

Abstract
AMP-activated protein kinase (AMPK) is the central metabolic regulator of the cell and controls energy consumption based upon nutrient availability. Due to its role in energy regulation, AMPK has been implicated as a barrier for cancer progression and is suppressed in multiple cancers. To examine whether AMPK regulates bladder cancer cell growth, HTB2 and HT1376 bladder cells were treated with an AMPK activator, AICAR. AICAR treatment reduced proliferation and induced the expression of p27Kip1 (CDKN1B), which was mediated through an mTOR-dependent mechanism. Interestingly, AMPKα2 knockdown resulted in reduced p27 levels, whereas AMPKα1 suppression did not. To further determine the exact mechanism by which AMPKa2 regulates p27, HTB2 and HT1376 cells were transduced with a shRNA targeting AMPKα2. Stable knockdown of AMPKα2 resulted in increased proliferation and decreased p27 protein. The reduced p27 protein was determined to be dependent upon SKP2. Additionally, loss of AMPKα2 in a xenograft and a chemical carcinogen model of bladder cancer resulted in larger tumors with less p27 protein and high SKP2 levels. Consistent with the regulation observed in the bladder cancer model systems, a comprehensive survey of human primary bladder cancer clinical specimens revealed low levels of AMPKα2 and p27, and high levels of SKP2.
Implications
These results highlight the contribution of AMPKα2 as a mechanism for controlling bladder cancer growth by regulating proliferation through mTOR suppression and induction of p27 protein levels, thus indicating how AMPKα2 loss may contribute to tumorigenesis.
Keywords: Bladder cancer, AMPK, p27, SKP2
Graphical abstract
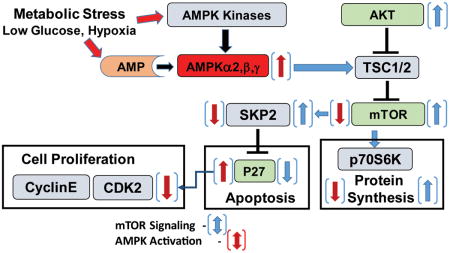
Introduction
Bladder cancer is currently the fifth most commonly diagnosed cancer with average age of onset of 71 years old. It is also one of the most expensive cancers to treat due to lifelong surveillance and invasive procedures (1, 2). Adenosine monophosphate activated protein kinase (AMPK) is a critical protein in the cell that signals to control proliferation and protein translation under cellular stress (3, 4). Once activated by an upstream kinase such as STK11, AMPK signaling negatively regulates MTOR (mTOR) to halt protein translation and proliferation when energy availability is low (5-7). AMPK is composed of an α, β and γ subunit. The α subunit which is composed of two isoforms (PRKAA1/AMPKα1 or PRKAA2/AMPKα2), functions as the catalytic subunit while the β and γ subunit function as a regulatory and AMP sensing subunit respectively (8, 9).
Due to its role in responding to cellular energy and controlling the switch from anabolic to catabolic processes in response to low nutrients, AMPK has long been thought to function to inhibit tumorigenesis. In many cancers the activation of AMPK results in decreased proliferation and survival of cancer cells (10-12). Additionally, it has been reported that AMPK activation and/or AMPKα2 protein or mRNA is diminished in breast cancer, hepatocellular carcinoma and gastric cancer (13-17). Although there is little data on the selective functions of the AMPKα isoforms, emerging evidence suggests that the proteins may indeed have some selective functions. For example, AMPKα2 has been shown to localize to the nucleus and can phosphorylate nuclear targets whereas AMPKα1 does not (18, 19). Additionally, Phoenix et al. demonstrated that AMPKα2-/- mouse embryonic fibroblasts that were transformed with H-RasV12 formed tumors in a xenograft while the AMPKα1 -/- mouse embryonic fibroblasts did not (20). AMPKα2 -/- vascular smooth muscle cells also displayed increased proliferation and a reduction in CDKN1B (p27) protein (21). All of these data demonstrate that AMPKα2 may be more selective for controlling p27 protein levels and proliferation than AMPKα1.
The cell cycle inhibitor protein p27 is a critical protein for controlling cellular proliferation and its loss has been heavily implicated in tumorigenesis. Low expression of p27 protein in many different cancer tissues including bladder cancer has been shown to be associated with worse survival and invasive disease (22-24). Under normal conditions most urothelial cells express p27 due to their slow proliferative rate. In a chemical carcinogenesis model of bladder cancer, p27-/- mice develop bladder cancer at a much earlier time point than their wild-type counter parts due to the critical role of p27 in controlling urothelial proliferation (25). These experiments demonstrate how crucial p27 is in controlling bladder cancer cell growth. The exact mechanism for loss of p27 function in bladder cancer is currently unknown. One of the major mechanisms governing p27 regulation is S-phase kinase-associated protein 2 (SKP2) mediated degradation (26). Although there are no studies correlating the expression status of SKP2 and p27 in bladder cancer, independent studies have shown that p27 is down-regulated in invasive bladder cancer and SKP2 is up-regulated (27). Therefore, this study seeks to determine the impact of AMPKα activation on bladder cancer proliferation and whether this is dependent on, or mediated in part by SKP2 mediated degradation and control of p27 protein.
Materials and Methods
Cell Lines and Reagents
Cell lines were purchased and maintained according to the American Type Culture Collection (ATCC). Cells obtained from ATCC were frozen down within 5 passages of being received and aliquots were not cultured for more than 15 passages. Rapamycin and 5-Aminoimidazole-4-carboxamide ribonucleotide (AICAR) were obtained from Cayman Chemical. Compound C was obtained from EMD millipore. Propidium iodide and RNase A were obtained from Invitrogen (Carlsbad, CA). Antibodies targeting AMPKα1 and AMPKα2 were obtained from US Biological (Swampscott, MA), total AMPKα, pAMPKαThr172, SKP2 and pS6 were obtained from Cell Signaling Technology (Beverly, MA), p27 was obtained from BD Pharmingen, KI67 was obtained from DAKO and β-actin was obtained from Abcam (Cambridge, MA).
HTB2 and HT1376 stable cell transfection, selection and maintenance
HTB2 and HT1376 cells were transduced with lentivirus containing shGFP and shAMPKα2 in the LKO.1-puromycin vector (Sigma, St. Louis, MO) as described previously (20). Briefly, HEK-293 FT cells were plated and transfected the next day with the LKO.1 vector (shGFP or shAMPKα2) and vectors containing RRE, REV and VSV-G. Lentiviral particles were harvested 72 hours post transfection and used to transduce HTB2 and HT1376 cells. HTB2 and HT1376 cells were transduced for 24 hours followed by 24 hours of recovery before being selected with puromycin for 72 hours. Stable expression was assessed post selection.
Cell Culture Growth Assays
Cells were plated in a 24 well dish (20,000 cells/well) and treated with AICAR the next day in triplicate. Live cell counts were taken on day 0 and at indicated experimental times. Briefly, cells were trypsinized and triturated to form a single cell suspension. Cells, in triplicate, were counted using a Coulter Counter®, (Beckman Coulter, Brea, CA) and normalized to day 0 cell counts to obtain percent growth. Graphs are representative data of one experiment out of a total of 3 distinct experiments.
Cell Cycle Analysis
HTB2 and HT1376 cells (1×106) were plated in p60 dishes. The next day the medium was aspirated and replaced with fresh medium or fresh medium containing 1mM AICAR. Cells were treated for a total of 24 hours and subsequently fixed in 70% ethanol. Fixed cells were incubated with propidium iodide (PI) staining solution (50μg/ml PI and 100μg/ml RNase A diluted in PBS) for 30 minutes before analysis by flow cytometry.
AICAR, Compound C and Rapamycin Treatments and Immunoblot Analysis
HTB2 and HT1376 cells were plated and eighteen hours later subsequently re-fed and treated with 0.5 mM AICAR, 5nM rapamycin or 5μM compound C for a total of 24 hours. Whole cell lysates were then harvested and analyzed by immunoblot as described previously (14). Immunoblots were visualized using ECL reagents (Millipore, Billerica, MA) and developed on a Kodak Multimodal Imager (2000MM). Signal was quantified by selecting each band and measuring total photon counts using the Kodak imaging software. Immunoblots are representative of at least 3 distinct experiments. All blots signals are normalized to the β-actin representative for that experiment.
Immunofluorescence
HTB2 and HT1376 cells transduced with either shGFP or shAMPKα2 were plated in a 6 well dish containing glass cover slips and allowed to grow for 48 hours. Cells were then fixed and stained using a p27 antibody. Nuclei were visualized by staining with DAPI (4′,6-diamidino-2-phenylindole). Cover slips were mounted with Prolong gold antifade reagent (Invitrogen, Carlsbad, CA) and visualized on a Zeiss Axio Observer.Z1 with a axiocam MRm camera using Zen software at 400× magnification.
siRNA transfection
Lipofectamine 2000™ (life technologies, Carlsbad, CA) was used to transfect HTB2 or HT1376 cells with 100nM of either siLuciferase, siAMPKα1, siAMPKα2 (Dharmacon) or siSKP2 (Cell signaling). Cells were lysed 72 hours post transfection and further analyzed by immunoblot.
