

Abstract
Programmed cell death is an essential aspect of animal development. Mutations in vertebrate genes that mediate apoptosis only mildly perturb development, suggesting that other cell death modes likely have important roles. Linker cell-type death (LCD) is a morphologically conserved cell death form operating during the development of Caenorhabditis elegans and vertebrates. We recently described a molecular network governing LCD in C. elegans, delineating a key role for the transcription factor heat-shock factor 1 (HSF-1). Although HSF-1 functions to protect cells from stress in many settings by inducing expression of protein folding chaperones, it promotes LCD by inducing expression of the conserved E2 ubiquitin-conjugating enzyme LET-70/UBE2D2, which is not induced by stress. Following whole-genome RNA interference and candidate gene screens, we identified and characterized four conserved regulators required for LCD. Here we show that two of these, NOB-1/Hox and EOR-1/PLZF, act upstream of HSF-1, in the context of Wnt signaling. A third protein, NHR-67/TLX/NR2E1, also functions upstream of HSF-1, and has a separate activity that prevents precocious expression of HSF-1 transcriptional targets. We demonstrate that the SET-16/mixed lineage leukemia 3/4 (MLL3/4) chromatin regulation complex functions at the same step or downstream of HSF-1 to control LET-70/UBE2D2 expression. Our results identify conserved proteins governing LCD, and demonstrate that transcriptional regulators influence this process at multiple levels.
Cell death is commonplace in development, homeostasis and disease. Although apoptotic cell death is extensively studied, inactivation of mouse apoptosis genes fails to block most developmental cell death.1, 2 Furthermore, many cells that die during vertebrate development do not exhibit apoptotic morphology.3, 4 Thus, other pathways likely account for most developmental cell culling.
Although non-apoptotic death pathways are described in cell culture and pathological settings, these do not appear to function during normal development.5 We previously reported the existence of a caspase-independent, non-apoptotic cell death program operating during Caenorhabditis elegans development.6, 7 Over 36 h, the male-specific linker cell guides gonad elongation from the ventral midbody to the cloaca.6, 8 The cell dies during the fourth larval stage (L4)-to-adult transition, allowing for gonad–cloaca tube fusion, enabling sperm exit/male fertility.8, 9 Thus, the mechanisms governing linker cell death (LCD) are likely under strong selective pressure.
Electron- and light-microscopy studies reveal that linker cell corpse morphology differs from that of apoptotic cells. Dying linker cells exhibit lack of chromatin condensation, crenellated nuclei, and swollen mitochondria and endoplasmic reticulum.7 Similar morphology accompanies some developmental vertebrate cell deaths,10 and is seen in degenerating neurons of patients with and mouse models of polyglutamine disease.11, 12 We refer to this cell death process as linker cell-type cell death (LCD).
LCD in C. elegans is unaffected by mutations blocking apoptosis, autophagy or necrosis.7, 13, 14 Similarly, LCD morphology is exhibited by some dying vertebrate cells carrying mutations in caspases/caspase regulators.4, 15 Thus, LCD represents a novel non-apoptotic cell death program.
Three regulatory modules promote LCD onset in C. elegans (Supplementary Figure S1). In one module, two opposing Wnt ligands, EGL-20/Wnt and LIN-44/Wnt, promote and inhibit LCD, respectively, and are detected by different transduction machineries within the cell.16 A second module, consisting of C. elegans heterochronic developmental timing pathway gene products, including the transcription factor LIN-29, works in parallel to Wnt signaling.16 A third module, consisting of the scaffold protein TIR-1/Sarm, the mitogen-activated protein kinase kinase (MAPKK) SEK-1, and the glutamine-rich protein PQN-41,17, 18 also promotes LCD in parallel to the Wnt and LIN-29 pathways.16 Importantly, mouse and Drosophila Sarm mutations block axon distal segment degeneration following axotomy,19 suggesting that this protein's function in non-apoptotic cell death may be conserved.
All three LCD modules function upstream of or in parallel to the heat-shock factor 1 (HSF-1) transcription factor. Although HSF-1 can protect cells from stressors,20 it is required for LCD by inducing expression of the conserved E2 ubiquitin-conjugating enzyme LET-70/UBE2D2 (Supplementary Figure S1). Other ubiquitin proteasome system (UPS) components, including E3 ligase components CUL-3, RBX-1, BTBD-2 and SIAH-1, are also required for LCD.16
To identify additional transcriptional regulators of LCD that function with HSF-1, we performed a genome-wide RNA interference screen and examined candidate gene mutations. Here, we characterize the roles of four transcriptional regulators. We show that NOB-1/Hox and EOR-1/PLZF are required cell autonomously for death, and that both function as part of the Wnt signaling module controlling LCD onset. We also show that Tailless/TLX-related NHR-67 functions cell autonomously in the linker cell and has two different activities: one promotes LCD upstream of HSF-1, and the other prevents precocious expression of LET-70/UBE2D2. Finally, we demonstrate that the SET-16/MLL3/4 H3K4 methyl transferase, as well as other components of this complex, functions together with or downstream of HSF-1 to control LET-70/UBE2D2 expression and cell death.
Our results support the framework for LCD in C. elegans that we previously described,16 uncover conserved proteins that function to promote LCD and demonstrate that transcription factors control LCD at distinct levels.
Results
NOB-1/Hox functions in Wnt signaling to promote LCD
The linker cell dies just anterior to the male tail, where Wnt signals governing LCD are specifically expressed. We wondered whether other spatially confined cues may control cell death. Hox genes are expressed in spatially restricted domains throughout metazoans. We examined whether mutations in five midbody/posterior Hox genes result in inappropriately surviving linker cells. As shown in Figure 1a, mutations in lin-39, mab-5 and php-3 do not affect LCD. A strong egl-5/Hox mutation has a weak effect (~10% survival); however, egl-5 is not expressed in the linker cell,21, 22, 23 and mutant males exhibit disorganized tails,24, 25 suggesting an indirect role in LCD. However, 31% of nob-1(os6) adult males exhibit linker cell survival (Figure 1a), suggesting a key role in LCD.
