromb Vasc Bio. 1999;19:1938–1944. doi: 10.1161/01.atv.19.8.1938. [DOI] [PubMed] [Google Scholar]
- 28.Foks AC, Engelbertsen D, Kuperwaser F, Alberts-Grill N, Gonen A, Witztum JL, Lederer J, Jarolim P, DeKruyff RH, Freeman GJ, Lichtman AH. Blockade of Tim-1 and Tim-4 Enhances Atherosclerosis in Low-Density Lipoprotein Receptor-Deficient Mice. Arterioscl Thromb Vasc Bio. 2016;36:456–465. doi: 10.1161/ATVBAHA.115.306860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Engelbertsen D, Foks AC, Alberts-Grill N, Kuperwaser F, Chen T, Lederer JA, Jarolim P, Grabie N, Lichtman AH. Expansion of CD25+ Innate Lymphoid Cells Reduces Atherosclerosis. Arterioscl Thromb Vasc Bio. 2015;35:2526–2535. doi: 10.1161/ATVBAHA.115.306048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kamala T. Hock immunization: a humane alternative to mouse footpad injections. J Immunol Meth. 2007;328:204–214. doi: 10.1016/j.jim.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lyons AB, Doherty KV. Flow cytometric analysis of cell division by dye dilution. Curr Protoc Cytom. 2004 doi: 10.1002/0471142956.cy0911s27. Chapter 9: Unit 9 11. [DOI] [PubMed] [Google Scholar]
- 32.Monti P, Mercalli A, Leone BE, Valerio DC, Allavena P, Piemonti L. Rapamycin impairs antigen uptake of human dendritic cells. Transplantation. 2003;75:137–145. doi: 10.1097/00007890-200301150-00025. [DOI] [PubMed] [Google Scholar]
- 33.Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, Pittet MJ. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007;117:195–205. doi: 10.1172/JCI29950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Swirski FK, Pittet MJ, Kircher MF, Aikawa E, Jaffer FA, Libby P, Weissleder R. Monocyte accumulation in mouse atherogenesis is progressive and proportional to extent of disease. Proc Nat Acad Sci USA. 2006:40–10345. doi: 10.1073/pnas.0604260103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Getz GS, Reardon CA. Animal models of atherosclerosis. Arteriosclerosis, thrombosis, and vascular biology. 2012;32:1104–1115. doi: 10.1161/ATVBAHA.111.237693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Swirski FK, Weissleder R, Pittet MJ. Heterogeneous in vivo behavior of monocyte subsets in atherosclerosis. Arterioscl Thromb Vasc Bio. 2009;29:1424–1432. doi: 10.1161/ATVBAHA.108.180521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Witztum JL, Lichtman AH. The influence of innate and adaptive immune responses on atherosclerosis. Ann Rev Path. 2014;9:73–102. doi: 10.1146/annurev-pathol-020712-163936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hoyer KK, Dooms H, Barron L, Abbas AK. Interleukin-2 in the development and control of inflammatory disease. Immunological reviews. 2008;226:19–28. doi: 10.1111/j.1600-065X.2008.00697.x. [DOI] [PubMed] [Google Scholar]
- 39.Zernecke A. Dendritic cells in atherosclerosis: evidence in mice and humans. Arterioscl Thromb Vasc Bio. 2015;35:763–770. doi: 10.1161/ATVBAHA.114.303566. [DOI] [PubMed] [Google Scholar]
- 40.Fang H, Lin J, Wang L, Xie P, Wang X, Fu J, Ai W, Chen S, Chen F, Zhang F, Su Y, Li D. Kruppel-like factor 2 regulates dendritic cell activation in patients with acute coronary syndrome. Cell Physiol Biochem. 2013;32:931–941. doi: 10.1159/000354496. [DOI] [PubMed] [Google Scholar]
- 41.Bista P, Mele DA, Baez DV, Huber BT. Lymphocyte quiescence factor Dpp2 is transcriptionally activated by KLF2 and TOB1. Mol Immunol. 2008;45:3618–3623. doi: 10.1016/j.molimm.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Buckley AF, Kuo CT, Leiden JM. Transcription factor LKLF is sufficient to program T cell quiescence via a c-Myc-dependent pathway. Nat Immunol. 2001;2:698–704. doi: 10.1038/90633. [DOI] [PubMed] [Google Scholar]
- 43.Dekker RJ, Boon RA, Rondaij MG, Kragt A, Volger OL, Elderkamp YW, Meijers JC, Voorberg J, Pannekoek H, Horrevoets AJ. KLF2 provokes a gene expression pattern that establishes functional quiescent differentiation of the endothelium. Blood. 2006;107:4354–4363. doi: 10.1182/blood-2005-08-3465. [DOI] [PubMed] [Google Scholar]
- 44.Cella M, Scheidegger D, Palmer-Lehmann K, Lane P, Lanzavecchia A, Alber G. Ligation of CD40 on dendritic cells triggers production of high levels of interleukin-12 and enhances T cell stimulatory capacity: T-T help via APC activation. J Exp Med. 1996;184:747–752. doi: 10.1084/jem.184.2.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 46.Frleta D, Lin JT, Quezada SA, Wade TK, Barth RJ, Noelle RJ, Wade WF. Distinctive maturation of in vitro versus in vivo anti-CD40 mAb-matured dendritic cells in mice. J Immunother. 2003;26:72–84. doi: 10.1097/00002371-200301000-00008. [DOI] [PubMed] [Google Scholar]
