

Abstract
Cocaine self-administration induces dysfunctional neuroadaptations in the prefrontal cortex that underlie relapse to cocaine-seeking. Cocaine self-administration disturbs glutamatergic transmission in the nucleus accumbens that is prevented by infusion of brain-derived neurotrophic factor (BDNF) into the prelimbic area of the prefrontal cortex. Intra-prelimbic infusion of BDNF decreases cocaine-seeking in a TrkB-ERK MAP kinase-dependent manner. Neuronal activity triggers an interaction between TrkB receptors and NMDA receptors, leading to ERK activation. In the present study, infusion of the GluN2A-containing NMDA receptor antagonist, TCN-201, or the GluN2B-containing NMDA receptor antagonist, Ro-25-6981, into the prelimbic cortex of rats blocked the suppressive effect of BDNF on cocaine-seeking. During early withdrawal from cocaine self-administration, tyrosine phosphorylation of ERK, GluN2A, and GluN2B in the prelimbic cortex was reduced and this reduction of phospho-proteins was prevented by intra-prelimbic BDNF infusion. TCN-201 infusion into the prelimbic cortex inhibited the BDNF-mediated increase in pERK and pGluN2A whereas Ro-25-6981 infusion into the prelimbic cortex blocked BDNF-induced elevation of pERK and pGluN2B, indicating that both GluN2A- and GluN2B-containing NMDA receptors underlie BDNF-induced ERK activation. These data demonstrate that BDNF-mediated activation of GluN2A- and GluN2B-containing NMDA receptors underlies ERK activation in the prelimbic cortex during early withdrawal, preventing subsequent relapse to cocaine-seeking.
Keywords: Addiction, Brain-derived neurotrophic factor, Extracellular signal-regulated kinase, NMDA receptors, Prefrontal cortex, Reinstatement
1. Introduction
The glutamatergic pathway arising from prelimbic cortex that innervates the nucleus accumbens (NAc) core mediates reinstatement of cocaine-seeking in rats with a cocaine self-administration (SA) history (McLaughlin and See, 2003; McFarland et al. 2003). Brain-derived neurotrophic factor (BDNF) and its receptor, TrkB, are expressed in cortical pyramidal neurons where they modulate synaptic plasticity and glutamatergic neurotransmission in an activity-dependent manner (Bramham and Messaoudi, 2005; Gomes et al. 2006; McGinty et al. 2010). BDNF exerts many effects on synaptic plasticity in models of substance use disorders depending on the brain area and the phase of addiction (reviewed in McGinty et al., 2010, 2015; Li and Wolf, 2015). An intra-prelimbic BDNF infusion reverses cocaine-induced ERK and CREB dephosphorylation in prelimbic cortex within 2 hr of the end of cocaine SA (Whitfield et al. 2011). How the BDNF-induced reversal of the ERK-CREB shutoff in prelimbic cortex in early withdrawal underlies the ability of BDNF to suppress cocaine-seeking 1–3 weeks later (Berglind et al. 2007) is not known. However, intervention immediately after cocaine self-administration ends is critical because phospho-ERK levels in the prelimbic cortex do not differ from yoked-saline control values one day or one week after the end of cocaine self-administration (Whitfield et al., J Neurosci 2011-supplementary figure 1). Moreover, phospho-CREB levels are normal at 24 hr and elevated one week later and the latter increase is mediated by PKA, not ERK (Sun et al., Addiction Biol 2014). Further, because the suppressive effect of BDNF on cocaine-seeking is ERK-dependent (Whitfield et al., 2011), if BDNF is infused one week after the end of cocaine self-administration when ERK levels in PFC are normal, BDNF has no effect on reinstatement (Berglind et al., 2007). These dynamic changes in prefontal phospho-proteins over time support our hypothesis that neuroadaptations change according to the addiction phase (during self-administration, during early hours of abstinence, after prolonged abstinence, and after relapse). Thus, different treatment strategies that target different protein changes depending on the phase of addiction may be required to treat addiction.
Because glutamatergic neuronal activity enhances the duration of BDNF-induced ERK activity in cortical cultures (Matsumoto et al. 2006), we investigated whether NMDA receptor activity mediates BDNF-induced suppression of cocaine-seeking and elevation of ERK phosphorylation in a short access SA-reinstatement model. The rationale for investigating the link between BDNF-TrKB and NMDA receptor subtypes is based on the fact that BDNF increases the open probability time of NMDARs in a TrkB-dependent manner (Levine et al. 1998). Regulation of ERK activity is mediated by GluN2A- and GluN2B-containing NMDARs that are more highly expressed in the cerebral cortex than other NMDAR regulatory subunits (Ivanov et al. 2006; Yashiro and Philpot, 2008). BDNF activation of TrkB promotes the association of TrkB with Src family kinases (SFK), leading to SFK activation and tyrosine phosphorylation of GluN2A and GluN2B (Goebel-Goody et al. 2009; Huang and McNamara, 2010; Iwasaki et al. 1998; Tezuka et al. 1999). SFK-mediated phosphorylation of GluN2B at Y1472 increases the expression of the subunit in postsynaptic density-enriched fractions (Nakazawa et al. 2001; Cheung and Gurd, 2001; Goebel-Goody et al. 2009). Moreover, SFK-mediated phosphorylation of GluN2A at Y1325 leads to the potentiation of NMDA-stimulated whole cell currents (Taniguchi et al. 2009) whereas SFK-mediated phosphorylation of GluN2B enhances GluN2B surface expression (Roche et al. 2001; Prybylowski et al. 2005).
Based on the tyrosine phosphorylation-dependent interaction between NMDARs and TrkB, an important objective of this study was to examine the effect of infusing a GluN2A-selective antagonist, TCN-201, or a GluN2B-selective antagonist, Ro-25-6981, into the prelimbic cortex before BDNF to examine the effect of TCN-201 or Ro-25-6981 on BDNF-mediated tyrosine phosphorylation of ERK, GluN2A, or GluN2B in the prelimbic cortex in the presence or absence of cocaine. In addition, the role of the GluN2 subunits in BDNF’s ability to suppress cocaine-seeking was examined. Our results demonstrate that both GluN2A- and GluN2B-containing NMDARs underlie the ability of BDNF to prevent cocaine-induced ERK dephosphorylation in the prelimbic cortex during early withdrawal, suppressing subsequent relapse to cocaine-seeking.
2. Experimental procedures
2.1 Experimental design
All experimental designs are illustrated in Figure 1A–C. In all experiments, intra- prelimbic cortical infusions were performed immediately after the last of 12–14 short access (2 hr) daily cocaine self-administration sessions. In Experiment 1 (Fig 1A), vehicle (2%DMSO/PBS), TCN-201, or Ro-25-6981 was infused into the prelimbic cortex 20 min before BDNF or PBS. Rats underwent 6 d of homecage abstinence followed by a 2-hr post-abstinence (PA) relapse test under extinction conditions followed by daily extinction training to criterion before a cue-induced reinstatement test. In Experiment 2 (Fig 1B), the prelimbic cortex of rats was infused with BDNF or PBS 2 hr before decapitation to determine/confirm whether BDNF prevented a cocaine SA-induced decrease in the phosphorylation of ERK, GluN2A, and GluN2B. In Experiments 3 and 4 (Fig 1C), the selective GluN2A antagonist, TCN-201, or the selective GluN2B antagonist, Ro-25-6981, was infused into the prelimbic cortex 20 min before BDNF or PBS and the rats were decapitated 2 hr later to determine whether inhibition of GluN2A or GluN2B-containing NMDARs blocked BDNF’s ability to prevent the cocaine SA-induced decrease in phospho-proteins in prelimbic cortex.