Figure 1.

NOB-1 promotes LCD through Wnt signaling. For all images, arrow, linker cell. Scale bar, 10 μm. For all histograms, error bar, S.E.M., numbers within bars, no. of animals, *P<0.05. (a) nob-1 is the main Hox gene required for LCD. (b) NOB-1::GFP is expressed in both migrating (left) and dying (right) linker cells. (c) egl-20 is not required for expression of a nob-1::GFP reporter. (d) nob-1 acts upstream of or in parallel to the Wnt signaling pathways required for LCD. (e) bar-1, sek-1, lin-29, nhr-67, set-16 and egl-20 are not required for nob-1 expression. (f) nob-1 is required for let-70::GFP expression
The nob-1/Hox locus encodes two alternatively spliced mRNAs, nob-1A and nob-1B. The predicted NOB-1B protein contains a DNA-binding homeodomain helix, including the conserved motif RRMKXKK not present in NOB-1A. The nob-1(os6) allele truncates this domain, suggesting that NOB-1B is likely relevant for death. To test this directly, and to confirm that the nob-1(os6) lesion is indeed responsible for linker cell survival, we generated animals expressing a nob-1B complementary DNA (cDNA) under the control of the linker cell-specific mig-24 promoter. Three independent mig-24p::nob-1B transgenes restored LCD to nob-1(os6) mutants (Figure 1a). This demonstrates that nob-1 is required for LCD, that the NOB-1B isoform is likely functional and that NOB-1 functions cell autonomously.
To further examine the NOB-1 site of action, we followed expression of a 15-kb genomic transgene, containing the nob-1 locus fused to green fluorescent protein (GFP).26, 27 As shown in Figure 1b, nob-1 is strongly expressed in the linker cell nucleus from the L3 larval stage onward, supporting a cell autonomous role. NOB-1 is thought to function as a posterior Hox gene, and is expressed in many tail cell nuclei (Figure 1b); however, linker cell expression is already evident when the cell is in the midbody region of the animal. Thus, although the linker cell is born in the midbody, it acquires posterior fate characteristics earlier.
The tail-restricted EGL-20 Wnt signal promotes LCD,16 raising the possibility that EGL-20/Wnt may control nob-1 expression. To test this, we examined expression of the 15-kb nob-1::GFP transgene in egl-20(n585) null mutants. As shown in Figure 1c, nob-1 expression is unaffected by the egl-20/Wnt lesion. Furthermore, although nob-1 mutations show additive interactions with EGL-20/Wnt and LIN-29 mutations (Figure 1d), nob-1 is properly expressed in the linker cells of bar-1/beta-catenin, sek-1 and lin-29 mutants as well as egl-20; sek-1 double mutants (Figure 1e). Thus, NOB-1 is unlikely to function downstream of Wnt, SEK-1/MAPKK or the developmental timing pathway.
Supporting this, we previously showed that lin-44/Wnt pathway mutations restore LCD to egl-20/Wnt and bar-1/beta-catenin mutants16 (Supplementary Figure S1). We found that a lin-44/Wnt mutation also restores LCD to nob-1 mutants (Figure 1d). Thus, nob-1 likely functions upstream of the lin-44/Wnt pathway. A mutation in the Wnt-antagonizing kinase gene gsk-3/GSK3β,21, 28 which functions downstream of EGL-20/Wnt16 (Supplementary Figure S1), also restores LCD to nob-1 mutants (Figure 1d).
Consistent with these observations, an HSF-1(R145A)-expressing transgene can restore LCD to egl-20/Wnt and bar-1/beta-catenin mutants, demonstrating that HSF-1 acts downstream of this signaling pathway16 (Supplementary Figure S1). As shown in Figure 1d, HSF-1(R145A) also restores death to nob-1(os6) mutants. Furthermore, nob-1(os6) mutants fail to induce expression of a let-70::GFP transgene tracking expression of the HSF-1 target LET-70/UBE2D2 (Figure 1f).
Together, these observations suggest that nob-1 functions upstream of EGL-20/Wnt signaling to promote LCD.
EOR-1/PLZF also functions with Wnt signals to promote LCD
The eor-1 gene encodes a protein similar to the promyelocytic leukemia zinc-finger transcription factor PLZF, which is fused to retinoic-acid receptor alpha in human acute promyelocytic leukemia.29 eor-1 mutations were suggested to regulate cell-specific death in C. elegans through an unknown mechanism independent of the Bcl-2 homology domain 3 (BH3)-only apoptotic regulator EGL-1.30 We therefore wondered whether eor-1 is also required for LCD. As shown in Figure 2a, systemic RNA interference (RNAi) against eor-1 modestly affects linker cell survival, with 14% of cells surviving inappropriately. Three other eor-1 mutations have stronger defects, with the strongest allele, eor-1(cs28), exhibiting 40% survival, suggesting an important role for EOR-1 protein in LCD.
Figure 2.

EOR-1 promotes LCD through Wnt. For all histograms, error bars, S.E.M., numbers within bars, number of animals scored. *P<0.0001, Fisher's exact test. (a) eor-1/eor-2 promote LCD. Error bars, S.E.M., Fisher's exact test. (b) An EOR-1 genomic fusion to GFP is expressed in the nucleus of the linker cell during death (left) and migration (right). Arrow: linker cell. Scale bar, 10 μm. (c) eor-1 mutants show reduced let-70p::let-70::gfp expression in surviving linker cells. (d) eor-1 acts with the UPS to promote LCD. (e) eor-1 acts with known LCD genes to promote death. R, eor-1(RNAi)
Linker cell-specific eor-1 RNAi subtly affects LCD, suggesting a cell autonomous role. We tested this directly by introducing a linker cell-expressing mig-24p::eor-1 cDNA transgene into eor-1(cs28) mutants. Death was restored in transgenic animals (Figure 2a), demonstrating that EOR-1 protein indeed functions cell autonomously. An eor-1::GFP fusion transgene expressed under the control of endogenous eor-1 regulatory sequences shows constitutive linker cell expression, and EOR-1::GFP protein localizes to the linker cell nucleus, among other nuclei (Figures 2a and b), consistent with the idea that EOR-1 regulates transcription cell autonomously.
