

Abstract
Unfavorable thermodynamics often render furans reluctant to engage in high-yielding Diels-Alder (DA) cycloaddition reactions. Here we report the highly efficient conversion of the bio-sourced reactants itaconic anhydride (IA) and furfuryl alcohol (FA) to a single DA adduct. The free energy advantages provided by anhydride ring-opening and crystal lattice energy of the product overcome the loss of aromaticity of the furanoid diene. Detailed 1H NMR studies provided valuable insights about relevant kinetic and thermodynamic features.
Graphical abstract
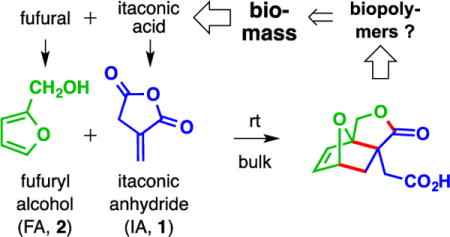
Itaconic acid1 and furfural2 are two chemicals abundantly available from biomass. The first arises by the classical citric acid (or tricarboxylic or Krebs3) cycle; the second by acid-catalyzed dehydration of 5-carbon sugars prevalent in, for example, corncobs (see Volume 1 of Organic Syntheses4). For a century it has been known that each of itaconic anhydride (1, IA) and furfuryl alcohol (2, FA) is readily available by simple conversions (dehydration5 and reduction,6 respectively) of these readily available precursors.
The ability of IA to function as a dienophile in Diels–Alder (DA) cycloaddition reactions was described even in the first publication by Diels and Alder.7 The use of furan as a diene was reported one year later in their second paper on the subject of “hydroaromatic synthesis”.8 Remarkably, we can find no reports of IA (1) or itaconic acid (or its esters) ever having been reacted with any furan derivative in the intervening >85 years. This is in spite of the fact that DA adducts of furans with many other types of dienophiles have been appropriated as strategically valuable and enabling intermediates in numerous syntheses of complex molecules.9
With an eye toward the preparation of novel reactive monomers from furans for use in sustainable polymer synthesis,10 we have studied the reactions of IA (1) with various furans and report our findings here. A hallmark of furans as participants in DA cycloadditions is the often-unfavorable enthalpic change upon product formation. This frequently results in only low equilibrium conversion to the [4+2] adduct because of loss of heteroaromaticity in the diene and ring strain in the 7-oxanorbornene product.11 This is all the more true for 1,1-disubstituted dienophiles analogous to 1 (e.g., methacrolein,12 methacrylates,12a–c and methacrylonitrile12d), where the equilibrium concentration of product ranges from 7–36% even in the presence of 20-fold excess of furan, at low temperature, and/or under high pressure.
We first describe reactions between IA (1) and a variety of simple furans, starting with furan (3) itself. In one experiment IA was dissolved in 20 equivalents of furan and held at ambient temperature. Aliquots were periodically withdrawn and dissolved in CDCl3 to monitor reaction progress. It was important in this kind of analysis that the spectral data be recorded soon after sample preparation, because the retro-Diels-Alder reaction was also operative at room temperature, and dilution (here, from neat to NMR sample concentration) shifts the equilibrium composition of this bimolecular-to-unimolecular process toward the starting pair of reactants (here, 1 + 3). Two diastereomeric products, 4-endo and 4-exo, are produced (Figure 1a and Table 1, entry 1). Even at early time points, they formed at nearly identical rates. After 40 hours the system had essentially reached its equilibrium state, which comprises a ratio of 73% of the initial IA (1) and 27% of the sum of the two DA adducts. At equilibrium, there was a very slight predominance of 4-endo over the amount of 4-exo. These diastereomeric DA adducts were sufficiently stable to be isolable by rapid chromatographic separation on silica gel, even though some retro-DA reaction was occurring as the solutions were being manipulated. Isolated solid-state samples of each isomer were considerably more stable. Upon dissolution in CDCl3 or C6D6, each isomer began to slowly revert to 1 and 3 (ca. 50% conversion after 6 h), consistent with the rate of their formation and final equilibrium ratios.
Figure 1.