Figure 1.

Schematic representations of the experimental designs (A–C) and cannula placements (D). A) Effects of intra-prelimbic cortex infusions of TCN-201 or Ro-25-6981 on the suppressive effects of BDNF on cocaine-seeking. (B) Effects of intra- prelimbic cortex infusion of BDNF on phospho-protein expression in the prelimbic cortex 2 hr after infusions. (C) Effects of intra-prelimbic cortex infusions of TCN-201 or Ro-25-6981 on BDNF-induced elevation of phospho-proteins in the prelimbic cortex 2 hr after infusions. (D) Illustration of cannula placements and tissue punches. In Experiment 1, the tips of the intracranial infusion cannulae within the target area of the ventral anterior cingulate and prelimbic cortex are shown as small filled dots. For rats in Experiments 2–4, cannula placements within the prelimbic cortex were verified by inspection during brain punching (shown as large, unfilled circles). Two additional rats were routinely assigned to BDNF-infused groups (n=10) than PBS-infused groups (n=8) to allow for any infusion failures in the course of the study; however, none were lost for this reason. The following rats were omitted from the study: rats which lost their head-caps with implanted guide cannulae before PA relapse test or cue test (n=2), rats in which the cannula placement was inaccurate (n=2), rats in TCN experiment did not reinstate during PA relapse test or cue test (n=3, TCN-PBS and n=3, TCN-BDNF groups). PA=post-abstinence
2.2. Animals and surgery
Adult male Sprague Dawley rats (Charles River Laboratories, Raleigh, NC; 275–300g on arrival) were singly-housed and fed ad libitum in a temperature- and humidity-controlled room on a reversed light/dark cycle for 4 days before surgery. All procedures were conducted with the approval of the MUSC Institutional Animal Care and Use Committee and the NIH Guide for the Care and Use of Laboratory Animals (2012).
On the day of surgery, rats were anesthetized with a mixture of ketamine (66 mg/kg, i.p.) and xylazine (1.33 mg/kg,i.p.) followed by equithesin (0.5 ml/kg, i.p.) and ketorolac (2.0 mg/kg, i.p.). One end of a catheter was placed into the right jugular vein and the other end was threaded subcutaneously to a back mount cannula pedestal (Med Associates, Inc., St. Albans, VT). Immediately after catheterization, the skull was prepared to receive 26 gauge bilateral guide cannulae (Plastics One, Roanoke, VA) placed 1 mm above the prelimbic cortex [anterior-posterior (AP) relative to bregma, +3.0; medial-lateral (ML), +/− 0.6; dorsoventral (DV), −1.6 relative to dural surface] (Paxinos and Watson 2005). Guide cannulae were affixed to the skull with cranioplastic cement and steel screws. Stylets the same length as the guide cannulae (Plastics One) were inserted into the cannulae. Following surgery, rats were infused intravenously with 0.1 ml of cefazolin (33.3 mg/kg, 10 mg/0.1ml) as well as an anti-microbial lock solution TCS (0.1 ml; Access Technologies, Skokie, IL) twice daily for 5 days. Two rats lost their headcaps and were omitted from the study.
2.3. Cocaine self-administration
In all experiments, after at least 5 days of surgical recovery, rats were food-restricted (20 g/day) and trained to self-administer cocaine on a FR1 reinforcement schedule (2hr/day x 12–14d) as described (Berglind et al. 2007; Whitfield et al. 2011; Sun et al. 2013). Rats pressing the active (right) lever were infused with cocaine hydrochloride (0.2 mg/infusion; NIDA, Research Triangle Park, NC) paired with a light and tone cue. For Experiments 2–4, each rat in a yoked-saline group was matched to a rat in the cocaine SA group and infused with 0.9% saline when its paired rat received a cocaine infusion. Rats were fed ad libitum after the end of self-administration.
2.4. Micro-infusion of drugs into prelimbic cortex
All reagents were infused (0.25 μl/min) into prelimbic cortex through bilateral 33 gauge injectors (Plastics One) protruding 1 mm below the tip of the guide cannulae for 2 min. TCN-201 and Ro-25-6981 were purchased from Tocris Bioscience (Bristol, UK) and BDNF was purchased from R&D Systems, Inc. (Minneapolis, MN). TCN-201 was dissolved in 2% DMSO/10mM PBS; Ro-25-6981 and BDNF (0.75 μg/0.5 μl/side) were dissolved in 10 mM PBS.
Concentrations of GluN2A and GluN2B antagonists for infusion into the prelimbic cortex were based on the literature and confirmed by pilot experiments (Supplementary Figures 1 and 2). Gipson et al. (2013) demonstrated that 0.01 and 0.1 nmol TCN-201 (equivalent to 0.005 and 0.05 μg) in 0.5 μl infused into nucleus accumbens immediately before cue-induced reinstatement suppressed cocaine-seeking in a concentration-dependent manner. Zhao et al. (2005) demonstrated that 2 μg/0.5 μl/ Ro-25-6981 infused into the anterior cingulate cortex before fear conditioning decreased subsequent freezing to the auditory fear stimulus. Confirmation that 0.05 and 0.005 μg/0.5 μl of TCN-201 or 0.2 and 2 μg/0.5 μl of Ro-25-6981 infused 20 min before BDNF (0.75 μg/0.5 μl/side) prevented the ability of BDNF to increase phospho-ERK expression in a dose-dependent manner two hr later is presented in Supplementary Figures 1 and 2. Basal pERK levels in the prelimbic cortex of rats infused with vehicle or either dose of TCN-201 or Ro-25-6981 20 min before PBS did not differ. Based on these studies, the most effective concentration of TCN-201 (0.05 μg/0.5 μl/side in 2% DMSO/PBS) or Ro-25-6981 (2 μg/0.5 μl/side in PBS) was infused into the prelimbic cortex at the end of self-administration or yoked-saline sessions.
2.5. Post-abstinence relapse test, extinction, and cue-induced reinstatement test
In Experiment 1 (Fig 1A), after the end of cocaine SA and intracranial infusions, rats underwent 6 days of home cage abstinence before a 2-hr post-abstinence relapse test under extinction conditions followed by daily extinction training (to a criterion of <15 active lever presses per session) before a cue-induced reinstatement test was performed as described (Berglind et al. 2007; Whitfield et al. 2011). Six control rats did not reinstate (>1.5 SD from the mean) in either the post-abstinence or cue tests and were removed from the study.
2.6. Histology
All rats were decapitated without anesthesia and their brains were extracted and frozen. In Experiment 1, rats were euthanized after cue-induced reinstatement and 40 μm coronal sections were cut in a cryostat to confirm cannula placements by Nissl-staining. In Experiments 2–4, a 2 mm-thick AP punch was taken with a 3 mm diameter biopsy punch (Braintree Scientific, Inc., Braintree, MA) centered on the midline in the prelimbic cortex. Brains were cut coronally in a cryostat to AP 4.7 and cannula placements were verified visually.