Quantitative RT-PCR
RNA isolation and cDNA synthesis were carried out using the RNeasy mini kit (Qiagen, Valencia, CA) and iScript cDNA synthesis kit (Bio-Rad, Hercules, CA), respectively according to the manufacturer protocol. Primers are as follows: AMPKα1 forward (5′-AGGAGAGCTATTTGATTATATCTGTAAGAATG-3′), AMPKα1 reverse (5′-ACACCAGAAAGGATCTGTTGGAA-3′), AMPKα2 forward (5′-CGGCTCTTTCAGCAGATTCTGT-3′) AMPKα2 reverse (5′-ATCGGCTATCTTGGCATTCATG-3′), Cyclophilin A forward (5′-CTGGACCCAACACAAATGGTT-3′), Cyclophilin A reverse (5′-CCACAATATTCATGCCTTCTTTCA-3′). Quantitative PCR was then performed with SYBR green fluorescence on a Bio-Rad thermocycler with a MyIQ detection system.
Xenograft Model
HT1376 shGFP and HT1376 shAMPKα2 were injected in the flank (2×106 per site) region of 6-8 week old male NSG mice. Tumors were measured using an external caliper twice a week over 34 days. The estimate ellipsoid volume equation (length × width × height × 0.52) was used to calculate tumor volume.
BBN Chemical carcinogenesis model
Male wild-type and AMPKα2-/- C57BL/6 (28) at the age of 8 weeks were supplemented with 0.05% N-Butyl-N-(4-hydroxybutyl)nitrosamine (BBN) in their drinking water twice weekly for 20 weeks. After 20 weeks mice were harvested and their bladders were removed, weighed and processed for histological analysis.
Histological assessment
Bladder cancer tissue array (BL1002) was obtained from US Biomax (Rockville, MD). Immunohistochemistry was performed on paraffin sections and quantified as described previously(14). Staining was quantified from 5 40× images per section using Image Pro Plus or through manual counting. Histoscore was determined based scoring from three independent reviewers. Samples that contained no staining were scored as 0, while samples that contained staining were scored from a range of 1-3, where 3 is representative of the strongest staining. Samples were also given a percent coverage for each score and then the numbers were multiplied to obtain a range of 0-300. Images were taken on a Zeiss Axioplan 2 microscope with AxioCam MRc camera and AxioVision software (Oberkochen, Germany) at 400× magnification.
Statistical Analysis
Data from each experiment is represented as the mean +/- standard error of the mean (SEM). Statistical analysis was performed using the 2-tailed student t-test or ANOVA with tests for equal variance where appropriate. Significance is represented as *p<0.05, **p<0.01, *** p<0.001.
Results
AMPK activation reduces proliferation in human bladder cancer cells through mTOR-mediated control of p27
In order to determine the effect of AMPK activation in bladder cancer, HTB2 (representative of low grade bladder cancer) and HT1376 (representative of high grade bladder cancer) bladder cancer cell lines were treated with 0.5 mM 5-Aminoimidazole-4-carboxamide ribonucleotide (AICAR), an AMPK activator, and proliferation was determined by cell count 72 hours post-treatment. Treatment with AICAR led to a significant decrease in proliferation in both cell lines (Fig.1A-B). In order to determine the mechanism by which AMPK activation reduces proliferation, cell cycle analysis was performed on HTB2 and HT1376 cells treated with 1mM AICAR for 24 hours. Cell cycle analysis revealed that both cell lines presented with a significant increase in the proportion of cells in G1 indicating that AMPK activation controls proliferation of bladder cancer cells through inducing G1 arrest (Fig. 1 C-D). To better understand the signaling mechanisms responsible for AMPK mediated suppression of proliferation, both HTB2 and HT1376 cells were treated with 0.5mM AICAR for 24 hours and immunoblots were performed. Both cells lines displayed potent activation of AMPK in response to AICAR as demonstrated by AMPKαThr172 phosphorylation and ACACA (ACC) phosphorylation (direct substrate of AMPKα). Furthermore, both cell lines demonstrated inhibition of mTOR activation as assessed by a decrease in phosphorylation of ribosomal protein S6 (S6). Additionally, HTB2 cells displayed a 110% up-regulation of p27 and HT1376 cells displayed a 27% increase in p27 levels in this experiment when normalized to β-actin in response to AICAR (Fig. 1 E-F and Supplemental Fig. S1 A-B). AICAR treatment did not alter CCNE1 (Cyclin E) or CDK2 levels and resulted in only minor suppression of CCND1 (Cyclin D1) and CDK4 in HT1376 cells (Supplemental Fig. S2 A-B). Taken together this data demonstrates the importance of AMPK signaling in maintaining cellular proliferation of bladder cancer cells by regulating p27 protein levels and thus causing G1 arrest. Since AMPK is a major negative regulator of mTOR activity through activation of the upstream TSC1/2 complexes, we next analyzed whether mTOR inhibition could replicate the AMPK dependent regulation of p27 by using rapamycin. HTB2 and HT1376 cells were treated with 5 nM rapamycin for 24 hours and immunoblot analysis was performed. In both cell lines rapamycin significantly inhibited the phosphorylation of ribosomal protein S6 (S6), indicating that mTOR activation was blocked. Additionally, rapamycin treatment induced the expression of p27 by 57% and 43% when normalized to β-actin in this experiment, consistent with the effects of AICAR on the HTB2 and HT1376 cells, respectively (Fig. 1 G-H and Supplemental Fig. S 1 C-D). To determine more specifically if AMPK mediated suppression of mTOR is responsible for the observed up-regulation of p27 protein and cellular growth, HTB2 and HT1376 cells were treated with rapamycin and compound C, an AMPK inhibitor. In both cell lines compound C resulted in a down-regulation of p27 protein and an up-regulation of p-S6. Additionally, rapamycin inhibition of mTOR resulted in reduced p-S6 and coordinate up-regulation of p27. The combination of compound C and rapamycin demonstrated that by blocking AMPK activation the up-regulation of p27 caused by mTOR inhibition with rapamycin alone was diminished indicating that effect is in part mediated through mTOR. (Fig. 1 I-J and Supplemental Fig. S1 E-F). Taken together this data demonstrates that AMPK mediated control of p27 is dependent upon its ability to inhibit mTOR.
Figure 1. AMPK activation reduces proliferation through mTOR mediated control of p27.