EOR-1 binds to and functions with the uncharacterized protein EOR-2 to control gene expression in several contexts in C. elegans.31, 32, 33 Consistent with this, eor-2 mutants also possess surviving linker cells, a defect that can be rescued by linker cell-specific eor-2 expression (Figure 2a). Thus, EOR-1 likely functions with EOR-2 in LCD.
To understand how EOR-1 functions in the context of LCD, we examined expression of a let-70::GFP transgene in eor-1(cs28) mutants. Surviving linker cells fail to induce the reporter, indicating that eor-1 acts upstream of let-70 to promote its expression (Figure 2c). Epistasis experiments corroborate this, as double mutants harboring lesions in both eor-1 and UPS components required for LCD16 show insignificant enhancement in survival compared with single mutants alone (Figure 2d) (Supplementary Figure S1).
To further evaluate the context of EOR-1 function, we examined the effects of hsf-1(R145A) on EOR-1 activity. As shown in Figure 2e, although an eor-1 mutation synergizes with a weak hsf-1 loss-of-function lesion, eor-1(cs28); hsf-1(R145A) double mutants have more LCD than eor-1(cs28) mutants alone, demonstrating that the hsf-1(R145A) lesion can bypass EOR-1 deficits. Thus, EOR-1 likely functions upstream of or in parallel to hsf-1, consistent with its effects on LET-70/UBE2D2 expression (Figure 2c), and therefore may function with one of the three upstream modules.
To address whether EOR-1 functions with the MAPKK module, we attempted to examine the effects of simultaneously inhibiting eor-1 and components of this module (Figure 2e). However, eor-1(cs28); sek-1(ag1) double mutants exhibit embryonic lethality, and many eor-1(cs28); pqn-41(ns294) mutants die as larvae or are sterile. As eor-1(cs28) and pqn-41(ns294) are strong loss-of-function alleles, this suggests that EOR-1 and the MAPKK module work in parallel in some contexts. This may also be the case in the linker cell, as eor-1(RNAi) in sek-1 and pqn-41 mutants promotes additional survival compared with either alone.
To further address this issue, we examined genetic interactions between eor-1 and the Wnt module. lin-44/Wnt or wrm-1/beta-catenin mutations restore LCD to eor-1(cs28) mutants, as is the case for nob-1(os6) mutants (Figure 2e). Consistent with this, egl-20(n585null); eor-1(RNAi) double mutants show no significant increase in linker cell survival compared with egl-20(n585null) mutants alone, and a similar result is seen with bar-1/beta-catenin mutants (Figure 2e). Thus, eor-1 likely acts within the Wnt module to promote death. eor-1(cs28); nob-1(os6) double mutants show synergistic cell survival, suggesting that they function in parallel in Wnt signaling.
NHR-67/TLX functions in parallel to Wnt signaling and upstream of HSF-1
A genome-wide RNAi screen for LCD-promoting genes18 revealed that inactivation of nhr-67, encoding a Tailless/Tlx-like nuclear hormone receptor, prevents LCD. In all, 72% of first larval stage (L1) males, fed continuously on bacteria expressing dsRNA against nhr-67, exhibit linker cell survival as adults (Figures 3a and b). Accompanying this defect is a fully penetrant linker cell migration defect, consistent with previous observations.34, 35 To confirm these results, we examined animals containing genetic nhr-67 lesions. Although animals carrying strong nhr-67 loss-of-function mutations are inviable,34 nhr-67(pf159) mutants, lacking 484 bp of nhr-67 upstream regulatory sequences,36 survive. In 67% of these males, the linker cell survives inappropriately 2 h past the L4-to-adult transition (Figure 3b) with a fully penetrant migration defect. Furthermore, 40% of 24-h old nhr-67(pf159) adult males exhibit healthy, migration defective linker cells (n=82), suggesting that nhr-67 lesions block LCD.
Figure 3.

NHR-67 has opposing functions during LCD. For all images, arrow, linker cell, asterisk, cloaca. Scale bar, 10 μm. For all histograms, error bar, S.E.M., *P<0.05. (a) Top: nhr-67(RNAi) linker cells survive inappropriately and fail to migrate to the cloaca. Bottom: nhr-67(RNAi) surviving linker cell in 24 h old adult. (b) nhr-67 is required cell autonomously for LCD. EV, empty vector. Numbers within bars, no. of animals. (c) nhr-67 is expressed in the linker cell. Top: a 484 bp fragment upstream of nhr-67 driving GFP is expressed in the linker cell. Bottom: a 2-kb fragment upstream of nhr-67 driving mCherry is expressed in the linker cell. (d) bar-1 and lin-29 are not required for nhr-67::GFP expression. (e) nhr-67 mutants in combination with other LCD mutants causes a synergistic increase in linker cell survival. (f) nhr-67 causes inappropriate, early expression of UPS-GFP reporters. *P<0.05
To determine whether nhr-67 possesses independent migration and death roles, we fed L1 animals on bacteria expressing dsRNA against nhr-67 for only 7 h before transferring them to their regular food source. We found that the linker cell completed migration in about 50% of these males, and 20% of these exhibited linker cell survival (Figure 3b). This suggests that the nhr-67 LCD role is more sensitive to nhr-67 depletion, and that this role is independent of nhr-67 migration function.
To determine where nhr-67 is expressed, we examined animals containing a transgene fusing 2 kb of nhr-67 upstream regulatory sequences to mCherry. As shown in Figure 3c, this transgene is continuously expressed in the linker cell. Similarly, a transgene containing the 484-bp sequence deleted in the nhr-67(pf159) allele fused to a pes-10 minimal promoter and GFP is also robustly expressed in the linker cell until death. Thus, NHR-67 likely functions cell autonomously for LCD. To test this directly, we examined nhr-67(pf159) mutants expressing a mig-24p::nhr-67 cDNA transgene specifically in the linker cell. This transgene rescues inappropriate linker cell survival (Figure 3b), confirming that NHR-67 functions cell autonomously for death. Further supporting this, linker cell-specific RNAi against nhr-67 results in inappropriate cell survival (Figure 3b).