2.7. Immunoblotting
Tissue punches from Experiments 2–4 were processed and immunoblotting was performed as described (Sun et al., 2013). Separate PVDF membranes were incubated overnight at 4°C with primary antibodies against pERK (Cell Signaling Technology Cat# 9101S RRID:AB_331646, 1:4000), pGluN2A (Y1325) (Abcam Cat# ab106590 RRID:AB_10861125, 1:500), or pGluN2B (Y1472) (Sigma-Aldrich Cat# M2442 RRID:AB_262150, 1:500), exposed to IgG-HRP, ECL Plus, and developed. Membranes were stripped and re-probed with primary antisera against total ERK (Cell Signaling Technology Cat# 9102S RRID:AB_10695746 1:6000), total GluN2A (Millipore Cat# 07-632 RRID:AB_310837, 1:2000), or total GluN2B (LifeSpan Cat# LS-C7350-200 RRID:AB_610416, 1:500). The integrated density of each protein band was measured using Image J software (NIH). To confirm whether equal amounts of protein were loaded in each lane, the total protein membranes were stripped again and incubated with anti-calnexin (CNXN) antibody (Enzo Life Sciences Cat# ADI-SPA-860-F RRID:AB_11178981, 1:6000) and the ratio of total protein/CNX in each group was calculated and compared among groups. As there was no difference in total protein/CNX among groups, the ratio of each phosphoprotein/total protein was used for statistical analysis.
2.8. Statistical analysis
One- or two-way ANOVAs followed by Student–Newman–Keuls (SNK) pairwise comparison tests were used for the analysis of the behavioral data and immunoblotting data for four or more groups using GraphPad Prism Software (La Jolla, CA). Unpaired t-tests were used to analyze data from two groups. Comparisons were considered statistically significant at p<0.05.
3. Results
3.2. Histology
The cannula placements for rats in Experiment 1 and the AP extent of the 2 mm-thick punches in prelimbic cortex in Experiments 2–4 are shown in Figure 1D. Rats in which cannula tips were located outside the ventral anterior cingulate or prelimbic cortex (n=2) were excluded from the schematic mapping and data analysis.
3.3. Activity of GluN2A- and GluN2B-containing receptors is necessary for the effect of intra- prelimbic BDNF on cocaine-seeking
Figure 2 illustrates the data from Experiment 1. A one-way ANOVA showed that the average number of inactive and active lever presses, respectively, was not significantly different between the two vehicle groups (PBS or 2% DMSO) infused 20 min before PBS or BDNF during the last 3 days of cocaine SA, the post-abstinence relapse test, the last 2 days of extinction training, or the cue-induced reinstatement test (data not shown). Therefore, the two vehicle groups infused 20 min before PBS were combined into one control group and the two vehicle groups infused 20 min before BDNF were combined into another group.
Figure 2.

Infusion of 0.05 μg/0.5 μl/side TCN-201 (TCN), or 2 μg/0.5 μl/side Ro-25-6981 (Ro) into the prelimbic cortex blocked BDNF’s suppressive effect on cocaine-seeking during the post-abstinence (PA) relapse test and cue-induced reinstatement test. (A) Left: The average number of inactive lever presses during the last 3d of cocaine SA was similar among groups. Right: During the last 3d of cocaine SA, the average number of active lever presses was similar among all rats subsequently assigned to one of six infusion groups (n=17–19 in Veh-P or Veh-B; n=6–10 in other groups). (B) Left: The average number of inactive lever presses during the PA test was similar among groups. Right: TCN or Ro effects on PA test responding after one week of abstinence. Veh-BDNF rats showed significantly less pressing on the previously active levers than Veh-PBS controls (***p < 0.001), but rats infused with TCN or Ro before BDNF pressed significantly more on the active levers than rats infused with vehicle-BDNF (##p < 0.01; #p < 0.05) (n=17–19 in vehicle-infused groups; n=6–10 in other groups). (C) Left: The average number of inactive lever presses during the last 2d of extinction training was similar among groups. Right: During the last 2d of extinction training, the average number of active lever presses was similar among groups (n=17–18 in vehicle-infused groups; n=6–9 in other groups). (D) Left: The average number of inactive lever presses during the cue-induced reinstatement was similar among groups. Right: TCN or Ro effects on cue-induced reinstatement after extinction training. Vehicle-BDNF- treated rats exhibited significantly less active lever pressing than Vehicle-PBS controls (***p<0.001). Infusion of TCN or Ro before BDNF significantly inhibited the ability of BDNF to suppress cocaine-seeking compared to the Vehicle-BDNF group (##p< 0.01) (n=17–18 in vehicle-infused groups; n=6–9 in other groups). The bar graphs indicate the mean ± SEM; Veh= vehicle, TCN=TCN-201, Ro=Ro-25-6981, P=PBS, B= BDNF.
A one-way ANOVA showed that the average number of inactive (F(5,60)= 0.43, p= 0.83; Fig. 2A) and active lever presses (F(5,60)=0.90, p= 0.49; Fig. 2A), respectively, did not differ among groups during the last 3 days of cocaine SA before intracranial infusions. Whereas a one-way ANOVA showed that the average number of inactive lever presses was similar among all rats during the post-abstinence relapse test (F(5,60)= 0.36, p= 0.88; Fig. 2B), a two-way ANOVA showed that during the post-abstinence relapse test (Fig. 2B), there was a significant interaction between the number of active lever presses for treatment #1 (antagonist vs. vehicle infusion) and treatment #2 (BDNF vs. PBS infusion) (F(2,60)=6.16, p=0.004). SNK multiple comparison tests revealed that rats infused with vehicle/BDNF pressed the active levers significantly less than vehicle/PBS-infused rats (p<0.001), TCN-201/BDNF-infused rats (p<0.01), or Ro-25-6981/BDNF-infused rats (p<0.05). Further, there was no significant difference in active lever pressing between vehicle/PBS-infused rats, TCN-201/PBS-infused rats, or Ro-25-6981/PBS-infused rats (p>0.05), confirming that the doses of these drugs blocked BDNF-mediated behavioral changes only, leaving cocaine-induced responding unaffected.
A one-way ANOVA showed that during the last two days of extinction after the post-abstinence relapse test (Fig. 2C), the average number of inactive (F(5,56)= 0.42, p= 0.83), and active lever presses (F(5,56)= 0.63, p= 0.68), respectively, was not different among the groups. Whereas a one-way ANOVA showed that the average number of inactive lever presses was similar among all rats during the cue-induced reinstatement test (F(5,56)= 0.30, p= 0.91; Fig. 2D), a two-way ANOVA showed that there was a significant interaction between the number of active lever presses for treatment #1 (antagonist vs. vehicle) and treatment #2 (BDNF vs. PBS)(F(2,56)= 7.83, p=0.001) during the cue-induced reinstatement test (Fig. 2D). SNK tests revealed that vehicle/BDNF-infused rats reinstated significantly less than vehicle/PBS-infused rats (p<0.001) and that rats infused with TCN-201/BDNF or Ro-25-6981/BDNF reinstated significantly more than those infused with vehicle/BDNF (p<0.01). Further, there was no significant difference in active lever pressing between rats infused with vehicle/PBS, TCN-201/PBS, or Ro-25-6981/PBS (p>0.05), further confirming that the doses of these drugs blocked BDNF-mediated suppression only, leaving cue-induced reinstatement in PBS-infused rats unaffected.