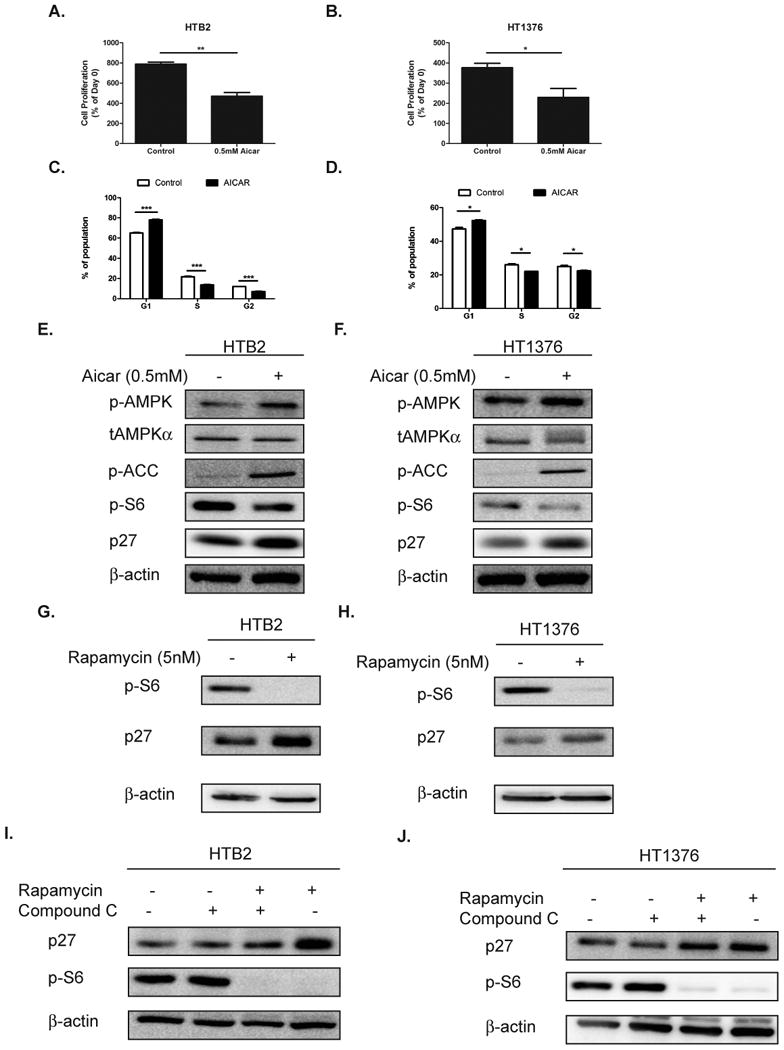
A-B HTB2 (A.) and HT1376 (B.) bladder cancer cells were treated with 0.5mM AICAR for 72 hours and cell proliferation was assessed by direct cell count. C-D. Cell cycle analysis of HTB2 (C.) and HT1376 (D.) cells treated with 1mM AICAR for 24 hours. E-F. HTB2 (E.) and HT1376 (F.) were treated with 0.5mM AICAR for 24 hours and immunoblot analysis for p-AMPKα, p-ACC, p-S6 and p27 was performed. β-actin was used as a loading control. G-H. HTB2 (G.) and HT1376 (H.) bladder cancer cells were treated with 5nM rapamycin for 24 hours and immunoblot for p-S6, p27 and β-actin was performed. I-J. HTB2 (I.) and HT1376 (J.) were treated simultaneously with Rapamycin (5nM) and compound C (5μM) for 24 hours and immunoblot for p27, p-S6 and β-actin was performed. Statistical significance indicated as; *p<0.05, **p<0.01, ***p<0.001
AMPK mediated control of p27 is dependent on the AMPKα2 isoform
AMPK is a heterotrimeric kinase composed of 3 subunits (α, β and γ) with the α subunit containing the kinase domain. In order to determine which of the two α subunits were responsible for the observed regulation of p27, HTB2 cells were transfected with 100nM of siRNA against luciferase as a control, AMPKα1, or AMPKα2 and analyzed by immunoblot. As depicted in Figure 2A, transfection of siAMPKα1 in HTB2 cells caused a 50% reduction of AMPKα1 protein, but did not alter the levels of p27 protein. However, transfection of siAMPKα2 resulted in a 55% reduction in AMPKα2 levels and a caused a 45% reduction in p27 protein levels (Supplemental Fig. 3A). Transfection of HT1376 cells with siAMPKα1 resulted in a 48% reduction in AMPKα1 but also reduced p27 protein levels by 38%. However, transfection with siAMPKα2 resulted in a 51% reduction in AMPKα2 levels and a 51% reduction in p27 protein levels (Fig. 2B and Supplemental Fig. 3B). Taken together this data suggests that AMPKα2 may be more selective in regulating the levels of p27 in bladder cancer cells.
Figure 2. Knockdown of AMPKα2 in bladder cancer cells leads to a reduction in p27 levels.
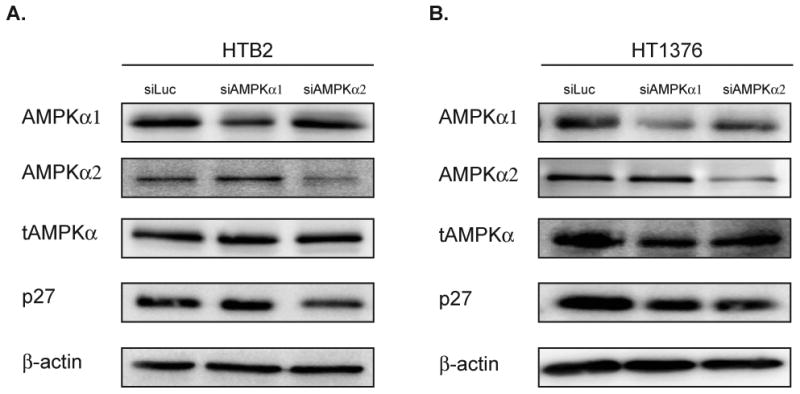
A-B. HTB2 and HT1376 cells were transfected with siRNA (100nM) against luciferase, AMPKα1 or AMPKα2 and assessed by immunoblot 72 hours post transfection for AMPKα1, AMPKα2, total AMPK (α1/α2), p27 and β-actin.
AMPKα2 regulates cellular proliferation in bladder cancer through p27
To determine if AMPKα2 is indeed responsible for the AMPK mediated control of cell proliferation, bladder cancer cells that express the AMPKα2 protein were transduced with lentiviral-shRNA to AMPKα2 or GFP (as a control). The HTB2 and HT1376 stably selected clones were analyzed by immunoblot. HTB2 shAMPKα2 cells displayed a 56% reduction in AMPKα2 levels and a 13% reduction in total AMPK alpha 1 and 2 (tAMPKα1/α2) levels (Fig. 3 A). HT1376 shAMPKα2 cells displayed a 46% reduction in AMPKα2 levels and a 28% reduction in tAMPKα1/α2 levels (Fig. 3 B). Quantitative real time PCR analysis of these cells also revealed corresponding reductions in AMPKα2 mRNA in both HTB2 (77%) and HT1376 (54%) cells (Fig. 3 C-D). Further analysis of the AMPKα2 knockdown cells revealed that p27 protein levels were down-regulated by 50% in the HTB2 shAMPKα2 cells (Fig. 3 E) and 41% in the HT1376 shAMPKα2 cells (Fig. 3 F). In order to determine more specifically what could be causing the down-regulation of p27, SKP2 levels were analyzed. SKP2 is a subunit of the F-box ubiquitin ligase complex which targets p27 (29). In both the HTB2 shAMPKα2 and the HT1376 shAMPKα2 cells, SKP2 levels were up-regulated by 40% and 81% respectively (Fig. 3 E-F). In both cell lines, shAMPKa2 did not affect the cell cycle proteins Cyclin D1, Cyclin E, or CDK2, however did induce a moderate increase in CDK4 (Supplemental Fig. S4 A-B). Furthermore, to determine if the down-regulation of p27 also affected its nuclear accumulation, immunofluoresence for p27 was performed on HTB2 shAMPKα2, HT1376 shAMPKα2 and their respective control cells (Fig. 3 G). Quantification of the percentage of nuclear p27 cells revealed that there were significantly fewer p27 positive cells in the shAMPKα2 cell lines (Fig. 3 H). Consistent with the down-regulation of total p27 and nuclear p27, the HTB2 and HT1376 shAMPKα2 cells displayed a significant increase in proliferation as assessed by direct cell count over 7 days (Fig. 3 I-J). Further analysis of HTB2 and HT1376 cells transduced with a different shRNA targeting AMPKα2 confirmed these results (Supplemental Fig. S4 C-D & S5 A-I). Together, these data demonstrate the importance of AMPKα2 in controlling bladder cell proliferation by regulating the levels of p27 potentially through SKP2 mediated mechanisms.
Figure 3. AMPKα2 knockdown reduces p27 through a SKP2-mediated mechanism and results in a proliferative advantage.
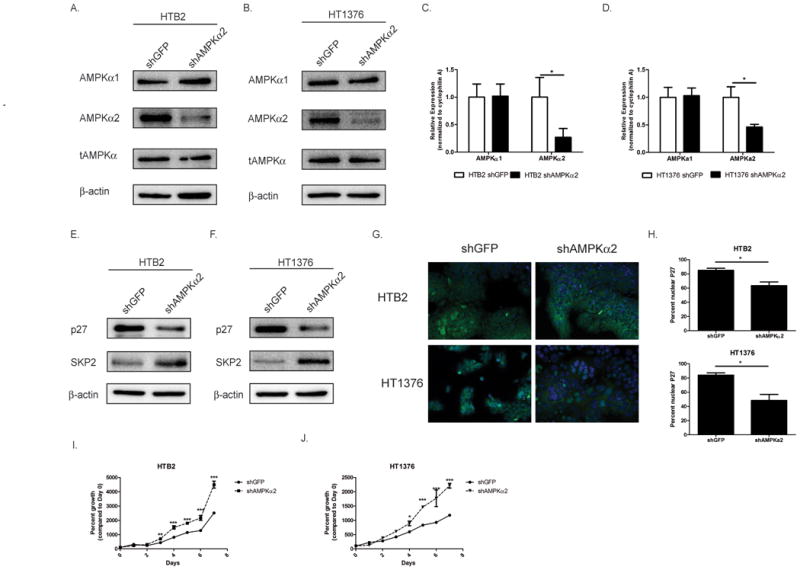
A-B. HTB2 (A.) and HT1376 (B.) cells were transduced with shRNA to GFP or AMPKα2 and stable cell lines were created. HTB2 and HT1376 stable cell lines were assessed by immunoblot for AMPKα1, AMPKα2, total AMPK (α1/α2) and β-actin. C-D. Transduced cells (see 3A &3B) were assessed by qRT-PCR for AMPKα1 and AMPKα2 mRNA levels. E-F. HTB2 shGFP, HTB2 shAMPKα2, HT1376 shGFP and HT1376 shAMPKα2 cells were assessed by immunoblot analysis for p27, SKP2 and β-actin. G. HTB2 and HT1376 stable cell lines were stained with p27 (green) and DAPI (blue). H. Quantification of staining in G. I-J. HTB2 and HT1376 stable cells were plated and cell counts were taken daily over 7 days. Graphs represented as percent growth of Day 0 cell count. Statistical significance indicated as; *p<0.05, **p<0.01, ***p<0.001
AMPK mediated control of p27 is SKP2 dependent
Due to the observed up-regulation of SKP2 in the AMPKα2 knockdown cell lines, cells were transfected with 100nM siRNA to either luciferase as a control or SKP2 for 72 hours to determine if AMPKα2 mediated control of p27 is indeed mediated though SKP2. Knockdown of SKP2 in both HTB2 and HT1376 indeed relieved the repression of p27 that occurred in the AMPKα2 knockdown cells (Fig. 4 A-B). In order to determine if this affect was biologically important, proliferation assays were performed simultaneously with immunoblot analyses. In both HTB2 and HT1376 cells, knockdown of SKP2 significantly negated the proliferative advantage of the shAMPKα2 cells (Fig. 4C-D). Furthermore, knockdown of SKP2 in HTB2 and HT1376 cells transduced with the second shRNA clones confirmed these results (Supplemental Fig. S6 A-D). This data signifies that the effect that AMPKα2 has on p27 and its role in controlling cellular proliferation is dependent on SKP2 regulation.