To determine how NHR-67 interacts with known LCD machinery components, we noted that expression of the 484-bp nhr-67::pes-10-minimal promoter::GFP construct is not perturbed by bar-1/beta-catenin or lin-29 loss-of-function (Figure 3d), suggesting that nhr-67 likely works in parallel to the Wnt and developmental timing modules. Furthermore, nhr-67(RNAi, 7 h) synergizes with egl-20(null), strong eor-1, or strong lin-29 mutations to enhance cell survival (Figure 3e). Similarly, nhr-67(RNAi, continuous) synergizes with a strong pqn-41/glutamine-rich mutation to enhance survival. Finally, lin-44/Wnt mutations cannot restore death to animals with reduced nhr-67 function (Figure 3e), and nhr-67 lesions do not affect nob-1::GFP expression (Figure 1e). Thus, NHR-67 appears to function in parallel to the Wnt, developmental timing, and MAPKK modules.
Importantly, although weak nhr-67 and hsf-1 lesions synergistically block LCD, hsf-1(R145A) restores death to animals with reduced nhr-67 function (Figure 3e); thus, NHR-67 acts upstream of or in parallel to HSF-1. Supporting a parallel function, nhr-67 RNAi does not affect hsf-1p::hsf-1::gfp expression in the linker cell or elsewhere (no RNAi: 77% GFP+ linker cells, n=22; nhr-67 RNAi: 84% GFP+ linker cells, n=67; P=0.53, Fisher's exact test), although NHR-67 could also function indirectly to affect HSF-1 activity.
NHR-67/TLX paradoxically inhibits LET-70/UBE2D2 expression
UPS components are the most downstream known elements of the C. elegans LCD pathway.16 We therefore sought to examine the relationship of NHR-67 to these components. We found that nhr-67(RNAi, 7 h) strongly enhances linker cell survival in animals carrying the let-70(ns770) mutation (Figure 3e), consistent with the idea that NHR-67 functions through or in parallel to the UPS. Surprisingly, we found that nhr-67(RNAi) animals express let-70::GFP precociously in L3 animals, and let-70::GFP accumulates to higher levels than in wild type (Figure 3f). Notably, ChIP-seq experiments conducted by the C. elegans ModENCODE project37 identify NHR-67 binding peaks within 500-bp upstream of the let-70 start site in vivo (Supplementary Figure S3).
This is consistent with the idea that early on, NHR-67 prevents inappropriate linker cell expression of UPS components. Later, NHR-67 promotes LCD upstream of or in parallel to HSF-1, perhaps through direct let-70 gene promoter binding. The notion that NHR-67 has distinct temporally regulated linker cell functions is consistent with previous observations regarding linker cell migration.34, 35
The SET-16/MLL3/4 HMT complex functions with HSF-1 to promote LCD
Our RNAi screen also revealed a role for swd-2.2 in LCD. Two different swd–2.2 RNAi constructs promote linker cell survival (Figure 4a), whereas RNAi against the closely related swd-2.1 has no effect (Supplementary Figure S2). Thus, swd-2.2 is specifically required for LCD.
Figure 4.

MLL3/4 complex components promote LCD. For all histograms, error bars, S.E.M., numbers within bars, no. of animals. (a) swd-2.2 and set-16 are required cell autonomously for LCD. *P<0.0001, Fisher's exact test. (b) MLL3/4 complex components are required for LCD. (c) swd-2.2p::gfp (left) and a set-16-GFP fosmid (right) are expressed in the linker cell (top right, DIC, bottom right, GFP). Arrow, linker cell. Scale bar, 10 μm. (d) MLL3/4 complex components are required for expression of UPS components. *P<0.0001, Fisher's exact test. (e) set-16 acts with known LCD genes to promote death
SWD-2.2 homologs have been identified in complex with SET-domain histone methyl transferases (HMT), and as subunits of the cleavage and polyadenylation factor (CPF) involved in transcription termination. To determine in which complex SWD-2.2 functions for death, we depleted components of each. RNAi against CPF components does not affect LCD (Supplementary Table S1). However, RNAi against the SET complex genes utx-1, ash-2, rbbp-5, wdr-5 and pis-1 does perturb LCD (Figure 4b); and RNAi against the catalytic subunit gene set-16 produces extensive survival (Figure 4a). C. elegans encodes two conserved SET domain proteins. SET-16 is most similar in structure to mammalian MLL3/4, whereas SET-2 resembles Set1/Set1A/B. We found that RNAi against set-2, or other complex genes (hcf-1, dpy-30 or cpf-1) fails to induce linker cell survival (Supplementary Figure S2). Together, these results show that an MLL3/4-related HMT complex is required for LCD.
Both set-16 and swd-2.2 are expressed in the linker cell (Figure 4c), suggesting that they may function cell autonomously for LCD. Supporting this idea, expressing a silently mutated RNAi-resistant swd-2.2 cDNA derived from the nematode C. brenneri under the control of a linker cell-specific promoter restores death to swd-2.2 RNAi animals (Figure 4a). Furthermore, linker cell-specific RNAi against set-16 also promotes inappropriate cell survival (Figure 4a). We conclude that the SET-16 HMT complex acts cell autonomously to promote LCD.
As shown in Figure 4d, ubq-1::GFP and let-70::GFP reporter expression decreases strongly in set-16(RNAi) and swd-2.2(RNAi) animals; thus, these genes may promote LCD by inducing UPS component expression. set-16 RNAi enhances survival in let-70(ns770), pqn-41(ns294), lin-29(n333) or egl-20(n585) mutants (Figure 4e), and does not affect NOB-1 expression (Figure 1e). In contrast to most LCD regulators we have studied, hsf-1(R145A) does not restore death to set-16(RNAi) surviving cells (Figure 4e).