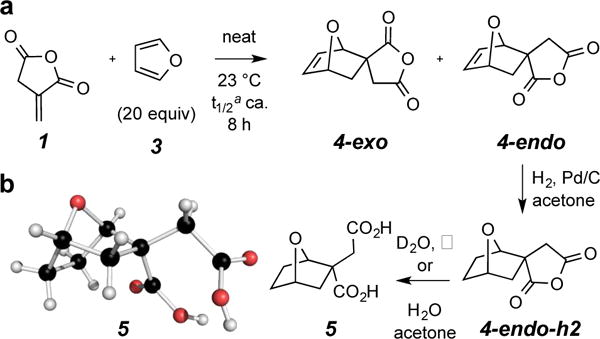
a. Rate and final equilibrium resting state for the DA reaction between IA (1) and furan (3). b. Conversion of 4-endo to 5 via 4-endo-h2 and 3D structure of 5 from a single crystal X-ray analysis.
a This is the half-time for reaching the final equilibrium mixture (this ratio for 1:4-exo:4-endo was 73:13:14).
Table 1.
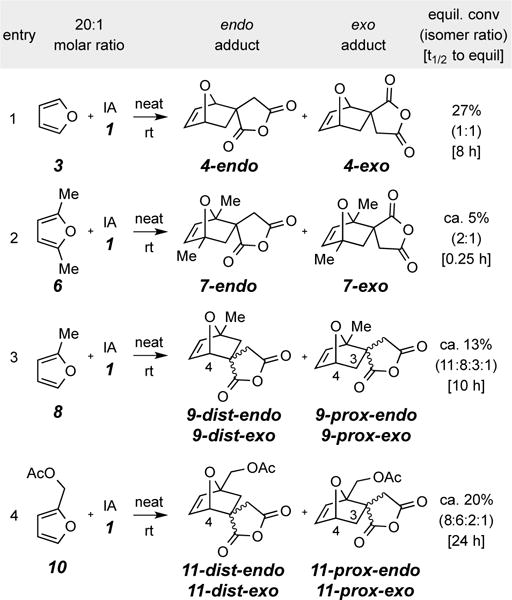
Reactions of itaconic anhydride (1) with furans 3, 6, 8, and 10 (20 equiv) at ambient temperature
|
Our assignment of the diastereomeric relationship within each of 4-exo and 4-endo was initially based on detailed analyses of NMR data [see Supporting Information (SI)]. This was subsequently confirmed by an X-ray structure of the diacid 5 (Figure 1b) obtained by catalytic hydrogenation of 4-endo to the derivative 4-endo-h2, which was then hydrolyzed to the crystalline diacid 5. Diagnostic features in the 1H NMR spectral data of each diastereomer of 4 were then useful in assessing the relative configuration of the DA adducts prepared from additional furan derivatives (cf. 4, 7, 9, and 11 in Table 1); details again are provided in the SI.
2,5-Dimethylfuran (6) was the next diene studied, again in an experiment where it was used as the reaction solvent and in ca. 20-fold excess over IA (1). The results are summarized in Table 1 (entry 2). The reaction of 6 with 1 was notably faster than that of furan (t1/2 ~15 min vs. ~8 h at 23 °C); apparently the greater electron density in diene 6 is a more important factor than the increase in its steric bulk. However, the reaction proceeded to a considerably lower equilibrium conversion (ca. 5%) of the sum of DA adducts 7-endo and 7-exo, which reflects the greater steric compression between the substituents on the spirocyclic carbon and the adjacent (proximal) bridgehead methyl group present in adducts 7 vis-à-vis adducts 4. At an intermediate time point (10 min, ca. 3% conversion), the formation of the major isomer had outpaced that of the minor to the extent of ca. 2:1, a ratio that remained essentially constant thereafter.
We next examined the reaction of IA with 2-methylfuran (8), a desymmetrized diene that could give rise to four isomeric, NMR-distinguishable DA adducts. These are a pair of endo and exo diastereomers for each of a distal (9-dist-endo and 9-dist-exo) vs. a proximal (9-prox-endo and 9-prox-exo) pair of constitutional isomers. Again, we used an excess of the furan component as the solvent in this ambient temperature experiment and monitored the reaction progress over time. We observed: (i) that the proximal isomers are formed more quickly than the distal, a reasonable event given the electronic polarization imparted by the donor methyl group in 8, (ii) that the distal isomers eventually predominated over the proximal, again reasonable, now on the basis of minimization of steric compression, and (iii) that the equilibrium amounts of the four products (Table 1, entry 3) are in line with the observed equilibrium ratios of DA adducts 4 and 7. Finally, the overall extent of conversion was intermediate between that of furan itself and the 2,5-dimethylated derivative 6.
2-Acetoxymethylfuran (10) was another diene substrate that proved informative (Table 1, entry 4). At equilibrium, the four IA DA adducts 11 were formed, again to an extent intermediate between that of 4 vs. 7. This was observed to be the slowest of all reactions we studied, consistent with the acetoxymethyl substituent having a weakly electron withdrawing character. As was the case for 9, at equilibrium the distal isomers predominated. The assignments of structure to the distal vs. proximal substitution patterns among the isomers of 9 and 11 were based on the difference in coupling patterns of the resonances for the bridgehead protons (at C4) in each (see SI). HMQC and HMBC NMR analyses also were consistent with the assignments.