Figure 4. Knockdown of SKP2 negates the effects of AMPKα2 mediated control of p27 and cell proliferation.
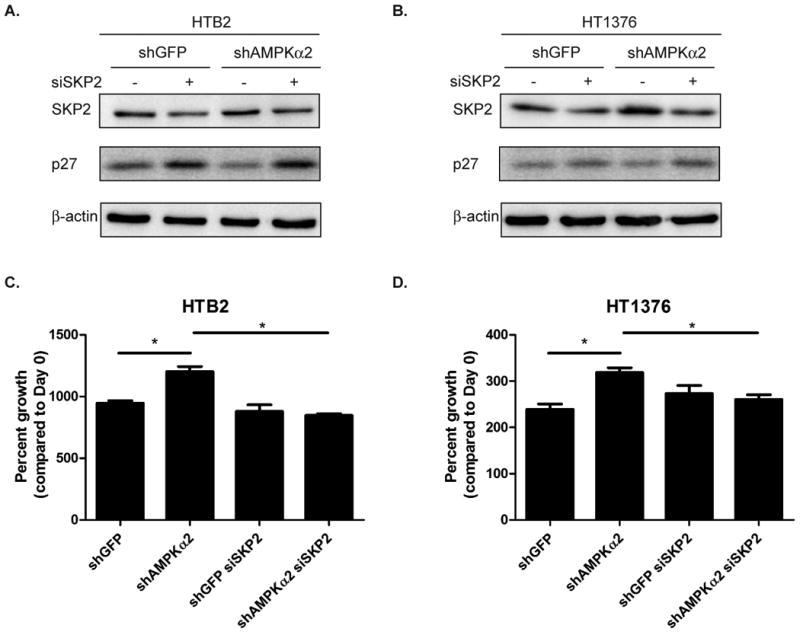
A-B. HTB2 shGFP, HTB2 shAMPKα2, HT1376 shGFP and HT1376 shAMPKα2 cells were transfected with siRNA(100nM) to luciferase or SKP2 and assessed for SKP2, p27 and β-actin levels by immunoblot 72 hours post transfection. C-D. Day 3 cell counts compared to day 0 cell counts of transduced cells transfected with siLuc or siSKP2. Statistical significance indicated as; *p<0.05
Knockdown of AMPKα2 results in larger tumor formation in a xenograft model
To determine whether AMPKα2 and its regulation of p27 could affect overall tumorigenicity, the bladder cancer cells, HT1376 shGFP and HT1376 shAMPKα2 cells were injected (2×106 cells) into the flank region of Nod Scid gamma (NSG) mice and tumors were allowed to grow for 34 days. On day 34, HT1376 shAMPKα2 tumors were 56% larger (p<0.05) than the shGFP control tumors (Fig. 5A) as determined by external caliper measurements and were subsequently collected and fixed for immunohistochemical analysis. In order to establish if the same signaling cascade was occurring in the xenograft model that occurred in vitro, the levels of phosphorylated AMPK were assessed by immunohistochemistry in the shGFP and the shAMPKα2 tumors. Indeed, knockdown of AMPKα2 significantly decreased p-AMPKThr172 levels by 23% in the xenograft model (Fig.5B). Consistent with the decreased AMPK activity in the shAMPKα2 tumors, it was also observed that there was a concomitant activation of mTOR by 48% in these tumors as determined by p-S6 levels (Fig. 5D). Additionally, knockdown of AMPKα2 in the xenograft model significantly increased proliferation (45% vs. 57%), as assessed by KI67 staining (Fig. 5C). Furthermore, p27 levels were significantly diminished by 55% (33% vs. 14%) in the shAMPKα2 group when compared to the shGFP group (Fig. 5E), with a corresponding increase in SKP2 levels (19% vs. 32%) (Fig. 5 F). Taken together these data demonstrate that the AMPKα2 mediated control of cellular proliferation in vitro is also seen in an in vivo model of bladder cancer.
Figure 5. Knockdown of AMPKα2 in HT1376 cells promotes tumor growth in a xenograft model.
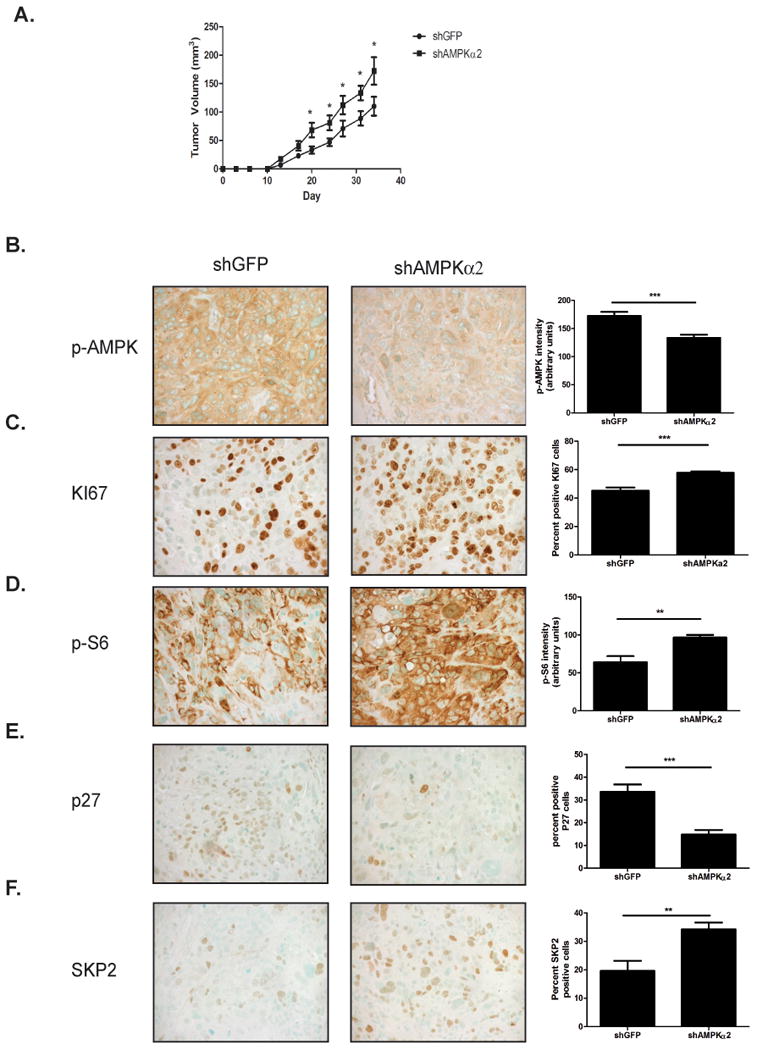
A. HT1376 shGFP and HT1376 shAMPKα2 (n=9/group) cells were injected into the flank of NSG mice. Tumor volume was measured twice weekly. B-F. pAMPK(B.), KI67(C.), p-S6(D.), p27(E.) and SKP2(F.) immunohistochemical staining representative images and corresponding quantification (n=9/group). Statistical significance indicated as; *p<0.05, **p<0.01, ***p<0.001
Genetic deletion of AMPKα2 results in larger bladder tumors in a chemical carcinogenesis model
In order to better establish the impact that AMPKα2 has on bladder tumorigenesis in an endogenous system, AMPKα2 knockout mice were utilized in a chemical carcinogenesis model. Age matched wild-type and AMPKα2-/- mice were given N-Butyl-N-(4-hydroxybutyl)nitrosamine (BBN) at a concentration of 0.05% for 20 weeks in drinking water. After 20 weeks of exposure, mice were euthanized and the bladders harvested were weighed as a surrogate marker for tumor burden. AMPKα2-/- bladders were significantly larger (44%, p<0.05) than the wild-type control bladders (Fig. 6A). Additionally, bladder tumors from AMPKα2-/- displayed a significant 52% reduction (p<0.05) in AMPK activation as assessed by p-AMPKThr172 when compared to wild-type mice (Fig. 6B-C), but only a 19% reduction in tAMPKα1/α2 (Fig. 6D-E). AMPKα2-/- bladder tumors also displayed a 33% increase in mTOR activation as assessed by p-S6 (Fig. 6H-I). Consistent with the in vitro cell proliferation and xenograft model, AMPKα2-/- tumors demonstrated a significant increase in proliferation as assessed by KI67 staining and quantification (Fig. 6F-G). Furthermore, AMPKα2-/- bladder tumors had significantly less p27 positive cells (p<0.05) (Fig. 6J-K) and a significant increase in SKP2 expression (p<0.05) (Fig. 6L-M), when compared to the wild-type control bladder tumors. Together these data demonstrate that AMPKα2 functions to control bladder cancer growth through regulating SKP2 mediated degradation of p27.