Thus, unlike other LCD regulators, set-16 likely acts downstream of or in concert with hsf-1 to promote death. Our previous studies suggest that HSF-1 binds sequences in the let-70 5'-regulatory region.16 We propose that the SET-16 HMT complex functions with HSF-1 to regulate LET-70/UBE2D2 expression, and perhaps other UPS components.
Discussion
The results presented here are consistent with a model in which LCD is transcriptionally regulated at multiple levels (Figure 5). The known downstream elements of LCD in C. elegans include let-70, encoding an E2 ubiquitin-conjugating enzyme, which acts with other UPS components to effect LCD.16 HSF-1 likely directly induces let-70/E2 expression, and the studies presented here suggest that the SET-16 HMT complex may function with HSF-1 to control this expression. Indeed, set-16/MLL3/4 inactivation cannot be rescued by the hsf-1(R145A) gain-of-function allele; and, like hsf-1 loss-of-function alleles, set-16 is required for let-70 linker cell induction during death. Recently, chromatin-modifying factors have been found to cooperate with HSF-1 to regulate transcription during the L4-to-adult transition,38 supporting a similar effect on LCD. Furthermore, HSFs have been shown to recruit MLL-type complexes as co-activators in other contexts.39, 40
Figure 5.
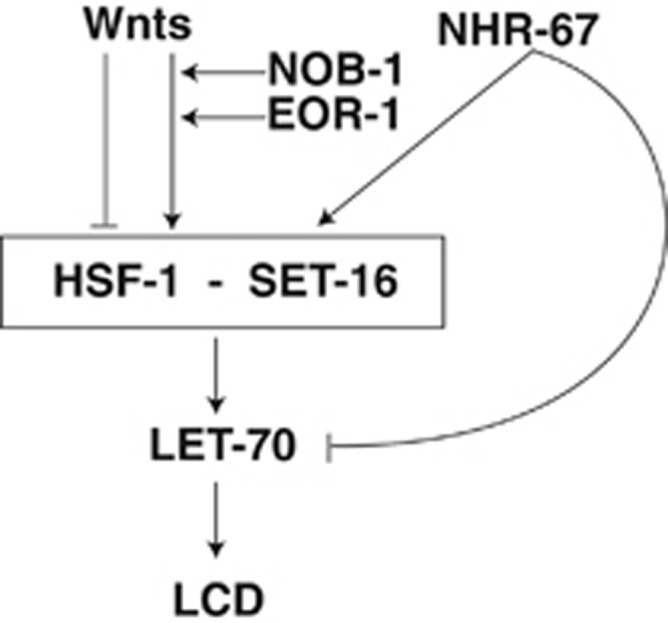
A model for transcriptional control of LCD. NOB-1/Hox and EOR-1/PLZF function in Wnt signaling upstream of HSF-1. NHR-67/TLX/NR2E1 functions upstream of HSF-1, but also blocks precocious LET-70/UBE2D2 expression. The SET-16 H3K4 methyl transferase complex promotes LCD together with HSF-1
Although HSF-1 and SET-16/MLL3/4 appear to directly upregulate LET-70/E2, our results suggest that NHR-67/TLX can antagonize this activity, as nhr-67 mutations induce precocious, increased LET-70 linker cell expression. Cooperative interactions between MLL3/4 and nuclear hormone receptors have been recognized in the literature in several settings.41, 42 Given our genetic/gene expression studies, we suspect that NHR-67 may directly bind DNA in the vicinity of the let-70 gene to counteract HSF-1 function early during the life of the cell. Testing this idea in detail, however, is difficult, as single-cell analysis of protein–DNA interactions in C. elegans is currently unreliable.
Our results show that NHR-67 also paradoxically promotes LCD. This activity appears to be genetically upstream of and in parallel to HSF-1 function. One possibility is that to induce death, NHR-67 protein functions on let-70 DNA, but acquires a new activity through protein modification, or through locus chromatin remodeling. Alternatively, the effects of NHR-67 in this context may be indirect. The genetics of NHR-67 function suggest that it may function in an incoherent feed-forward loop, a network regulatory structure in which a transcription factor activates both target gene transcription and transcription of a target inhibitor. Such modules are ubiquitous in signaling pathways,30, 31, 32 and cause a pulsed, biphasic response, perhaps accounting for the highly specific control of LCD onset. Careful observation of NHR-67 modification dynamics could uncover evidence for such a role.
Our studies also demonstrate the importance of NOB-1/HOX and EOR-1/PLZF in controlling Wnt signaling leading to LCD activation. Hox genes have previously been shown to act in parallel to or upstream of Wnt pathways,37, 38 and during C. elegans vulval development, EOR-1/PLZF acts with Wnts and in parallel to Hox genes.33 These observations support a role for the Wnt module as a regulatory entity, with different targets depending on the specific cell and developmental context.
EOR-1/PLZF has been implicated in the apoptotic death of the HSN neuron in C. elegans males.30 The association with sex-specific death in separate contexts is intriguing, and suggests the possibility of a common mode of function. Furthermore, although the role of EOR-1 in HSN neuron death is not understood, it functions independently of the EGL-1/BH3-only cell death regulator. Further studies of EOR-1/PLZF regulation and targets may reveal aspects of cell death common to both apoptosis and LCD.
What event controls LCD onset? Our data suggest that HSF-1 expression induction is unlikely to be the trigger, as HSF-1 exists in the linker cell well before death. Our previous work suggests that HSF-1 has two activities: one regulates the heat-shock response, the other promotes LCD. It is possible that toggling of HSF-1 between these two roles—at the level of transcript splicing, or at the post-translational level—determines linker cell viability. Whether upstream transcription factors directly or indirectly regulate this transition is unknown.
Finally, it is intriguing that vertebrate homologs of three of the transcriptional regulators we identified, EOR-1/PLZF, NHR-67/TLX and SET-16/MLL3/4, are implicated in the control of cancer in humans.43, 44, 45 Our findings that these factors control cell death onset in C. elegans raises the possibility that mutating their human counterparts blocks cell death, leading to tumor progression.