We then turned to the reaction between the bio-derived FA (2) and IA (1). Initially, we monitored the behavior of an equimolar mixture of this diene/dienophile pair in CDCl3 solution (~1.5 M). After being held at 55 °C for 10 minutes, a few percent of total conversion to a mixture of four DA adducts was seen, which is well in line with the behavior (rate and ratio) observed for the reaction between the acetate 10 and 1. By analogy then, we presumed these four compounds to be a mixture of the four isomeric anhydrides 12 (Figure 2a). When this reaction solution was examined after 7 days, a new, fifth, component was seen to emerge at about 15% relative to the unconsumed IA (1). The appearance of (i) a broad downfield resonance and (ii) a pair of downfield doublets (δ 4.62 and 4.83, Jab = 10.8 Hz) in this new, dominant compound suggested that a carboxylic acid lactone had formed; it is reasonable to anticipate a conversion of one of 12-prox-exo or 12-prox-endo to a ring-opened lactone acid by one of the four pathways implied by arrows “a” or “b” in 12-prox-exo or “a’ ” or “b’ ” in 12-prox-endo (Figure 2a). The favorable free energy change associated with anhydride opening provides a driving force for DA adduct formation.
Figure 2.
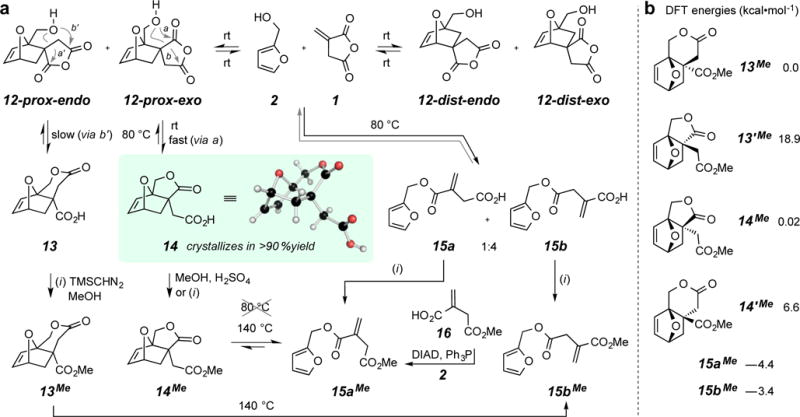
a. Reaction manifold showing the equilibration among 1 + 2, four isomeric anhydrides 12, lactone acids 13 and 14, and mono-furfuryl itaconates 15. b. DFT [M06-2X/6-31+G(d,p)(SMD:CHCl3), see SI for details] free energies (kcal•mol−1) of the four isomeric lactone esters from ring opening of 12-prox-endo and 12-prox-exo and the (more stable) mono-furfuryl itaconate esters 15a and 15b.
The significant breakthrough was achieved when an equimolar mixture of 1 and 2 was allowed to react in the bulk. A suspension of solid 1 (95% grade) in liquid 2 (98% grade) at ambient temperature changed over time in consistency. After about 3.5 h the initial heterogeneous slurry could no longer be magnetically stirred; we could identify in the NMR spectrum of an aliquot the presence of all four isomeric anhydrides 12 (Figure 3), albeit to the total extent of only ca. 5%, along with a significant amount of the same fifth component mentioned above. After 10 h the composition of the bulk mixture was that of a paste and after 18 h it had turned to a solid mass. This material was now comprised of largely a single component, having the same spectral properties as those of the new, fifth component that had appeared after 7 days in the homogenous CDCl3 solution experiment described above. The 13C NMR spectrum of this compound showed a carbonyl resonance at δ 177.8 ppm, suggestive that it contained a 5-membered butyrolactone ring.13 X-ray diffraction showed the structure to be that of the lactone acid 14, arising therefore from 12-prox-exo, presumably by path “a.”
Figure 3.