Figure 6. Genetic deletion of AMPKα2 results in larger tumors in a BBN carcinogen-induced bladder tumor model.
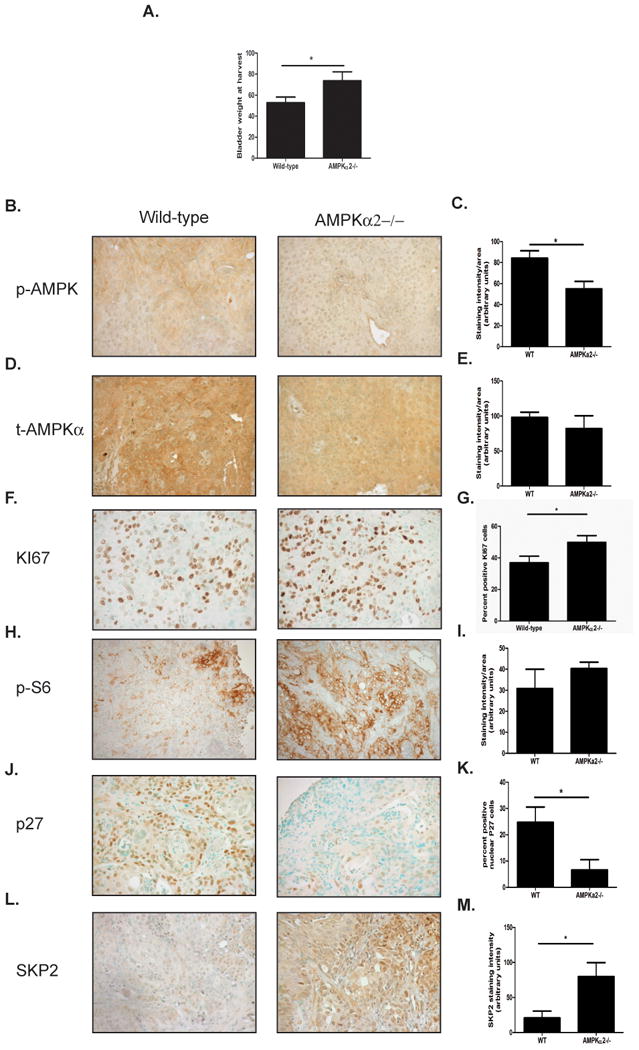
A. Bladder weights of wild-type and AMPKα2-/- mice after 20 weeks of continuous BBN supplementation in drinking water (n=20/group). B. Representative images of p-AMPKα staining. C. Quantification of p-AMPKα staining in wild-type and AMPKα2-/- bladders. D. Representative images of tAMPKα staining. E. Quantification of tAMPKα staining in wild-type and AMPKα2-/- bladders. F. Representative images of KI67 staining. G. Quantification of KI67 staining in wild-type and AMPKα2-/- bladders. H. Representative images of p-S6 staining. I. Quantification of p-S6 staining in wild-type and AMPKα2-/- bladders. J. Representative images of p27 staining. K. Quantification of p27 staining in wild-type and AMPKα2-/- bladders. L. Representative images of SKP2 staining. M. Quantification of SKP2 staining in wild-type and AMPKα2-/- bladders. Statistical significance indicated as; *p<0.05
AMPKα2 is suppression in human bladder cancer correlates with decreased p27
AMPKα expression has been reported to be altered in many different cancers (13-15). In order to determine if AMPKα2 levels are altered in bladder cancer, immunohistochemistry was performed on a tissue array obtained from Biomax Inc. which consisted of 10 adjacent non-tumor tissue samples and 40 bladder tumor samples. AMPKα2 protein levels were suppressed by 59% in bladder tumor tissue when compared to adjacent non-tumor tissue as control, and was statistically significant (Fig. 7A-B). In order to rule out patient variability in expression of AMPKα2, 15 adjacent non-tumor bladder and patient matched bladder tumor tissue was immunostained for AMPKα2. Analysis of high grade patient matched samples confirmed the results of the bladder tissue array indicating that AMPKα2 protein is significantly suppressed by 68% in bladder cancer (Fig. 7 C-D). Furthermore, analysis of AMPKα2 mRNA from The Cancer Genome Atlas (TCGA) database revealed that AMPKα2 was significantly suppressed at the mRNA level by 82% in bladder cancer (Fig. 7 E). To avoid patient variation, AMPKα2 mRNA expression was assessed in patient matched samples from the TCGA dataset and this revealed that AMPKα2 mRNA is significantly suppressed by 76% in bladder cancer (Fig. 7 F). Since AMPKα2 was found to be suppressed in bladder cancer it was important to determine if the expression of p27 and/or SKP2 were altered in bladder cancer. The expression of both p27 and SKP2 was assessed in bladder cancer using the array from Biomax Inc. Immunostaining for p27 in adjacent non-tumor and bladder tumor tissue revealed that p27 was significantly suppressed by 73% in bladder cancer (Fig. 7 G). In order to determine if the decrease in p27 in bladder cancer was due to an increase in SKP2 expression in bladder cancer, the levels of SKP2 were assessed and found to be significantly elevated in bladder cancer (Fig. 7 H). In order to recapitulate the results from the in vitro and in vivo studies demonstrating that lack of AMPKα2 results in an increase in SKP2 and a decrease in p27, the expression of AMPKα2, SKP2 and p27 were assessed in the same tissue sections. This analysis revealed that there was a significant negative correlation between p27 and SKP2, as expected since SKP2 functions to target p27 for degradation, and that there was a significant correlation between AMPKα2 and p27 further demonstrating that AMPKα2 contributes to the regulation of p27 in bladder cancer (Fig. 7 I). Together this data demonstrates for the first time that AMPKα2 protein levels are reduced in bladder cancer when compared to non cancerous tissue and that this reduction is correlated with an increase in SKP2 expression with a concomitant reduction in p27 protein levels.
Figure 7. AMPKα2 suppression in bladder cancer correlates with p27.
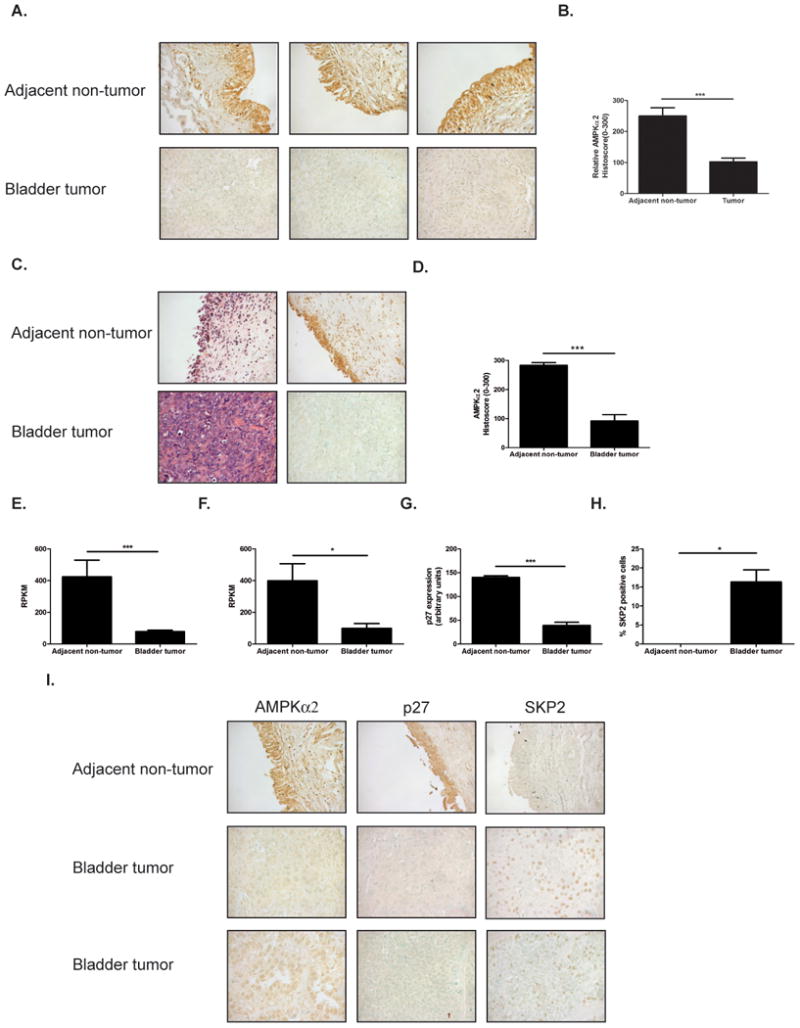
A. Representative 40× images of adjacent non-tumor and bladder tumor tissue stained for AMPKα2. Images represent an example of low AMPKα2 expression and high AMPKα2 expression in bladder tumor samples. B. Quantification of AMPKα2 expression in adjacent non-tumor and bladder tumor samples (n=10 adjacent non-tumor and n=40 tumor). C. Representative 40× images of patient matched adjacent non-tumor and bladder tumor tissue stained for AMPKα2. D. Quantification of patients matched samples (n=15). E. AMPKa2 mRNA in 19 adjacent non-tumor and 164 bladder tumor samples. (Represented as reads per kilobase of transcript per million mapped reads, RPKM). F. AMPKa2 mRNA in 15 patient matched adjacent non-tumor and bladder tumor samples (Represented as RPKM). G. Quantification of p27 staining from array (n=10 adjacent non-tumor and 40 bladder tumor) H. Quantification of SKP2 staining from array (n=10 adjacent non-tumor and 40 bladder tumor) I. Representative 40× images of AMPKα2, p27 and SKP2 staining in adjacent non-tumor and bladder tumor samples. Statistical significance indicated as; *p<0.05, ***p<0.001 The data presented in this figure is generated in part by data gathered by the TCGA Research Network.