3.4. Intra-prelimbic BDNF prevented cocaine SA-induced reduction of pERK, pGluN2A and pGluN2B levels in the prelimbic cortex during early withdrawal
Figure 3 illustrates the data from Experiment 2 in which the prelimbic cortex of rats was infused with BDNF (0.75 μg/0.5 μl/side) or 10 mM (PBS 0.5 μl/side) and the rats were decapitated 2 hr later as described previously (Berglind et al. 2007; Whitfield et al. 2011) to confirm whether BDNF prevented a cocaine SA-induced decrease in phospho (p)-ERK and to determine whether BDNF exerts a similar effect on pGluN2A and pGluN2B. A two-way ANOVA found that the i.v. drug (cocaine vs saline; F(1,25)=4.61, p<0.05) and the intracranial infusion (BDNF vs PBS; F(1,25)=14.61, p<0.001) had significant main effects on pERK (Fig. 3A) but there was no interaction between drug and infusion (F(1,25)=1.01, p=0.32) because BDNF increased pERK in the presence or absence of cocaine. However, as previously reported (Whitfield et al., 2011), SNK multiple comparison tests revealed a significant reduction in the pERK level in the prelimbic cortex of cocaine SA rats compared to yoked-saline rats infused with PBS (C-P vs S-P, p<0.05) and BDNF infusion not only prevented the pERK decrease in cocaine SA rats (C-P vs C-B, p<0.01) but augmented the level of pERK in yoked-saline rats (S-P vs S-B, p<0.05). Similarly (Fig. 3B), a two-way ANOVA found that the i.v. drug (cocaine vs saline; F(1,26)=4.73, p<0.05) and intracranial infusion (BDNF vs PBS; F(1,26)=20.33, p < 0.001) had significant main effects on pGluN2A but there was no interaction between drug and infusion (F(1,26)=0.12, p=0.74). SNK multiple comparison tests revealed that there was a profound reduction in pGluN2A expression in the prelimbic cortex of cocaine SA rats compared to yoked-saline rats infused with PBS (C-P vs S-P; p<0.05) and BDNF infusion not only prevented this decrease in cocaine SA rats (C-P vs C-B, p<0.05) but augmented the level of pGluN2A in yoked-saline rats (S-P vs S-B, p<0.01). Further (Fig. 3C), a two-way ANOVA found that the i.v. drug (cocaine vs saline; F(1,26) =7.75, p<0.05) and intracranial infusion (BDNF vs PBS; F(1,26)=21.45, p<0.001) had a significant main effect on the level of pGluN2B but there was no interaction between drug and infusion (F(1,26)=0.60, p=0.44). SNK multiple comparison tests revealed that there was a significant reduction in pGluN2B expression in the prelimbic cortex of cocaine SA rats compared to yoked-saline treated rats infused with PBS (C-P vs S-P, p<0.05) and that BDNF infusion not only prevented this decrease in cocaine SA rats (C-P vs C-B, p<0.01) but augmented the level of pGluN2B in yoked-saline rats (S-P vs S-B, p<0.05). A one way ANOVA found that the levels of total ERK (F(3,25)=1.85, p=0.16), total GluN2A (F(3,26)=2.44, p=0.09) and total GluN2B (F(3,26)=0.79, p=0.51) were not significantly different among the four experimental groups when normalized to the calnexin loading control (supplemental Table 1).
Figure 3.
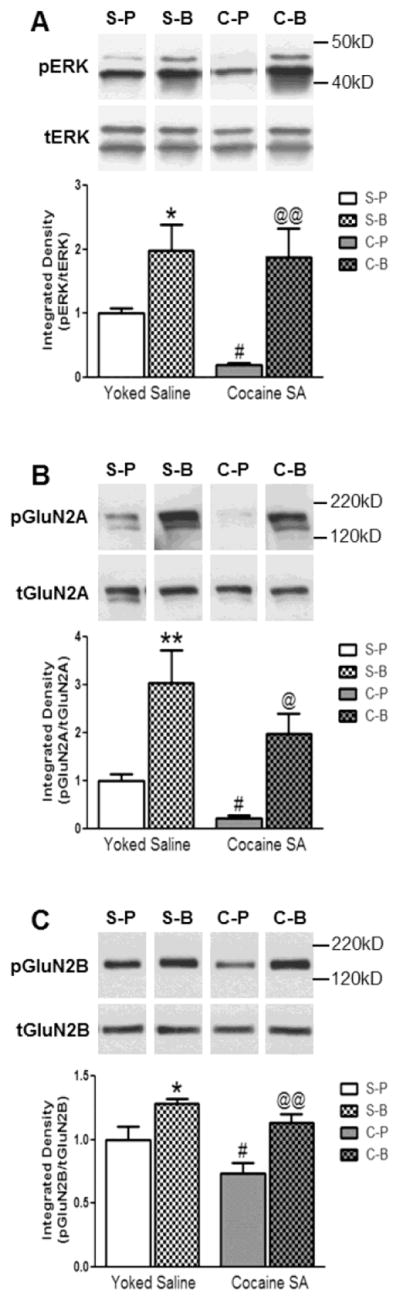
The effect of intra-prelimbic BDNF or PBS on pERK, pGluN2A and pGluN2B levels in rats with a cocaine SA or yoked-saline history 2 hr after infusion. (A) Cocaine SA caused a significant reduction in the pERK level in the prelimbic cortex of PBS-infused rats compared to yoked-saline rats infused with PBS (S-P vs C-P, #p<0.05) and BDNF infusion not only prevented the pERK decrease in cocaine SA rats (C-P vs C-B, @@p<0.01) but augmented the level of pERK in yoked-saline rats (S-P vs S-B, *p<0.05). (B) Cocaine SA reduced pGluN2A expression in the prelimbic cortex of PBS-infused rats compared to yoked-saline rats infused with PBS (S-P vs C-P; #p<0.05) and BDNF infusion not only prevented this decrease in cocaine SA rats (C-P vs C-B, @p<0.05) but augmented the level of pGluN2A in yoked-saline rats (S-P vs S-B, **p<0.01). (C) Cocaine SA caused a significant reduction in pGluN2B expression in the prelimbic cortex of PBS-infused rats compared to yoked-saline treated rats infused with PBS (S-P vs C-P, #p<0.05) and that BDNF infusion not only prevented this decrease in cocaine SA rats (C-P vs C-B, @@p<0.01) but augmented the level of pGluN2B in yoked-saline rats (S-P vs S-B, *p<0.05). Representative immunoblot images above bar graphs were prepared using images taken from different parts of the same gel. The positions and sizes of the protein molecular weight markers (kDa) are indicated on the right. The bar graphs indicate the mean ± SEM. S=Yoked-Saline, C=Cocaine SA, P=PBS, B=BDNF. N=6–9 per group.
3.5. GluN2A subunit-containing NMDA receptors are required for the effect of intra-prelimbic BDNF on pERK and pGluN2A but not pGluN2B levels in the prelimbic cortex during early withdrawal
Because TCN-201 had no effect on relapse to cocaine-seeking in PBS-infused rats in Experiment 1 and no effect on pERK in PBS-infused rats in the pilot experiments (Fig S1), all rats were treated with TCN-201 in this experiment. Thus, in yoked saline rats, the yoked-saline group infused with TCN-201 followed by PBS served as the baseline control. TCN-201 had no effect on the ability of cocaine to decrease pERK (Fig. 4A), pGluN2A (Fig. 4B), or pGluN2B (Fig. 4C) levels in the prelimbic cortex of PBS-infused rats; there was a significant main effect of cocaine vs. yoked saline in each case (pERK: F(1,25)=100.90, p<0.001; pGluN2A: F(1,25)=322.52, p<0.001; pGluN2B: F(1,25)=17.49, p<0.001). However, TCN-201 prevented the ability of BDNF to increase pERK and pGluN2A levels; i.e., there was no significant main effect of intracranial infusion (BDNF vs PBS) (pERK: F(1,25)=0.17, p=0.68; pGluN2A: F(1,25)=0.20, p=0.66). In the presence of TCN-201, SNK multiple comparison tests confirmed that there was a significant reduction in pERK and pGluN2A expression in the prelimbic cortex of cocaine SA rats compared to yoked-saline rats in the presence or absence of BDNF (p<0.001).