- 47.Ouaaz F, Arron J, Zheng Y, Choi Y, Beg AA. Dendritic cell development and survival require distinct NF-kappaB subunits. Immunity. 2002;16:257–270. doi: 10.1016/s1074-7613(02)00272-8. [DOI] [PubMed] [Google Scholar]
- 48.Wong BR, Josien R, Lee SY, Sauter B, Li HL, Steinman RM, Choi Y. TRANCE (tumor necrosis factor [TNF]-related activation-induced cytokine), a new TNF family member predominantly expressed in T cells, is a dendritic cell-specific survival factor. J Exp Med. 1997;186:2075–2080. doi: 10.1084/jem.186.12.2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Inaba K, Turley S, Iyoda T, Yamaide F, Shimoyama S, Reis e Sousa C, Germain RN, Mellman I, Steinman RM. The formation of immunogenic major histocompatibility complex class II-peptide ligands in lysosomal compartments of dendritic cells is regulated by inflammatory stimuli. J Exp Med. 2000;191:927–936. doi: 10.1084/jem.191.6.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.De Smedt T, Pajak B, Muraille E, Lespagnard L, Heinen E, De Baetselier P, Urbain J, Leo O, Moser M. Regulation of dendritic cell numbers and maturation by lipopolysaccharide in vivo. J Exp Med. 1996;184:1413–1424. doi: 10.1084/jem.184.4.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gommerman JL, Summers deLuca L. LTbetaR and CD40: working together in dendritic cells to optimize immune responses. Immunol Rev. 2011;244:85–98. doi: 10.1111/j.1600-065X.2011.01056.x. [DOI] [PubMed] [Google Scholar]
- 52.van Tits LJ, Stienstra R, van Lent PL, Netea MG, Joosten LA, Stalenhoef AF. Oxidized LDL enhances pro-inflammatory responses of alternatively activated M2 macrophages: a crucial role for Kruppel-like factor 2. Atherosclerosis. 2011;214:345–349. doi: 10.1016/j.atherosclerosis.2010.11.018. [DOI] [PubMed] [Google Scholar]
- 53.Herter JM, Grabie N, Cullere X, Azcutia V, Rosetti F, Bennett P, Herter-Sprie GS, Elyaman W, Luscinskas FW, Lichtman AH, Mayadas TN. AKAP9 regulates activation-induced retention of T lymphocytes at sites of inflammation. Nature communications. 2015;6:10182. doi: 10.1038/ncomms10182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Alberts-Grill N, Denning TL, Rezvan A, Jo H. The role of the vascular dendritic cell network in atherosclerosis. Am J Physiol Cell Phys. 2013 doi: 10.1152/ajpcell.00017.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bobryshev YV, Lord RS. Mapping of vascular dendritic cells in atherosclerotic arteries suggests their involvement in local immune-inflammatory reactions. Cardiovasc Res. 1998;37:799–810. doi: 10.1016/s0008-6363(97)00229-0. [DOI] [PubMed] [Google Scholar]
- 56.Bobryshev YV, Lord RS. Structural heterogeneity and contacting interactions of vascular dendritic cells in early atherosclerotic lesions of the human aorta. J Submicrosc Cytol Pathol. 1996;28:49–60. [PubMed] [Google Scholar]
- 57.Bobryshev YV, Lord RS. Ultrastructural recognition of cells with dendritic cell morphology in human aortic intima. Contacting interactions of Vascular Dendritic Cells in athero-resistant and athero-prone areas of the normal aorta. Arch Histol Cytol. 1995;58:307–322. doi: 10.1679/aohc.58.307. [DOI] [PubMed] [Google Scholar]
- 58.Martin S, Agarwal R, Murugaiyan G, Saha B. CD40 expression levels modulate regulatory T cells in Leishmania donovani infection. J Immunol. 2010;185:551–559. doi: 10.4049/jimmunol.0902206. [DOI] [PubMed] [Google Scholar]
- 59.Dai Z, Arakelov A, Wagener M, Konieczny BT, Lakkis FG. The role of the common cytokine receptor gamma-chain in regulating IL-2-dependent, activation-induced CD8+ T cell death. J Immunol. 1999;163:3131–3137. [PubMed] [Google Scholar]
- 60.Lenardo MJ. Interleukin-2 programs mouse alpha beta T lymphocytes for apoptosis. Nature. 1991;353:858–861. doi: 10.1038/353858a0. [DOI] [PubMed] [Google Scholar]
- 61.Richter GH, Mollweide A, Hanewinkel K, Zobywalski C, Burdach S. CD25 blockade protects T cells from activation-induced cell death (AICD) via maintenance of TOSO expression. Scand J Immunol. 2009;70:206–215. doi: 10.1111/j.1365-3083.2009.02281.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.