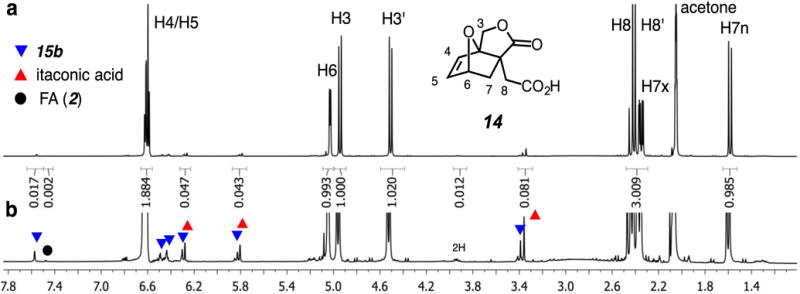
1H NMR spectrum in acetone-d6 of an aliquot of the bulk reaction mixture from 1:1 IA (1) and FA (2). a. Resonances from the major product 14 are assigned. b. Vertical scale increased 5x; the principal minor components are denoted; integration of all components indicates a 94% yield of 14. (see SI for full-page version of this graphic).
The fact that the rapidly formed, steady-state mixture of the four DA adducts 12 transformed to essentially the single component 14 indicates rapid reversibility of each of 12 back to IA (1) and FA (2). The 1H NMR spectrum of an aliquot taken from an equimolar mixture after 2 days is shown in Figure 3. Integration of key resonances indicated that the chemical yield for formation of 14 was 94%. The driving force for the conversion of IA+FA to 14 comes both from (i) the opening of the anhydride as well as (ii) the crystallization of the product from a dynamic, interconverting mixture of multiple components. Crystal lattice energies have been exploited for additional purposes in the arena of furan DA chemistry.14
A sample removed from the bulk mixture of 1 and 2 after just 30 minutes was immediately chromatographed on silica gel. The structures of three constituents, each obtained in <1% yield,15 were deduced. In dilute CDCl3 solution, each of the anhydrides 12-dist-endo and 12-dist-exo, assigned as such by comparative NMR analyses with some of the previous DA adducts, was observed to revert to 1 and 2 at room temperature in a matter of minutes. The third sample proved to be an acid lactone isomeric with 14. It contained ~20% of a second compound whose 1H NMR resonances suggested it to be the anhydride 12-prox-endo. Within a day in CDCl3, this anhydride had converted to the same, new acid lactone. By a series of correlation experiments (see below) we concluded the structure of this new lactone to be 13, arising by the attack indicated by “b’ ” in structure 12-prox-endo (Figure 2a).
Monitoring the thermal behavior of a CDCl3 solution of the lactone acid 14 was also informative. At 80 °C a 1:4 mixture of the two mono-furfuryl itaconate esters 15a and 15b (structure assignment discussed below) was formed. The isomer 15b cannot arise from direct retro-DA reaction of 14. Instead 14 apparently reverts to 12-prox-exo and, in turn 2 and 1, which then can repopulate the mixture of 15a and 15b. All of these intermediates were detectable (1H NMR, see SI). To probe whether 14 can lead to 15a directly by a retro-DA reaction, we converted 14 to the methyl ester 14Me. This compound was very stable at 80 °C in CDCl3 and only upon heating to 140 °C in the melt did it finally and solely revert to the ester 15aMe. An authentic sample of 15aMe was prepared by Mitsunobu esterification reaction between FA (2) and the commercially available mono-methyl itaconate 16. The rate of the retro-DA reaction of 14Me suggests that 14 does not proceed directly to 15a. A 1:1 mixture of IA and benzyl alcohol, a DA-silent mimic of FA (2), at 80 °C produced a mixture of mono-benzyl itaconates in which the major isomer was the analog of 15b.16
We returned to the isomeric lactone acid 13 and converted it to the methyl ester 13Me. Like its analog 14Me, this ester also showed clean retro-DA behavior when heated neat at 140 °C (partial reversion after 1 min and complete after 5 min). Only the mixed diester 15bMe was produced, verifying that 13 embodied a valero- rather than butyrolactone subunit.
In summary, detailed NMR analyses of an array of related Diels-Alder reactions between bio-derivable IA (1) and furans has provided an understanding of a number of the underlying kinetic and thermodynamic issues. We have discovered that the metastable lactone acid 14 can be produced in high yield (94%) under trivial reaction conditions [1:1 mixture of IA (1) and FA (2), neat, ambient temperature]. This opens the way for studying its further conversion into derivatives amenable to polymerization, a topic we are currently exploring.
Supplementary Material
Acknowledgments
Financial support for this research was provided by the Minnesota Corn Growers Association and the Center for Sustainable Polymers at the University of Minnesota, an NSF-supported Center for Chemical Innovation (CHE-1413862). NMR data were recorded on an instrument purchased with support of the NIH Shared Instrumentation Grant program (S10OD011952). Computations were performed with resources made available by the University of Minnesota Supercomputing Institute (MSI).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.orglett.6b00276.