Figure 4.

The effect of intra-prelimbic TCN-201 or Ro-25-6981 infusion before PBS or BDNF on (A, D) pERK, (B, F) pGluN2A, and (C, E) pGluN2B levels in the prelimbic cortex of rats with a cocaine SA or yoked-saline history 2 hr after infusion. (A–C) TCN-201 had no effect on the ability of cocaine to decrease levels of (A) pERK or (B) pGluN2A in the prelimbic cortex; (S-P vs. C-P; ###p<0.001). However, TCN-201 prevented the ability of BDNF to reverse the cocaine-induced decrease in pERK and pGluN2A levels (S-B vs. C-B, +++p<0.001). (C) TCN-201 did not prevent the ability of cocaine to decrease pGluN2B levels (S-P vs. C-P, ###p<0.001). However, in the presence of TCN-201, BDNF was able to increase pGluN2B levels in the prelimbic cortex of rats with a cocaine SA or yoked-saline history (C-P vs. C-B, @@@p<0.001 and S-P vs. S-B, **p<0.01, respectively). (D–F) Ro-25-6981 had no effect on the ability of cocaine to decrease (D) pERK and (E) pGluN2B levels in the prelimbic cortex (S-P vs. C-P; ##p<0.01). However, Ro-25-6981 prevented the ability of BDNF to reverse the cocaine-induced decrease in pERK and pGluN2B levels (S-B vs. C-B, ++p<0.01 and +++p<0.001, respectively). (F) Ro-25-6981 did not prevent the ability of cocaine to decrease pGluN2A (Y1375) levels (S-P vs. C-P, #p<0.05). However, in the presence of Ro-25-6981, BDNF was able to increase pGluN2A levels in the prelimbic cortex of rats with a cocaine SA or yoked-saline history (C-P vs. C-B, @@@p<0.001 and S-P vs. S-B, *p<0.05, respectively). Representative immunoblot images above bar graphs were prepared using images taken from different parts of the same gel. The positions and sizes of the protein molecular weight markers (kDa) are indicated on the right. The bar graphs indicate the mean ± SEM. S=Yoked-Sal, C=Cocaine SA, P=PBS, B=BDNF, T=TCN-201, Ro=Ro-25-6981. N=6–8 per group.
In contrast, a two-way ANOVA found that when TCN-201 was infused before BDNF or PBS, the drug (cocaine vs saline; F(1,25)=17.49, p<0.001) and the intracranial infusion (BDNF vs PBS; F(1,25)=54.24, p<0.001) had a significant main effect on pGluN2B but there was no interaction between the drug and the intracranial infusion (F(1,25)=3.59, p=0.07). In the presence of TCN-201, SNK multiple comparison tests found that there was a significant reduction in pGluN2B level in the prelimbic cortex of cocaine SA rats compared to yoked-saline rats infused with PBS (C-P vs S-P, p<0.001) and that a BDNF infusion not only prevented this decrease in cocaine SA rats (C-P vs C-B, p<0.001) but augmented the level of pGluN2B in yoked-saline-treated rats (S-P vs S-B, p<0.01). These data indicate that BDNF was still effective in blocking cocaine SA effects on GluN2B in the presence of TCN-201 and that the dose of TCN-201 used in the current study selectively blocked GluN2A, but not GluN2B, subunit-containing NMDARs.
A one-way ANOVA found that TCN-201 did not alter the expression of total ERK (F(3,25)=2.70, p=0.07), total GluN2A (F(3,25)=2.45, p=0.09) or total GluN2B (F(3,25)=2.12, p=0.12) among the four experimental groups. To further confirm whether an intra-prelimbic TCN-201 infusion affected the basal level of pERK, pGluN2A, or pGluN2B, prelimbic tissue from yoked-saline rats infused with PBS from experiment 2 was run with prelimbic tissue from saline-treated rats infused with TCN-201 before PBS from experiment 3. An unpaired t-test found that there was no significant difference in pERK (t10=0.27, p=0.79), pGluN2A (t10= 1.42, p=0.19), or pGluN2B (t10=1.26, p=0.24) levels between these two groups.
3.6. GluN2B subunit-containing NMDA receptors are required for the effect of intra-prelimbic BDNF on pERK and pGluN2B but not pGluN2A levels in the prelimbic cortex during early withdrawal
Because Ro-25-6981 had no effect on relapse to cocaine-seeking in PBS-infused rats in Experiment 1 and no effect on pERK in PBS-infused rats in the pilot experiments (Fig S2), all rats were treated with Ro-25-6981 in this experiment. Thus, the yoked-saline group infused with Ro-25-6981 followed by PBS served as the baseline control. Ro-25-6981 had no effect on the ability of cocaine to decrease pERK (Fig. 4D) or pGluN2B (Fig. 4E) levels in the prelimbic cortex of PBS-infused rats; i.e., there was a significant main effect of cocaine vs. saline in each case (pERK: F(1,22)=11.12, p<0.01; pGluN2B: F(1,22)=15.31, p<0.01). However, Ro-25-6981 prevented the ability of BDNF to reverse the cocaine-induced decrease in pERK and pGluN2B levels; i.e., there was no significant main effect of intracranial infusion (BDNF vs PBS) on pERK (F(1,22)=1.05, p=0.32) or pGluN2B (F(1,22) =0.76, p=0.39). In the presence of Ro-25-6981, SNK multiple comparison tests confirmed that there was a significant reduction in pERK and pGluN2B in the prelimbic cortex of cocaine SA rats compared to yoked-saline rats in the presence or absence of BDNF (pERK: p<0.01; pGluN2B: S-P vs C-P, p<0.01; S-B vs C-B, p<0.001).
To confirm the specificity of Ro-25-6981 on GluN2B subunit-containing NMDARs, the effect of Ro-25-6981 on pGluN2A levels was investigated. A two-way ANOVA found that when Ro-25-6981 was infused before BDNF or PBS (Fig. 4F), the intracranial infusion (BDNF vs PBS; F(1,22)=26.43, p<0.001), but not the drug (cocaine vs saline; F(1,22)=0.10, p=0.75) had a significant main effect on pGluN2A. In the presence of Ro-25-6981, SNK multiple comparison tests found that cocaine SA caused a significant reduction in pGluN2A level in the prelimbic of PBS-infused rats compared to yoked-saline rats infused with PBS (S-P vs C-P, p<0.05) and that a BDNF infusion significantly augmented the level of pGluN2A in yoked-saline-treated (p<0.05) and cocaine SA rats (p<0.001). These data indicate that the dose of Ro-25-6981 used in the current study selectively blocked GluN2B, but not GluN2A, subunit-containing NMDARs.