Discussion
AMPK is the central metabolic regulator of cells and regulates the critical anabolic and proliferative pathways largely through its inhibition of mTOR (6, 30, 31). Many reports have suggested that AMPK may function to inhibit tumorigenesis because of its involvement in regulating cell growth through canonical pathways such as protein translation and cell growth through regulating mTOR (8, 32-35). Although many reports have demonstrated that AMPK can function to reduce tumor cell growth there have also been some reports demonstrating the opposite. For instance, mouse embryonic fibroblasts null for both AMPKα1 and AMPKα2 that are transformed with H-RasV12 grew significantly slower than their wild-type counter parts in a xenograft model (36). Also, in glioblastoma it has been demonstrated that AMPKα activation facilitated tumor growth and that AMPKα activation was localized to areas of the tumor with high proliferation further indicating how AMPKα can function to promote tumor growth (37). This creates a situation where it is necessary to determine the impact of AMPK activation in different tumor types to avoid utilizing it as a therapeutic strategy in tumor types where it may promote growth.
Although there are many reports on the relevance of AMPK signaling and its anti-growth effects in different tumor types, few studies have addressed the relevance of AMPK signaling in bladder cancer (14, 20, 38-40). In bladder cancer specifically, activation of AMPK has been shown to result in translational inhibition through a decrease in mTOR activation (41). Additionally, Shorning et al. demonstrated that in a model of bladder cancer in which LKB1-/- (an upstream kinase of AMPK) and PTEN-/- mice were utilized, mTOR was drastically up-regulated in tumorigenesis and this was blocked by rapamycin treatment (42). These data suggest an importance of AMPK signaling in bladder cancer. We therefore assessed the effect of AMPK activity on bladder cancer cells. The AMPK activator AICAR was used to determine the impact of AMPK signaling on bladder cancer cell proliferation. This revealed that activation of AMPK causes an up-regulation of p27 and a reduction in proliferation. Two different reports have demonstrated that AMPK activation causes phosphorylation of p27 and induces p27 stability (43, 44). In order to determine if AMPK control of bladder cancer growth is due to a direct phosphorylation of p27 or due to downstream signaling, bladder cancer cells were treated with rapamycin, an mTOR inhibitor, and compound C, an AMPK inhibitor. This revealed that AMPK regulation of p27 was at least partially mTOR dependent.
AMPK is a heterotrimeric protein composed of α, β and γ subunits. The α subunit is responsible for the kinase activity of the protein and exists as one of two isoforms, α1 or α2. There have been conflicting reports to date as to whether the α isoforms function redundantly or whether they have additional specific or selective functions (32). A chemical genetic screen of AMPKα2 substrates defined several possible targets related to cell mitosis (19). Additionally, it has been demonstrated that only AMPKα2-/- mouse embryonic fibroblasts formed tumors upon H-Ras transformation in a xenograft model, suggesting that AMPKα1 and AMPKα2 may indeed have selective functions (14). Furthermore, knockdown of only AMPKα2 and not AMPKα1 in vascular smooth muscle cells resulted in a decrease of p27 protein and conferred a proliferative advantage to cells (21). To address the question as to whether AMPKα1, AMPKα2 or both isoforms can be responsible for the regulation of p27 protein levels, siRNA specific to each isoform was utilized and p27 levels were evaluated. Consistent with the study in vascular smooth muscle cells, only the knockdown of AMPKα2 resulted in a reduction of p27 protein indicating that the AMPKα2 isoform is a highly specific mechanism of bladder cancer proliferative control.
To better understand the impact that AMPKα2 may have in controlling bladder cancer cell growth, HTB2 and HT1376 cells were transduced with shRNA to AMPKα2. Although there was a significant decrease in AMPKα2 protein level post transduction, there was only a modest decrease in total AMPKα1/α2, indicating that the AMPKα1 isoform may be the major isoform expressed in bladder cancer. Despite being the minor isoform, knockdown of AMPKα2 had profound effects on increasing cellular proliferation and p27 protein levels. Consistent with decreased p27 protein levels, AMPKα2 knockdown resulted in decreased p27 nuclear accumulation. AMPKα2 knockdown also had an effect on CDK4 protein levels in HT1376 cells, however this was not observed in HTB2 cells and could be potentially attributed to the higher the loss of Rb protein observed in HT1376 cells (45). In order to specifically address what mechanism may be controlling AMPK mediated regulation of p27 protein, SKP2 levels were assessed due to its widely reported role in functioning as an E3 ubiquitin ligase targeting p27 and several reports demonstrating that activation of AMPK and/or inhibition of mTOR results in a decrease in SKP2 protein levels (24, 46-49). Not surprisingly, knockdown of AMPKα2 in bladder cancer cells resulted in a prominent up-regulation of SKP2 protein levels. Given that AMPKα2 knockdown in bladder cancer cells resulted in an up-regulation of SKP2 protein, siRNA to SKP2 was utilized to determine if SKP2 indeed was responsible for the down-regulation of p27 and the increased cell proliferation. Knockdown of SKP2 in both the control and AMPKα2 knockdown cells caused an increase in p27 protein levels. Consistent with this data, knockdown of SKP2 reversed the effects that AMPKα2 knockdown had on cellular proliferation, indicating that SKP2 is indeed responsible for the observed effects. Although further research is warranted to determine the direct impact that p27 expression on bladder cancer growth, this data suggests that a mechanism of proliferative control exists in bladder cancer, involving the cross-talk between metabolic and cell-cycle control pathways.
To further understand the impact that AMPKα2 may have on bladder cancer cell growth in vivo, a xenograft model utilizing HT1376 shGFP and HT1376 shAMPKα2 was employed. Consistent with the in vitro data, HT1376 shAMPKα2 cells resulted in larger tumors when compared to their control. Additionally, AMPK activation was reduced in the HT1376 shAMPKα2 which resulted in concomitant mTOR activation, an increase in proliferation, a decrease in nuclear p27 and an increase in SKP2 staining. Although the xenograft model showed an increase in tumor growth in vivo when AMPKα2 was reduced, a genetic chemical model of bladder cancer was employed to further investigate the importance of AMPKα2 in bladder tumorigenesis. In this model, wild-type and AMPKα2-/- mice received BBN in drinking water to induce bladder cancer formation and represents a model which more closely mimics that of human bladder cancer. In this model the genetic deletion of AMPKα2 resulted in larger tumor formation and increased proliferation. Furthermore the deletion of AMPKα2 resulted in decreased nuclear p27 staining and increased SKP2 staining. Although the deletion of AMPKα2 may have additional effects on tumorigenesis beyond its contribution to increasing mTOR, the results are consistent with the in vitro data and the xenograft model showing that loss of AMPKα2 contributes to a reduction in p27 protein with a concomitant increase in SKP2 expression.