Materials and Methods
Strains
C. elegans strains were cultured using standard methods46 and were grown at 20 °C. Wild-type animals were the Bristol N2 strain. The following alleles were crossed to qIs56 V,47 an integrated GFP linker cell reporter transgene, and him-8(e1489) IV or him-5(e1490) V,48 to increase the percentage of males in the population.
LGI: hsf-1(sy441),49 lin-44(n1792)50
LGII: lin-29(n333);51 rrf-3(pk1426),45 drSi28[hsf-1p:: hsf-1(R145A)-GFP]52
LGIII: pqn-41(ns294);18 nob-1(os6)53
LGIV: let-70(ns770);16 siah-1(tm1968) (Japanese National BioResource Project), eor-1(cs28;cs40;cs44)32
LGV: rde-1(ne219)54
RNAi experiments
RNAi was performed by feeding on the strains indicated.18 Briefly, synchronized L1 animals, generated by bleaching and hatching in M9 medium, were added to each RNAi plate and grown for approximately 48 h at 20–22 °C. In all, 0–2 h adults were scored using a fluorescent dissecting or compound microscope. Bacterial feeding RNAi clones were from the Ahringer library or the Vidal library (both Source Bioscience, Nottingham, UK). Clones not present in either library were generated by cloning into the L4440 vector.
Linker cell survival, migration and GFP expression assays
Late L4-stage males were picked off onto fresh plates, incubated at 20 °C, and scored 2 h later under a fluorescent compound microscope for the presence or absence of a linker cell. A surviving linker cell is described as an unrounded cell with a round nucleus. For GFP expression assays, 0–2 h adults containing the let-70 promoter::let-70::GFP (nsIs241) or ubq-1 promoter::ubq-1::GFP (nsIs386) transgenes were scored for the presence or absence of GFP expression in the linker cell. The fraction of animals expressing GFP was divided by the fraction of animals with surviving linker cells in order to obtain an accurate measure of linker cell expression. This method was verified by looking at GFP expression of reporters with a lag-2 promoter::mCherry coinjection marker; results using the two different methodologies showed similar expression patterns.16
Germline transformation and rescue experiments
Germline transformation was carried out as previously described.57 For GFP expression analysis, all plasmids were injected into unc-119(ed3) III; him-8(e1489) IV hermaphrodites with unc-119(+)52 as a transformation marker. All plasmids were injected at between 1 and 50 ng/ul. pBluescript (Agilent, Santa Clara, CA, USA) was used to adjust the DNA concentration of injection mixtures if necessary. For rescue studies, animals were picked under a fluorescent dissecting microscope (Leica, Wetzlar, Germany) the previous night as L3s with YFP- or mCherry-expressing linker cell to a new RNAi plate and scored the following day. For set-16 and swd-2.2, as the mig-24 promoter can cause a migration defect on its own, only correctly migrated linker cells were used in determining survival percentages.
swd-2.2 cDNA isolation of C. brenneri
Total RNA was extracted using TRIzol (Life Technologies, Carlsbad, CA, USA) using standard protocols.53 cDNAs were amplified from mixed stage C. brenneri and from day 1 adult C. briggsae using the SuperScript II Reverse Transcriptase (Life Technologies).
Microscopy
LCD and GFP expression were scored using conventional fluorescence microscopy. An Axioplan II compound microscope (Zeiss, Oberkochen, Germany) equipped with an AxioCam CCD camera (Zeiss) was used to take images on AxioVision software (Zeiss).
Statistical methods
Chi-square (Fisher's exact) tests were used for quantification of LCD experiments as well as quantification of GFP+ linker cells.
Acknowledgments
We thank members of the Shaham lab for discussions, and C Bargmann, M Sundaram, W Waterston, B Wightman and W Wood for strains and reagents. Some strains were provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440). JAM was supported by the Rockefeller Women and Science Fellowship Program and NIH training grant CA09673. MJK was supported by a Medical Scientist Training Program grant from the National Institute of General Medical Sciences of the National Institutes of Health under award number T32GM07739 to the Weill Cornell/Rockefeller/Sloan-Kettering Tri-Institutional MD-PhD Program. This work was supported by NIH grants HD078703 and NS081490 to SS.
Author contributions
JAM, MJK and SS designed experiments, and wrote the manuscript. MCA and ESB performed the RNAi screen and first demonstrated roles for nhr-67 and swd-2.2.