A one way ANOVA found that, in the presence of Ro-25-6981, total ERK (F(3,22)=2.05, p=0.14), total GluN2A (F(3,22)=2.21, p=0.12) and total GluN2B (F(3,22)=2.35, p=0.10) levels were similar among all four experimental groups. To further confirm whether an intra-prelimbic Ro-25-6981 infusion affected the basal level of pERK, pGluN2A, or pGluN2B, prelimbic tissue from yoked-saline rats infused with PBS from experiment 2 was run with prelimbic tissue from yoked-saline rats infused with Ro-25-6981 before PBS from experiment 4. An unpaired t-test found that there was no significant difference in pERK (t8=0.11, p=0.91), pGluN2A (t8=0.69, p=0.51), or pGluN2B (t8=1.54, p=0.16) levels between these two groups.
4. Discussion
4.1. Summary of findings
This study shows that intra-prelimbic infusion of TCN-201 or Ro-25-6981 immediately after the last cocaine SA session blocked the suppressive effect of BDNF on subsequent cocaine-seeking. Cocaine reduced pERK, pGluN2A, and pGluN2B levels in the prelimbic cortex 2 hr after cocaine SA, all of which were elevated by intra-prelimbic BDNF infusion. Furthermore, intra-prelimbic TCN-201 or Ro-25-6981 infused before BDNF blocked the ability of BDNF to rescue cocaine-mediated dephosphorylation of ERK (both GluN2 antagonists), pGluN2A (TCN-201), or GluN2B (Ro-25-6981) in the prelimbic cortex, which parallels the ability of TCN-201 or Ro-25-6981 to block BDNF-induced suppression of cocaine-seeking. Together, these data indicate that glutamatergic neuronal activity is essential for the ability of an intra-prelimbic infusion of BDNF to normalize cocaine-mediated dysfunctional neuroplasticity during early withdrawal that is associated with subsequent cocaine-seeking.
4.2. TCN-201 or Ro-25-6981 inhibits BDNF-mediated suppression of cocaine-seeking
This study demonstrated that the ability of an intra- prelimbic infusion of BDNF to suppress subsequent cocaine-seeking depends on preventing the hypo-phosphorylation of GluN2 subunit-containing NMDA receptors in the prelimbic cortex during early withdrawal. The importance of the TrkB-NMDAR interaction to cocaine-seeking has been reported in a different paradigm in which BDNF/TrkB enhanced the amplitude and prolonged the activation of NMDARs in the infralimbic cortex, facilitating extinction of a cocaine-induced conditioned place preference (CPP) that was blocked by the selective GluN2B antagonist, ifenprodil (Otis et al. 2014). Thus, BDNF-TrkB enhancement of NMDAR activity in heterogeneous regions of the prefrontal cortex, i.e., prelimbic and infralimbic, regulates different aspects of cocaine-seeking in the SA and CPP models. Importantly, an intra-prelimbic BDNF infusion at the end of cocaine SA does not merely enhance normal NMDAR phosphorylation that facilitates glutamate transmission but prevents a critical cocaine-induced deficit in NMDAR signaling that underlies subsequent relapse to cocaine-seeking. Further, such a BDNF infusion during early withdrawal normalizes extracellular glutamate levels in the nucleus accumbens core and prevents a cocaine prime-induced increase in glutamate levels in accumbens that is associated with suppression of cocaine-seeking three weeks after infusion (Berglind et al. 2009).
4.3. BDNF prevents a cocaine SA-induced suppression of pGluN2A and pGluN2B signaling in the prelimbic cortex
We confirmed that cocaine SA caused dephosphorylation of ERK and GluN2B in the prelimbic cortex (Whitfield et al. 2011; Sun et al. 2013) and showed concurrent suppression of pGluN2A levels 2 hr after the last cocaine SA session. Although the mechanisms underlying the cocaine SA-induced dephosphorylation of these phospho-proteins are not yet defined experimentally, our preliminary evidence points to the activation of striatal-enriched tyrosine phosphatase (STEP) in the prelimbic cortex 2 hr after the end of cocaine SA (Sun et al. 2013). STEP dephosphorylates and inactivates ERK and GluN2A/B directly as well as indirectly through dephosphorylation and inactivation of SFKs (Paul et al. 2003; Pulido et al. 1998; Nguyen et al. 2002; Huang and McNamara, 2010). STEP-mediated dephosphorylation of GluN2B at Y1472 induces endocytosis (Snyder et al., 2005), leading to the inhibition of NMDAR-mediated ERK phosphorylation in cortical neuronal cultures (Braithwaite et al. 2006). In contrast, SFK-mediated phosphorylation of GluN2A or GluN2B positively modulates NMDAR activity (see Introduction), leading to ERK activation (Ivanov et al. 2006). Therefore, suppression of pGluN2A, pGluN2B, and pERK levels within 2 hr of the end of cocaine SA suggests a profound, yet transient (Whitfield et al. 2011), cocaine-mediated hypofunction of synaptic activity in the prelimbic cortex. This discovery is consistent with reduced basal activity of the prelimbic cortex and reduced BDNF mRNA levels within 24 hr after cocaine SA (Sun and Rebec, 2006; McGinty et al. 2010).
The intra-prelimbic BDNF infusion immediately after the last cocaine SA session not only normalized the cocaine-mediated reduction in pERK as described (Whitfield et al. 2011), but also pGluN2A and pGluN2B, indicating that prevention of cocaine-induced hypo-phosphorylation of NMDA-ERK signaling in prelimbic cortex during early withdrawal has prolonged effects on subsequent cocaine-seeking. These data underline the fundamental importance of previous findings that activation of TrkB receptors facilitates NMDA receptor signaling in several different experimental paradigms (Huang and McNamara, 2010; Iwasaki et al. 1998; Lin et al. 1998; Narisawa-Saito et al. 1999; Tezuka et al. 1999; Xu et al. 2006).
4.4. TCN-201 and Ro-25-6981 block BDNF’s ability to reverse the cocaine SA-induced suppression of pGluN2A and pGluN2B signaling in the prelimbic cortex
TCN-201 infusion into the prelimbic cortex blocked the BDNF-mediated increase in pERK and pGluN2A, but not pGluN2B, levels whereas Ro-25-6981 blocked the BDNF-mediated increase in pERK and pGluN2B, but not pGluN2A levels in the prelimbic cortex in both yoked-saline and cocaine SA rats. These data indicate that the doses of TCN-201 and Ro-25-6981 used in the current study were selective for GluN2A and Glu2B, respectively, and that phosphorylation of both GluN2A- and GluN2B-containing NMDARs is required for the BDNF-induced enhancement of GluN2A and GluN2B signaling in the presence or absence of cocaine. BDNF-mediated upregulation of these phospho-proteins is likely to occur by interactions with GluN1/GluN2A and GluN1/GluN2B diheteromers not triheteromers because the inhibitory effect of TCN-201 and ifenprodil (Ro-25-6981 is a derivative of ifenprodil) on GluN1/GluN2A and GluN1/GluN2B diheteromers of NMDARs is much higher than their inhibitory effect on GluN1/GluN2A/GluN2B triheteromers of NMDARs (Hansen et al. 2014). TCN-201 binds to an allosteric site located at the interface of the GluN1/GluN2A agonist binding domains where it serves as a negative allosteric modulator of glycine binding (Hansen et al. 2012). In contrast, phenethanolamines, like Ro-25-6981, bind to a site at the interface between GluN1/GluN2B amino-terminal domains that regulates receptor assembly (Karakas et al. 2011). It is possible that binding of each antagonist to the extracellular domains of GluN2A or GluN2B receptors changes the receptor conformation so that the C-terminal tyrosines of each subunit are not accessible to SFK but this supposition awaits investigation. In support of this idea, the allosteric NMDA antagonist, ketamine, decreases the association between GluN2A and the SFK, Fyn, that is triggered by ischemia and reperfusion (Huo et al. 2002).