Experimental procedures and structural characterization data for all new compounds and 1H and 13C NMR spectra (PDF)
Notes
The authors have no competing financial interests to declare.
References
- 1.Medway AM, Sperry J. Green Chem. 2014;16:2084–2101. and references to earlier reviews therein. [Google Scholar]
- 2.Cai CM, Zhang T, Kumara R, Wymana CE. J Chem Technol Biotechnol. 2014;89:2–10. and references to earlier reviews therein. [Google Scholar]
- 3.The discoveries of key elements of the citric acid cycle were recognized by Nobel Prizes in Physiology or Medicine in 1937 (to Albert Szent-Györgyi) and 1953 (to Hans Adolf Krebs): (http://www.nobelprize.org/nobel_prizes/medicine/laureates/1937 and (http://www.nobelprize.org/nobel_prizes/medicine/laureates/1953 (each accessed Mar-16-2016).
- 4.Adams R, Voorhees V Furfural. Organic Syntheses. 1921;1:49–51. [Google Scholar]
- 5.Fittig R, Batt L, Nock K, Salomon H, Wernher G. J Liebigs Ann Chem. 1904;331:151–196. [Google Scholar]
- 6.Kaufmann WE, Adams R. J Am Chem Soc. 1923;45:3029–3044. and earlier citations therein. [Google Scholar]
- 7.Diels O, Alder K. J Liebigs Ann der Chem. 1928;460:98–122. [Google Scholar]
- 8.Diels O, Alder K. Ber Dtsch Chem Ges. 1929;62:554–562. [Google Scholar]
- 9.Raczko J, Jurczak J. Furan in the synthesis of natural products. In: Rahman Atta ur., editor. Studies in Natural Products Chemistry. Vol. 16. Elsevier Science; Amsterdam: 1995. pp. 639–685. [Google Scholar]
- 10.(a) Shiramizu M, Toste FD. Chem Eur J. 2011;17:12452–12457. doi: 10.1002/chem.201101580. [DOI] [PubMed] [Google Scholar]; (b) Williams CL, Chang CC, Do P, Nikbin N, Caratzoulas S, Vlachos DG, Lobo RF, Fan W, Dauenhauer PJ. ACS Catal. 2012;2:935–939. [Google Scholar]; (c) Pacheco JJ, Davis ME. Proc Natl Acad Sci USA. 2014;111:8363–8367. doi: 10.1073/pnas.1408345111. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Mahmoud E, Watson DA, Lobo RF. Green Chem. 2014;16:167–175. [Google Scholar]
- 11.(a) Vogel P, Cossy J, Plumet J, Arjona O. Tetrahedron. 1999;55:13521–13642. [Google Scholar]; (b) Bur S, Padwa A. [4+2] Cycloaddition chemistry of substituted furans. In: Nishiwaki N, editor. Methods and Applications of Cycloaddition Reactions in Organic Syntheses. First. Wiley; New York: 2014. pp. 355–406. [Google Scholar]
- 12.(a) Dauben WG, Krabbenhoft HO. J Am Chem Soc. 1976;98:1992–1993. [Google Scholar]; (b) Murai A, Takahashi K, Taketsuru H, Masamune T. Chem Commun. 1981:221–222. [Google Scholar]; (c) Hayashi Y, Nakamura M, Nakao S, Inoue T, Shoji M. Angew Chem Int Ed. 2002;41:4079–4081. doi: 10.1002/1521-3773(20021104)41:21<4079::AID-ANIE4079>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]; (d) Rimmelin J, Jenner G, Rimmelin P. Bull Soc Chim Fr. 1978:II-461–II-464. [Google Scholar]
- 13.Lambert JB, Wharry SM, Block E, Bazzi AA. J Org Chem. 1983;48:3982–3985. [Google Scholar]
- 14.(a) Guidi A, Theurillat-Moritz V, Vogel P. Tetrahedron: Asymmetry. 1996;7:3153–3162. [Google Scholar]; (b) Sevin A, Vogel P. J Org Chem. 1994;59:5920–5926. [Google Scholar]; (c) Theurillat-Moritz V, Vogel P. Tetrahedron: Asymmetry. 1996;7:3163–3168. [Google Scholar]
- 15.This example serves as a reminder of the value of the maxim that a 1% yield is infinitely greater than 0%. Importantly, the identification of these trace products revealed significant features about the energetics of several aspects of the reaction manifold.
- 16.Cheng X, Li L, Uttamchandani M, Yao SQ. Org Lett. 2014;16:1414–1417. doi: 10.1021/ol500206w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.