This study demonstrates for the first time the impact that AMPK, more specifically AMPKα2, has on bladder cancer growth through p27 regulation. AMPK has long been implicated as a potential tumor suppressor-like protein in cancer due to its multifaceted role in controlling cellular metabolism and recently it has been reported that AMPKα2 levels are altered in cancer. It has been reported that AMPKα2 protein is suppressed in breast cancer and hepatocellular carcinoma and suppressed in early stages of gastric cancer at the RNA level (13, 14, 17, 38). This data clearly implicates AMPKα2 specifically as an important protein in regulating cancer cell growth. To our knowledge there are no published studies of AMPK regulation in human bladder cancer to date. This study demonstrates that AMPKα2 mRNA and protein is suppressed in bladder cancer when compared to non-tumor urothelium. Previous reports have demonstrated that the AMPKα2 promoter is methylated and suppressed in hepatocellular carcinoma (17). Since AMPKα2 mRNA is suppressed in bladder cancer it is plausible that AMPKα2 is suppressed due to methylation, although further studies are required to validate this notion. Furthermore, our study demonstrates that p27 protein is down-regulated in bladder cancer and is correlated with AMPKα2 expression in the bladder. This further demonstrates the importance of AMPKα2 in regulating p27 protein levels in normal urothelium and in bladder cancer cells, where loss promotes proliferation and growth of tumors. AMPK is a multifaceted tumor suppressor-like protein and our study demonstrates how it is involved in controlling bladder cancer growth through regulation of p27, however further research is warranted to determine if the effects of AMPKα2 on bladder cancer growth are mediated through mTOR regulation or solely through the regulation of p27. It has been reported that p27 is often dysregulated in several types of cancer and that low expression is often linked to a more severe phenotype (24, 50). In bladder cancer specifically, it has been demonstrated that p27-/- mice are much more susceptible to tumorigenesis in the BBN model and that low p27 expression in human bladder cancer has been linked to metastatic disease and poor outcome (23, 25). It is therefore likely that a reduction of AMPKα2, which results in a reduction of p27, would have a profound impact on bladder cancer growth.
The direct targeting of AMPKα2 in bladder cancer would result in the repression of mTOR activity and would result in an increase in p27 protein which could limit bladder cancer growth (Graphical Abstract). However, due to the loss of AMPKα2 protein expression in bladder cancer it would not be feasible to target AMPKα2 as an effective mechanism for treating bladder cancer. Additionally, mTOR inhibited are currently being used in the clinic with mixed success due to off target effects and toxicity despite their promise in preclinical models. The results from this study demonstrate that an inhibitor of SKP2 could be an effective way of treating bladder cancer patients because inhibiting SKP2 would repress the proliferative advantage that the loss of AMPKα2 confers to bladder cancer. This would be a more direct mechanism for limiting the capacity of bladder cancer growth because the inhibition of SKP2 would lead to an increase in p27 protein levels.
Supplementary Material
Acknowledgments
The authors would like to thank Roderick Franczak and Shilpa Choudhary for their technical assistance and sample preparation. The authors would also like to thank Benoit Viollet for providing the AMPKα2-/- mice.
Footnotes
Conflict of Interest: The authors declare that they have no conflict of interest.
References
- 1.Degraff DJ, Cates JM, Mauney JR, Clark PE, Matusik RJ, Adam RM. When urothelial differentiation pathways go wrong: Implications for bladder cancer development and progression. Urol Oncol. 2011 doi: 10.1016/j.urolonc.2011.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosenberg JE, Hahn WC. Bladder cancer: modeling and translation. Genes Dev. 2009;23:655–659. doi: 10.1101/gad.1789109. [DOI] [PubMed] [Google Scholar]
- 3.Carling D, Viollet B. Beyond energy homeostasis: the expanding role of AMP-activated protein kinase in regulating metabolism. Cell Metab. 2015;21:799–804. doi: 10.1016/j.cmet.2015.05.005. [DOI] [PubMed] [Google Scholar]
- 4.Fay JR, Steele V, Crowell JA. Energy homeostasis and cancer prevention: the AMP-activated protein kinase. Cancer Prev Res (Phila) 2009;2:301–309. doi: 10.1158/1940-6207.CAPR-08-0166. [DOI] [PubMed] [Google Scholar]
- 5.Liang J, Mills GB. AMPK: a contextual oncogene or tumor suppressor? Cancer Res. 2013;73:2929–2935. doi: 10.1158/0008-5472.CAN-12-3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Steinberg GR, Kemp BE. AMPK in Health and Disease. Physiol Rev. 2009;89:1025–1078. doi: 10.1152/physrev.00011.2008. [DOI] [PubMed] [Google Scholar]
- 7.Dazert E, Hall MN. mTOR signaling in disease. Curr Opin Cell Biol. 2011;23:744–755. doi: 10.1016/j.ceb.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 8.Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009;9:563–575. doi: 10.1038/nrc2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Viollet B, Athea Y, Mounier R, Guigas B, Zarrinpashneh E, Horman S, Lantier L, Hebrard S, Devin-Leclerc J, Beauloye C, Foretz M, Andreelli F, Ventura-Clapier R, Bertrand L. AMPK: Lessons from transgenic and knockout animals. Front Biosci. 2009;14:19–44. doi: 10.2741/3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hadad SM, Hardie DG, Appleyard V, Thompson AM. Effects of metformin on breast cancer cell proliferation, the AMPK pathway and the cell cycle. Clin Transl Oncol. 2014;16:746–752. doi: 10.1007/s12094-013-1144-8. [DOI] [PubMed] [Google Scholar]
- 11.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhuang Y, Miskimins WK. Cell cycle arrest in Metformin treated breast cancer cells involves activation of AMPK, downregulation of cyclin D1, and requires p27Kip1 or p21Cip1. J Mol Signal. 2008;3:18. doi: 10.1186/1750-2187-3-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hadad SM, Baker L, Quinlan PR, Robertson KE, Bray SE, Thomson G, Kellock D, Jordan LB, Purdie CA, Hardie DG, Fleming S, Thompson AM. Histological evaluation of AMPK signalling in primary breast cancer. BMC Cancer. 2009;9:307. doi: 10.1186/1471-2407-9-307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fox MM, Phoenix KN, Kopsiaftis SG, Claffey KP. AMP-Activated Protein Kinase alpha 2 Isoform Suppression in Primary Breast Cancer Alters AMPK Growth Control and Apoptotic Signaling. Genes Cancer. 2013;4:3–14. doi: 10.1177/1947601913486346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang H, Zheng LY, Yang W, Wu FQ, Wang C, Yu L, Tang L, Qiu B, Li YQ, Guo L, Wu MC, Feng GS, Zou D. Prognostic significance of AMPK activation and therapeutic effects of metformin in hepatocellular carcinoma. Clin Cancer Res. 2013 doi: 10.1158/1078-0432.CCR-13-0203. [DOI] [PubMed] [Google Scholar]
- 16.Kim YH, Liang H, Liu X, Lee JS, Cho JY, Cheong JH, Kim H, Li M, Downey TJ, Dyer MD, Sun Y, Sun J, Beasley EM, Chung HC, Noh SH, Weinstein JN, Liu CG, Powis G. AMPKalpha modulation in cancer progression: multilayer integrative analysis of the whole transcriptome in Asian gastric cancer. Cancer Res. 2012;72:2512–2521. doi: 10.1158/0008-5472.CAN-11-3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee CW, Wong LL, Tse EY, Liu HF, Leong VY, Lee JM, Hardie DG, Ng IO, Ching YP. AMPK promotes p53 acetylation via phosphorylation and inactivation of SIRT1 in liver cancer cells. Cancer Res. 2012;72:4394–4404. doi: 10.1158/0008-5472.CAN-12-0429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Salt I, Celler JW, Hawley SA, Prescott A, Woods A, Carling D, Hardie DG. AMP-activated protein kinase: greater AMP dependence, and preferential nuclear localization, of complexes containing the alpha2 isoform. Biochem J. 1998;334(Pt 1):177–187. doi: 10.1042/bj3340177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Banko MR, Allen JJ, Schaffer BE, Wilker EW, Tsou P, White JL, Villen J, Wang B, Kim SR, Sakamoto K, Gygi SP, Cantley LC, Yaffe MB, Shokat KM, Brunet A. Chemical genetic screen for AMPKalpha2 substrates uncovers a network of proteins involved in mitosis. Mol Cell. 2011;44:878–892. doi: 10.1016/j.molcel.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Phoenix KN, Devarakonda CV, Fox MM, Stevens LE, Claffey KP. AMPKalpha2 Suppresses Murine Embryonic Fibroblast Transformation and Tumorigenesis. Genes Cancer. 2012;3:51–62. doi: 10.1177/1947601912452883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Song P, Wang S, He C, Wang S, Liang B, Viollet B, Zou MH. AMPKalpha2 deletion exacerbates neointima formation by upregulating Skp2 in vascular smooth muscle cells. Circ Res. 2011;109:1230–1239. doi: 10.1161/CIRCRESAHA.111.250423. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22.Shariat SF, Zlotta AR, Ashfaq R, Sagalowsky AI, Lotan Y. Cooperative effect of cell-cycle regulators expression on bladder cancer development and biologic aggressiveness. Mod Pathol. 2007;20:445–459. doi: 10.1038/modpathol.3800757. [DOI] [PubMed] [Google Scholar]