Together, our novel findings that GluN2A- and GluN2B-selective antagonists block BDNF-induced behavioral and neurochemical effects expand our knowledge of the molecular mechanisms by which BDNF induces prelimbic ERK activation. We postulate that BDNF-induced phosphorylation of GluN2A at Y1325 and GluN2B at Y1472 likely occurs via activation of the TrkB-SFK signaling pathway to oppose cocaine-induced dephosphorylation events. BDNF-induced pERK is a downstream signaling component directly associated with TrkB activation that is enhanced through TrkB-SKF mediated co-activation of synaptic NMDA receptors. Moreover, TrkB stimulation triggers degradation of STEP61 and phosphorylation of STEP substrates whereas decreased BDNF expression upregulates STEP (Xu et al. 2015). In the presence of GluN2 antagonists, BDNF would be unable to overcome a blockade of NMDARs and the dephosphorylating effects of cocaine would be unopposed. Future studies will examine the functional roles of STEP, mGluR5 and SFKs on the ability of intra-prelimbic infusion of BDNF during early withdrawal to suppress subsequent cocaine-seeking.
Supplementary Material
Acknowledgments
Role of Funding Sources
This work was supported by P50 DA15369, RO1 DA033479 (JFM), T32 DA007288, and F31 DA DA039709 (SMB). The study was conducted in a facility constructed with support from NIH C06 RR015455. The authors declare no competing financial interests. The funding sources had no role in study design, collection, analysis, or interpretation of data, writing the report, or decisions to submit this paper for publication.
We thank Wei-Lun Sun, Ph.D. and Stephen Saunier for technical assistance and Jeffrey Korte, Ph.D. for statistical consultation.
Footnotes
The authors report no conflicts of interest.
Contributors
BSG and JFM designed research; BSG and SMB performed research and analyzed data; BSG and JFM wrote the paper; JFM provided funding for the research. All authors contributed to and approved the final manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Berglind WJ, See RE, Fuchs RA, Ghee SM, Whitfield TW, Miller SW, McGinty JF. A BDNF infusion into the medial prefrontal cortex suppresses cocaine-seeking in rats. Eur J Neurosci. 2007;26:757–766. doi: 10.1111/j.1460-9568.2007.05692.x. [DOI] [PubMed] [Google Scholar]
- Berglind WJ, Whitfield TW, LaLumiere RJ, Kalivas PW, McGinty JF. A single intra-PFC infusion of BDNF prevents cocaine-induced alterations in extracellular glutamate within the nucleus accumbens. J Neurosci. 2009;29:3715–3719. doi: 10.1523/JNEUROSCI.5457-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braithwaite SP, Adkisson M, Leung J, Nava A, Masterson B, Urfer R, et al. Regulation of NMDA receptor trafficking and function by striatal-enriched tyrosine phosphatase (STEP) Eur J Neurosci. 2006;23:2847–2856. doi: 10.1111/j.1460-9568.2006.04837.x. [DOI] [PubMed] [Google Scholar]
- Bramham CR, Messaoudi E. BDNF function in adult synaptic plasticity: the synaptic consolidation hypothesis. Prog Neurobiol. 2005;76:99–125. doi: 10.1016/j.pneurobio.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Cheung HH, Gurd JW. Tyrosine phosphorylation of the N-methyl-d-aspartate receptor by exogenous and postsynaptic density-associated Src-family kinases. J Neurochem. 2001;78:524–534. doi: 10.1046/j.1471-4159.2001.00433.x. [DOI] [PubMed] [Google Scholar]
- Gipson CD, Reissner KJ, Kupchik YM, Smith AC, Stankeviciute N, Hensley-Simon ME, Kalivas PW. Reinstatement of nicotine seeking is mediated by glutamatergic plasticity. Proc Natl Acad Sci USA. 2013;110:9124–9129. doi: 10.1073/pnas.1220591110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebel-Goody SM, Davies KD, Alvestad Linger RM, Freund RK, Browning MD. Phospho-regulation of synaptic and extrasynaptic N-methyl-d-aspartate receptors in adult hippocampal slices. Neuroscience. 2009;158:1446–1459. doi: 10.1016/j.neuroscience.2008.11.006. [DOI] [PubMed] [Google Scholar]
- Gomes RA, Hampton C, El-Sabeawy F, Sabo SL, McAllister AK. The dynamic distribution of TrkB receptors before, during, and after synapse formation between cortical neurons. J Neurosci. 2006;26:11487–11500. doi: 10.1523/JNEUROSCI.2364-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KB, Ogden KK, Traynelis SF. Subunit-selective allosteric inhibition of glycine binding to NMDA receptors. J Neurosci. 2011;32:6197–6208. doi: 10.1523/JNEUROSCI.5757-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KB, Ogden KK, Yuan H, Traynelis SF. Distinct functional and pharmacological properties of triheteromeric GluN1/GluN2A/GluN2B NMDA receptors. Neuron. 2014;81:1084–1096. doi: 10.1016/j.neuron.2014.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YZ, McNamara JO. Mutual regulation of Src family kinases and the neurotrophin receptor TrkB. J Biol Chem. 2010;285:8207–8217. doi: 10.1074/jbc.M109.091041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov A, Pellegrino C, Rama S, Dumalska I, Salyha Y, Ben-Ari Y, Medina I. Opposing role of synaptic and extrasynaptic NMDA receptors in regulation of the extracellular signal-regulated kinases (ERK) activity in cultured rat hippocampal neurons. J Physiol. 2006;572:789–798. doi: 10.1113/jphysiol.2006.105510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki Y, Gay B, Wada K, Koizumi S. Association of the Src family tyrosine kinase Fyn with TrkB. J Neurochem. 1998;71:106–111. doi: 10.1046/j.1471-4159.1998.71010106.x. [DOI] [PubMed] [Google Scholar]
- Karakas E, Simorowski N, Furukama H. Subunit arrangement and phenylethanolamine binding in GluN1/GluN2B NMDA receptors. Nature. 2011;475:249–254. doi: 10.1038/nature10180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny PJ, Chartoff E, Roberto M, Carlezon WA, Jr, Markou A. NMDA receptors regulate nicotine-enhanced brain reward function and intravenous nicotine self-adminstration: role of the ventral tegmental area and central nucleus of the amygdala. Neuropsychopharmacol. 2009;34:266–281. doi: 10.1038/npp.2008.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine ES, Crozier RA, Black IB, Plummer MR. Brain-derived neurotrophic factor modulates hippocampal synaptic transmission by increasing N-methyl-D-aspartic acid receptor activity. Proc Natl Acad Sci USA. 1998;95:10235–10239. doi: 10.1073/pnas.95.17.10235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Wolf ME. Multiple faces of BDNF in cocaine addiction. Beh Brain Res. 2015;279:240–254. doi: 10.1016/j.bbr.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SY, Wu K, Levine ES, Mount HT, Suen PC, Black IB. BDNF acutely increases tyrosine phosphorylation of the NMDA receptor subunit 2B in cortical and hippocampal postsynaptic densities. Mol Brain Res. 1998;55:20–27. doi: 10.1016/s0169-328x(97)00349-5. [DOI] [PubMed] [Google Scholar]