Glossary
- bp
basepair
- BH3
Bcl-2 homology domain 3
- cDNA
complementary DNA
- ChIP-seq
chromatin immunoprecipitation with massively parallel DNA sequencing
- GFP
green fluorescent protein
- GSK3β
glycogen synthase kinase 3 beta
- HSF-1
heat-shock factor 1
- kb
kilobase
- LCD
linker cell-type death
- L1
first larval stage
- L4
fourth larval stage
- HMT
histone methyl transferase
- MAPKK
Map kinase kinase
- MLL3/4
mixed lineage leukemia 3/4
- ModENCODE
model organism encyclopedia of DNA elements
- n
number
- NR2E1
nuclear receptor subfamily 2, group E, member 1
- p
promoter
- PLZF
promyelocytic leukemia zinc-finger transcription factor
- RNAi
RNA interference
- TF
transcription factor
- TLX
tailless
- UBE2D2
ubiquitin-conjugating enzyme E2D 2
- UPS
ubiquitin proteasome system
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies this paper on Cell Death and Differentiation website (http://www.nature.com/cdd)
Edited by E Baehrecke
Supplementary Material
References
- Chautan M, Chazal G, Cecconi F, Gruss P, Golstein P. Interdigital cell death can occur through a necrotic and caspase-independent pathway. Curr Biol 1999; 9: 967–970. [DOI] [PubMed] [Google Scholar]
- Honarpour N, Du C, Richardson JA, Hammer RE, Wang X, Herz J. Adult Apaf-1-deficient mice exhibit male infertility. Dev Biol 2000; 218: 248–258. [DOI] [PubMed] [Google Scholar]
- Chu-Wang IW, Oppenheim RW. Cell death of motoneurons in the chick embryo spinal cord. I. A light and electron microscopic study of naturally occurring and induced cell loss during development. J Comp Neurol 1978; 177: 33–57. [DOI] [PubMed] [Google Scholar]
- Oppenheim RW, Flavell RA, Vinsant S, Prevette D, Kuan CY, Rakic P. Programmed cell death of developing mammalian neurons after genetic deletion of caspases. J Neurosci 2001; 21: 4752–4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Yuan J. Necroptosis in health and diseases. Semin Cell Dev Biol 2014; 35: 14–23. [DOI] [PubMed] [Google Scholar]
- Galvin BD, Kim S, Horvitz HR. Caenorhabditis elegans genes required for the engulfment of apoptotic corpses function in the cytotoxic cell deaths induced by mutations in lin-24 and lin-33. Genetics 2008; 179: 403–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham MC, Lu Y, Shaham S. A morphologically conserved nonapoptotic program promotes linker cell death in Caenorhabditis elegans. Dev Cell 2007; 12: 73–86. [DOI] [PubMed] [Google Scholar]
- Kimble J, Hirsh D. The postembryonic cell lineages of the hermaphrodite and male gonads in Caenorhabditis elegans. Dev Biol 1979; 70: 396–417. [DOI] [PubMed] [Google Scholar]
- Bloss TA, Witze ES, Rothman JH. Suppression of CED-3-independent apoptosis by mitochondrial betaNAC in Caenorhabditis elegans. Nature 2003; 424: 1066–1071. [DOI] [PubMed] [Google Scholar]
- Pilar G, Landmesser L. Ultrastructural differences during embryonic cell death in normal and peripherally deprived ciliary ganglia. J Cell Biol 1976; 68: 339–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman MJ, Shah AG, Fang Z-H, Ward EG, Warren ST, Li S et al. Polyglutamine domain modulates the TBP-TFIIB interaction: implications for its normal function and neurodegeneration. Nat Neurosci 2007; 10: 1519–1528. [DOI] [PubMed] [Google Scholar]
- Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA et al. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 1997; 90: 537–548. [DOI] [PubMed] [Google Scholar]
- Joshi P, Eisenmann DM. The Caenorhabditis elegans pvl-5 gene protects hypodermal cells from ced-3-dependent, ced-4-independent cell death. Genetics 2004; 167: 673–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denning DP, Hatch V, Horvitz HR. Programmed elimination of cells by caspase-independent cell extrusion in C.elegans. Nature 2012; 488: 226–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppenheim RW, Blomgren K, Ethell DW, Koike M, Komatsu M, Prevette D et al. Developing postmitotic mammalian neurons in vivo lacking Apaf-1 undergo programmed cell death by a caspase-independent, nonapoptotic pathway involving autophagy. J Neurosci 2008; 28: 1490–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinet MJ, Malin JA, Abraham MC, Blum ES, Silverman MR, Lu Y et al. HSF-1 activates the ubiquitin proteasome system to promote non-apoptotic developmental cell death in C. elegans. Elife 2016; 5: e12821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll M, Chalfie M. The mec-4 gene is a member of a family of Caenorhabditis elegans genes that can mutate to induce neuronal degeneration. Nature 1991; 349: 588–593. [DOI] [PubMed] [Google Scholar]
- Blum ES, Abraham MC, Yoshimura S, Lu Y, Shaham S. Control of nonapoptotic developmental cell death in Caenorhabditis elegans by a polyglutamine-repeat protein. Science (New York, NY) 2012; 335: 970–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterloh JM, Yang J, Rooney TM, Fox AN, Adalbert R, Powell EH et al. dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science (New York, NY) 2012; 337: 481–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X. HSF1 is required for extra-embryonic development, postnatal growth and protection during inflammatory responses in mice. EMBO J 1999; 18: 5943–5952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treinin M, Chalfie M. A mutated acetylcholine receptor subunit causes neuronal degeneration in C. elegans. Neuron 1995; 14: 871–877. [DOI] [PubMed] [Google Scholar]
- Kalis AK, Murphy MW, Zarkower D. EGL-5/ABD-B plays an instructive role in male cell fate determination in the C. elegans somatic gonad. Dev Biol 2010; 344: 827–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korswagen HC, Park JH, Ohshima Y, Plasterk RH. An activating mutation in a Caenorhabditis elegans Gs protein induces neural degeneration. Genes Dev 1997; 11: 1493–1503. [DOI] [PubMed] [Google Scholar]
- Hall DH, Gu G, García-Añoveros J, Gong L, Chalfie M, Driscoll M. Neuropathology of degenerative cell death in Caenorhabditis elegans. J Neurosci 1997; 17: 1033–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chisholm A. Control of cell fate in the tail region of C. elegans by the gene egl-5. Development 1991; 111: 921–932. [DOI] [PubMed] [Google Scholar]