- 23.Rabbani F, Koppie TM, Charytonowicz E, Drobnjak M, Bochner BH, Cordon-Cardo C. Prognostic significance of p27Kip1 expression in bladder cancer. BJU Int. 2007;100:259–263. doi: 10.1111/j.1464-410X.2007.06927.x. [DOI] [PubMed] [Google Scholar]
- 24.Bloom J, Pagano M. Deregulated degradation of the cdk inhibitor p27 and malignant transformation. Semin Cancer Biol. 2003;13:41–47. doi: 10.1016/s1044-579x(02)00098-6. [DOI] [PubMed] [Google Scholar]
- 25.Hikosaka A, Ogawa K, Sugiura S, Asamoto M, Takeshita F, Sato SY, Nakanishi M, Kohri K, Shirai T. Susceptibility of p27 kip1 knockout mice to urinary bladder carcinogenesis induced by N-butyl-N-(4-hydroxybutyl)nitrosamine may not simply be due to enhanced proliferation. Int J Cancer. 2008;122:1222–1228. doi: 10.1002/ijc.23249. [DOI] [PubMed] [Google Scholar]
- 26.Nickeleit I, Zender S, Kossatz U, Malek NP. p27kip1: a target for tumor therapies? Cell Div. 2007;2:13. doi: 10.1186/1747-1028-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Elsherif E, Elbaky TA, Elserafy F, Elkady N, Dawood M, Gaber MA, Badawy A, Gharabawy ME. beta-catenin and SKP2 proteins as predictors of grade and stage of non-muscle invasive urothelial bladder carcinoma. Chin Clin Oncol. 2016;5:6. doi: 10.3978/j.issn.2304-3865.2016.02.02. [DOI] [PubMed] [Google Scholar]
- 28.Viollet B, Andreelli F, Jorgensen SB, Perrin C, Geloen A, Flamez D, Mu J, Lenzner C, Baud O, Bennoun M, Gomas E, Nicolas G, Wojtaszewski JF, Kahn A, Carling D, Schuit FC, Birnbaum MJ, Richter EA, Burcelin R, Vaulont S. The AMP-activated protein kinase alpha2 catalytic subunit controls whole-body insulin sensitivity. J Clin Invest. 2003;111:91–98. doi: 10.1172/JCI16567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bochis OV, Irimie A, Pichler M, Berindan-Neagoe I. The role of Skp2 and its substrate CDKN1B (p27) in colorectal cancer. J Gastrointestin Liver Dis. 2015;24:225–234. doi: 10.15403/jgld.2014.1121.242.skp2. [DOI] [PubMed] [Google Scholar]
- 30.Hardie DG. AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev. 2011;25:1895–1908. doi: 10.1101/gad.17420111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Towler MC, Hardie DG. AMP-activated protein kinase in metabolic control and insulin signaling. Circ Res. 2007;100:328–341. doi: 10.1161/01.RES.0000256090.42690.05. [DOI] [PubMed] [Google Scholar]
- 32.Dasgupta B, Chhipa RR. Evolving Lessons on the Complex Role of AMPK in Normal Physiology and Cancer. Trends Pharmacol Sci. 2016;37:192–206. doi: 10.1016/j.tips.2015.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hardie DG. AMP-activated protein kinase: a cellular energy sensor with a key role in metabolic disorders and in cancer. Biochem Soc Trans. 2011;39:1–13. doi: 10.1042/BST0390001. [DOI] [PubMed] [Google Scholar]
- 34.Fogarty S, Ross FA, Vara Ciruelos D, Gray A, Gowans GJ, Hardie DG. AMPK Causes Cell Cycle Arrest in LKB1-Deficient Cells via Activation of CAMKK2. Mol Cancer Res. 2016;14:683–695. doi: 10.1158/1541-7786.MCR-15-0479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zadra G, Batista JL, Loda M. Dissecting the Dual Role of AMPK in Cancer: From Experimental to Human Studies. Mol Cancer Res. 2015;13:1059–1072. doi: 10.1158/1541-7786.MCR-15-0068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Laderoute KR, Amin K, Calaoagan JM, Knapp M, Le T, Orduna J, Foretz M, Viollet B. 5′-AMP-activated protein kinase (AMPK) is induced by low-oxygen and glucose deprivation conditions found in solid-tumor microenvironments. Mol Cell Biol. 2006;26:5336–5347. doi: 10.1128/MCB.00166-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rios M, Foretz M, Viollet B, Prieto A, Fraga M, Costoya JA, Senaris R. AMPK activation by oncogenesis is required to maintain cancer cell proliferation in astrocytic tumors. Cancer Res. 2013;73:2628–2638. doi: 10.1158/0008-5472.CAN-12-0861. [DOI] [PubMed] [Google Scholar]
- 38.Zheng L, Yang W, Wu F, Wang C, Yu L, Tang L, Qiu B, Li Y, Guo L, Wu M, Feng G, Zou D, Wang H. Prognostic significance of AMPK activation and therapeutic effects of metformin in hepatocellular carcinoma. Clin Cancer Res. 2013;19:5372–5380. doi: 10.1158/1078-0432.CCR-13-0203. [DOI] [PubMed] [Google Scholar]
- 39.Zakikhani M, Dowling R, Fantus IG, Sonenberg N, Pollak M. Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res. 2006;66:10269–10273. doi: 10.1158/0008-5472.CAN-06-1500. [DOI] [PubMed] [Google Scholar]
- 40.Dowling RJ, Zakikhani M, Fantus IG, Pollak M, Sonenberg N. Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res. 2007;67:10804–10812. doi: 10.1158/0008-5472.CAN-07-2310. [DOI] [PubMed] [Google Scholar]
- 41.Liu Z, Li X, Simoneau AR, Jafari M, Zi X. Rhodiola rosea extracts and salidroside decrease the growth of bladder cancer cell lines via inhibition of the mTOR pathway and induction of autophagy. Mol Carcinog. 2012;51:257–267. doi: 10.1002/mc.20780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shorning BY, Griffiths D, Clarke AR. Lkb1 and Pten synergise to suppress mTOR-mediated tumorigenesis and epithelial-mesenchymal transition in the mouse bladder. PLoS One. 2011;6:e16209. doi: 10.1371/journal.pone.0016209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liang J, Shao SH, Xu ZX, Hennessy B, Ding Z, Larrea M, Kondo S, Dumont DJ, Gutterman JU, Walker CL, Slingerland JM, Mills GB. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol. 2007;9:218–224. doi: 10.1038/ncb1537. [DOI] [PubMed] [Google Scholar]
- 44.Short JD, Dere R, Houston KD, Cai SL, Kim J, Bergeron JM, Shen J, Liang J, Bedford MT, Mills GB, Walker CL. AMPK-mediated phosphorylation of murine p27 at T197 promotes binding of 14-3-3 proteins and increases p27 stability. Mol Carcinog. 2010;49:429–439. doi: 10.1002/mc.20613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Goodrich DW, Chen Y, Scully P, Lee WH. Expression of the retinoblastoma gene product in bladder carcinoma cells associates with a low frequency of tumor formation. Cancer Res. 1992;52:1968–1973. [PubMed] [Google Scholar]
- 46.Hsu JD, Kao SH, Ou TT, Chen YJ, Li YJ, Wang CJ. Gallic acid induces G2/M phase arrest of breast cancer cell MCF-7 through stabilization of p27(Kip1) attributed to disruption of p27(Kip1)/Skp2 complex. J Agric Food Chem. 2011;59:1996–2003. doi: 10.1021/jf103656v. [DOI] [PubMed] [Google Scholar]
- 47.Vervoorts J, Luscher B. Post-translational regulation of the tumor suppressor p27(KIP1) Cell Mol Life Sci. 2008;65:3255–3264. doi: 10.1007/s00018-008-8296-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fan GH, Wang ZM, Yang X, Xu LP, Qin Q, Zhang C, Ma JX, Cheng HY, Sun XC. Resveratrol inhibits oesophageal adenocarcinoma cell proliferation via AMP-activated protein kinase signaling. Asian Pac J Cancer Prev. 2014;15:677–682. doi: 10.7314/apjcp.2014.15.2.677. [DOI] [PubMed] [Google Scholar]
- 49.Totary-Jain H, Sanoudou D, Dautriche CN, Schneller H, Zambrana L, Marks AR. Rapamycin resistance is linked to defective regulation of Skp2. Cancer Res. 2012;72:1836–1843. doi: 10.1158/0008-5472.CAN-11-2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Philipp-Staheli J, Payne SR, Kemp CJ. p27(Kip1): regulation and function of a haploinsufficient tumor suppressor and its misregulation in cancer. Exp Cell Res. 2001;264:148–168. doi: 10.1006/excr.2000.5143. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.