- Matsumoto T, Numakawa T, Yokomaku D, Adachi N, Yamaguchi S, Numakawa Y, Kunugi H, Taguchi T. Brain-derived neurotrophic factor-induced potentiation of glutamate and GABA release: Different dependency on signaling pathways and neuronal activity. Mol Cell Neurosci. 2006;31:70–84. doi: 10.1016/j.mcn.2005.09.002. [DOI] [PubMed] [Google Scholar]
- McFarland K, Lapish CC, Kalivas PW. Prefrontal glutamate release into the core of the nucleus accumbens mediates cocaine-induced reinstatement of drug-seeking behavior. J Neurosci. 2003;23:3531–3537. doi: 10.1523/JNEUROSCI.23-08-03531.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinty JF, Whitfield TW, Jr, Berglind WJ. Brain-derived neurotrophic factor and cocaine addiction. Brain Res. 2010;1314:183–193. doi: 10.1016/j.brainres.2009.08.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinty JF, Zelek-Molik A, Sun W-L. Cocaine self-administration causes signaling deficits in corticostriatal circuitry that are reversed by BDNF in early withdrawal. Brain Res. 2015;1628:82–87. doi: 10.1016/j.brainres.2014.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin J, See RE. Selective inactivation of the dorsomedial prefrontal cortex and the basolateral amygdala attenuates conditioned-cued reinstatement of extinguished cocaine-seeking behavior in rats. Psychopharmacology (Berl) 2003;168:57–65. doi: 10.1007/s00213-002-1196-x. [DOI] [PubMed] [Google Scholar]
- Nakazawa T, Komai S, Tezukama T, Hisatsune C, Umemori H, Semba K, Mishina M, Manabe T, Yamamoto T. Characterization of Fyn-mediated tyrosine phosphorylation sites on GluR epsilon 2 (NR2B) subunit of the N-methyl-D-aspartate receptor. J Biol Chem. 2001;276:693–699. doi: 10.1074/jbc.M008085200. [DOI] [PubMed] [Google Scholar]
- Narisawa-Saito M, Silva AJ, Yamaguchi T, Hayashi T, Yamamoto T, Nawa H. Growth factor-mediated Fyn signaling regulates alpha-amino-3- hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor expression in rodent neocortical neurons. Proc Natl Acad Sci USA. 1999;96:2461–2466. doi: 10.1073/pnas.96.5.2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TH, Liu J, Lombroso PJ. Striatal enriched phosphatase 61 dephosphorylates Fyn at phosphotyrosine 420. J Biol Chem. 2002;277:24274–24279. doi: 10.1074/jbc.M111683200. [DOI] [PubMed] [Google Scholar]
- Otis JM, Fitzgerald MK, Mueller D. Infralimbic BDNF/TrkB enhancement of GluN2B currents facilitates extinction of a cocaine-conditioned place preference. J Neurosci. 2014;34:6057–6064. doi: 10.1523/JNEUROSCI.4980-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul S, Nairn AC, Wang P, Lombroso PJ. NMDA-mediated activation of the tyrosine phosphatase STEP regulates the duration of ERK signaling. Nat Neurosci. 2003;6:34–42. doi: 10.1038/nn989. [DOI] [PubMed] [Google Scholar]
- Pierce RC, Bell K, Duffy P, Kalivas PW. Repeated cocaine augments excitatory amino acid transmission in the nucleus accumbens only in rats having developed behavioral sensitization. J Neurosci. 1996;16:1550–1560. doi: 10.1523/JNEUROSCI.16-04-01550.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prybylowski K, Chang K, Sans N, Kan L, Vicini S, Wenthold RJ. The synaptic localization of NR2B-containing NMDA receptors is controlled by interactions with PDZ proteins and AP-2. Neuron. 2005;47:845–857. doi: 10.1016/j.neuron.2005.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulido R, Zuniga A, Ullrich A. PTP-SL and STEP protein tyrosine phosphatases regulate the activation of the extracellular signal-regulated kinases ERK1 and ERK2 by association through a kinase interaction motif. EMBO J. 1998;17:7337–7350. doi: 10.1093/emboj/17.24.7337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche KW, Standley S, McCallum J, Dune Ly C, Ehlers MD, Wenthold RJ. Molecular determinants of NMDA receptor internalization. Nat Neurosci. 2001;4:794–802. doi: 10.1038/90498. [DOI] [PubMed] [Google Scholar]
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci. 2005;8:1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- Sun W, Rebec GV. Repeated cocaine self-administration alters processing of cocaine-related information in rat prefrontal cortex. J Neurosci. 2006;26:8004–8008. doi: 10.1523/JNEUROSCI.1413-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun WL, Coleman NT, Zelek-Molik A, Barry SM, Whitfield TW, Jr, McGinty JF. Relapse to cocaine-seeking after abstinence Is regulated by cAMP-dependent protein kinase A in the prefrontal cortex. Addiction Biology. 2014;19:77–86. doi: 10.1111/adb.12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun WL, Zelek-Molik A, McGinty JF. Short and long access to cocaine self-administration activates tyrosine phosphatase STEP and attenuates GluN expression but differentially regulates GluA expression in the prefrontal cortex. Psychopharmacology. 2013;229:603–163. doi: 10.1007/s00213-013-3118-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi S, Nakazawa T, Tanimura A, Kiyama Y, Tezuka T, Watabe AM, et al. Involvement of NMDAR2A tyrosine phosphorylation in depression-related behaviour. EMBO J. 2009;28:3717–29. doi: 10.1038/emboj.2009.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tezuka T, Umemori H, Akiyama T, Nakanishi S, Yamamoto T. PSD-95 promotes Fyn-mediated tyrosine phosphorylation of the N-methyl-D-aspartate receptor subunit NR2A. Proc Natl Acad Sci USA. 1999;96:435–440. doi: 10.1073/pnas.96.2.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitfield TM, Shi X, Sun WL, McGinty JF. The suppressive effect of an Intra-prefrontal cortical infusion of BDNF on cocaine-seeking is Trk receptor and extracellular signal-regulated protein kinase mitogen-activated protein kinase dependent. J Neurosci. 2011;31:834–842. doi: 10.1523/JNEUROSCI.4986-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu F, Plummer MR, Len GW, Nakazawa T, Yamamoto T, Black IB, Wu K. Brain-derived neurotrophic factor rapidly increases NMDA receptor channel activity through Fyn-mediated phosphorylation. Brain Res. 2006;1121:22–34. doi: 10.1016/j.brainres.2006.08.129. [DOI] [PubMed] [Google Scholar]
- Yashiro K, Philpot BD. Regulation of NMDA receptor subunit expression and its implications for LTD, LTP, and metaplasticity. Neuropharmacology. 2008;55:1081–1094. doi: 10.1016/j.neuropharm.2008.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao MG, Toyoda H, Lee YS, Wu LJ, Ko SW, Zhang XH, et al. Roles of NMDA NR2B subtype receptor in prefrontal long-term potentiation and contextual fear memory. Neuron. 2005;47:859–872. doi: 10.1016/j.neuron.2005.08.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.