- Hong K, Driscoll M. A transmembrane domain of the putative channel subunit MEC-4 influences mechanotransduction and neurodegeneration in C. elegans. Nature 1994; 367: 470–473. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Boyle TJ, Liu Z, Murray JI, Wood WB, Waterston RH. A negative regulatory loop between microRNA and Hox gene controls posterior identities in Caenorhabditis elegans. PLoS Genet 2010; 6: e1001089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlesinger A, Shelton CA, Maloof JN, Meneghini M, Bowerman B. Wnt pathway components orient a mitotic spindle in the early Caenorhabditis elegans embryo without requiring gene transcription in the responding cell. Genes Dev 1999; 13: 2028–2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi L, Gerstbrein B, Frøkjaer-Jensen C, Royal DC, Mukherjee G, Royal MA et al. The neurotoxic MEC-4(d) DEG/ENaC sodium channel conducts calcium: implications for necrosis initiation. Nat Neurosci 2004; 7: 1337–1344. [DOI] [PubMed] [Google Scholar]
- Hoeppner DJ, Spector MS, Ratliff TM, Kinchen JM, Granat S, Lin S-C et al. eor-1 and eor-2 are required for cell-specific apoptotic death in C. elegans. Dev Biol 2004; 274: 125–138. [DOI] [PubMed] [Google Scholar]
- Howell K, Arur S, Schedl T, Sundaram MV. EOR-2 is an obligate binding partner of the BTB-zinc finger protein EOR-1 in Caenorhabditis elegans. Genetics 2010; 184: 899–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocheleau CE, Howard RM, Goldman AP, Volk ML, Girard LJ, Sundaram MV. A lin-45 raf enhancer screen identifies eor-1, eor-2 and unusual alleles of Ras pathway genes in Caenorhabditis elegans. Genetics 2002; 161: 121–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard RM, Sundaram MV. C. elegans EOR-1/PLZF and EOR-2 positively regulate Ras and Wnt signaling and function redundantly with LIN-25 and the SUR-2 Mediator component. Genes Dev 2002; 16: 1815–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, Sternberg PW. The C. elegans tailless/Tlx homolog nhr-67 regulates a stage-specific program of linker cell migration in male gonadogenesis. Development 2009; 136: 3907–3915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz EM, Kato M, Sternberg PW. Functional transcriptomics of a migrating cell in Caenorhabditis elegans. Proc Natl Acad Sci USA 2012; 109: 16246–16251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verghese E, Schocken J, Jacob S, Wimer AM, Royce R, Nesmith JE et al. The tailless ortholog nhr-67 functions in the development of the C. elegans ventral uterus. Dev Biol 2011; 356: 516–528. [DOI] [PubMed] [Google Scholar]
- Celniker SE, Dillon LAL, Gerstein MB, Gunsalus KC, Henikoff S, Karpen GH et al. Unlocking the secrets of the genome. Nature 2009; 459: 927–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labbadia J, Morimoto RI. Repression of the heat shock response is a programmed event at the onset of reproduction. Mol Cell 2015; 59: 639–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Chen J, Yu J, Yang G, Temple E, Harbinski F et al. Identification of mixed lineage leukemia 1(MLL1) protein as a coactivator of heat shock factor 1(HSF1) protein in response to heat shock protein 90 (HSP90) inhibition. J Biol Chem 2014; 289: 18914–18927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ST, Petruk S, Sedkov Y, Cho E, Tillib S, Canaani E et al. Modulation of heat shock gene expression by the TAC1 chromatin-modifying complex. Nat Cell Biol 2004; 6: 162–167. [DOI] [PubMed] [Google Scholar]
- Lee S, Kim D-H, Goo Y-H, Lee YC, Lee S-K, Lee JW. Crucial roles for interactions between MLL3/4 and INI1 in nuclear receptor transactivation. Mol Endocrinol 2009; 23: 610–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedkov Y, Cho E, Petruk S, Cherbas L, Smith ST, Jones RS et al. Methylation at lysine 4 of histone H3 in ecdysone-dependent development of Drosophila. Nature 2003; 426: 78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Brand NJ, Chen A, Chen SJ, Tong JH, Wang ZY et al. Fusion between a novel Krüppel-like zinc finger gene and the retinoic acid receptor-alpha locus due to a variant t(11;17) translocation associated with acute promyelocytic leukaemia. EMBO J 1993; 12: 1161–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Khan MA, Weiler M, Blaes J, Jestaedt L, Geibert M et al. Targeting self-renewal in high-grade brain tumors leads to loss of brain tumor stem cells and prolonged survival. Stem Cell 2014; 15: 185–198. [DOI] [PubMed] [Google Scholar]
- Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 2011; 476: 298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics 1974; 77: 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegfried KR, Kimble J. POP-1 controls axis formation during early gonadogenesis in C. elegans. Development 2002; 129: 443–453. [DOI] [PubMed] [Google Scholar]
- Hodgkin J, Horvitz HR, Brenner S. Nondisjunction mutants of the nematode Caenorhabditis elegans. Genetics 1979; 91: 67–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajdu-Cronin YM, Chen WJ, Sternberg PW. The L-type cyclin CYL-1 and the heat-shock-factor HSF-1 are required for heat-shock-induced protein expression in Caenorhabditis elegans. Genetics 2004; 168: 1937–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman MA, Horvitz HR. The Caenorhabditis elegans gene lin-44 controls the polarity of asymmetric cell divisions. Development 1994; 120: 1035–1047. [DOI] [PubMed] [Google Scholar]
- Ambros V, Horvitz HR. Heterochronic mutants of the nematode Caenorhabditis elegans. Science (New York, NY) 1984; 226: 409–416. [DOI] [PubMed] [Google Scholar]
- Morton EA, Lamitina T. Caenorhabditis elegans HSF-1 is an essential nuclear protein that forms stress granule-like structures following heat shock. Aging Cell 2013; 12: 112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arata Y, Kouike H, Zhang Y, Herman MA, Okano H, Sawa H. Wnt signaling and a Hox protein cooperatively regulate psa-3/Meis to determine daughter cell fate after asymmetric cell division in C. elegans. Dev Cell 2006; 11: 105–115. [DOI] [PubMed] [Google Scholar]
- Tabara H, Sarkissian M, Kelly WG, Fleenor J, Grishok A, Timmons L et al. The rde-1 gene, RNA interference, and transposon silencing in C. elegans. Cell 1999; 99: 123–132. [DOI] [PubMed] [Google Scholar]
- Eisenmann DM, Maloof JN, Simske JS, Kenyon C, Kim SK. The beta-catenin homolog BAR-1 and LET-60 Ras coordinately regulate the Hox gene lin-39 during Caenorhabditis elegans vulval development. Development 1998; 125: 3667–3680. [DOI] [PubMed] [Google Scholar]
- Kim DH, Feinbaum R, Alloing G, Emerson FE, Garsin DA, Inoue H et al. A conserved p38 MAP kinase pathway in Caenorhabditis elegans innate immunity. Science (New York, NY) 2002; 297: 623–626. [DOI] [PubMed] [Google Scholar]
- Mello CC, Kramer JM, Stinchcomb D, Ambros V. Efficient gene transfer in C.elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J 1991; 10: 3959–3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.