

Abstract
Hydrogen peroxide (H2O2) is usually considered to be an important reagent in green chemistry since water is the only by-product in H2O2 involved oxidation reactions. Early studies show that direct synthesis of H2O2 by plasma-water interactions is possible, while the factors affecting the H2O2 production in this method remain unclear. Herein, we present a study on the H2O2 synthesis by atmospheric pressure plasma-water interactions. The results indicate that the most important factors for the H2O2 production are the processes taking place at the plasma-water interface, including sputtering, electric field induced hydrated ion emission, and evaporation. The H2O2 production rate reaches ~1200 μmol/h when the liquid cathode is purified water or an aqueous solution of NaCl with an initial conductivity of 10500 μS cm−1.
Hydrogen peroxide (H2O2) has found many applications in modern industry including acting as a strong oxidizer, a bleaching agent, disinfectant, and the propellant in rocketry1,2 etc. due to the specific property of its oxygen-oxygen single bond. At present, the industrial production of H2O2 is dominated by the anthraquinone process which needs multistep processes and consumes much energy3. Therefore, a direct synthesis of H2O2 is required to avoid the disadvantages of anthraquinone process. Obviously, H2O2 synthesis directly from its constituent elements, i.e., H2 and O2, is a simple idea4,5,6, but the mixture of H2 and O2 is explosive. Some alternative direct methods do not use H2 and O2 which avoid the explosive problem and enable the in-situ continuous synthesis of H2O2, such as electrochemical synthesis from O2 and water7,8,9,10,11, photocatalytic TiO2 in aqueous solutions12,13,14,15, and even sunlight-driven production from water and O2 although the production rate is relatively low (0.625 μmol/h)16.
It has been proven that H2O2 can be formed by the reaction of H2 + O2 → H2O2 in plasma containing H2 and O2 gases17,18,19. When plasma is in contact with water, the plasma-water interactions can entail many direct reactions at the plasma-water interface and indirect cascade reactions in the bulk water20,21,22,23,24,25. One important species at the plasma-water interface is hydroxyl radical (OH) produced by plasma-induced water reactions with electrons and ions. The exact OH formation pathways by plasma-water interactions are very complicated, and one can refer to a review paper26 for details. As the building blocks, the generated OH radicals combination contributes to the main process of H2O2 formation in plasma-water interactions. In addition, other less probable pathways such as OH + H2O* → H2O2 + H27,28 are also possible. In fact, there exist many reports on the H2O2 formation by discharge plasma operated over and inside aqueous solutions27,28,29,30,31,32,33,34,35. However, the determining factors which influence the H2O2 production from the plasma-water interactions remain unclear. To optimize the H2O2 production from plasma-water interactions, a better understanding of the H2O2 production process is desired. Herein, we present an insight into the understanding of H2O2 production by the plasma-water interactions. The results indicate that the sputtering, the electric field induced ion emission, and the evaporation at the water surface strongly influences the H2O2 production.
Results
Experimental approach and the H2O2 production
H2O2 was produced by the device schematically shown in Fig. 1 [see Supplementary Information (SI) for details]. A direct current Ar atmospheric pressure plasma was generated between a tungsten steel tube (1.02 mm and 6.35 mm in inner and outside diameters, respectively) and a liquid surface. The liquid acts as cathode or anode (positive or negative voltage applied to the tungsten steel electrode), and NaCl was used to adjust the initial conductivity of the liquid. The Ar flow rate, the discharge current, and the gap between the tungsten steel tube and the liquid surface were set to be 20 sccm, 30 mA, and 3 mm, respectively. To refresh the surface liquid, the total 400 ml liquid was circuited by a peristaltic pump at a flow rate of 200 ml/min. During the plasma-solution interactions, the H2O2 yield was measured at a given interval of 10 min, and the temperature, pH value, and the conductivity of the solution were also investigated.
Figure 1. Schematic diagram of the device for the H2O2 production.
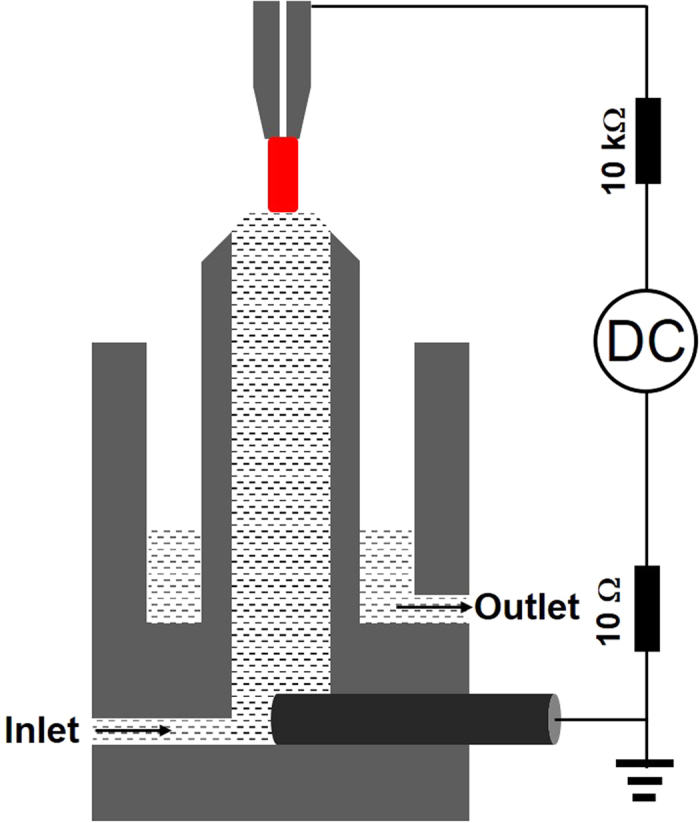
Figure 2 presents the H2O2 yields at different experimental condit ions. For liquids with low initial conductivities (1.60 μS cm−1 and 1440 μS cm−1), the H2O2 production rate (the slope of the yield curve) decreases with increasing time, while they are constants for the liquids with high initial conductivities (4800 μS cm−1 and 10500 μS cm−1) [Fig. 1(a)]. Even with an initial conductivity of 4800 μS cm−1, the H2O2 yield for NaOH solution is almost zero after 60 min plasma treatment [Fig. 1(b)]. As the discharge current increases, the H2O2 production rate is enhanced [Fig. 1(c)]. When the liquid acts as anode (negative voltage applied to the tungsten steel electrode), there is almost no H2O2 production [Fig. 1(d)]. These results guide us to a question: What is the underlying mechanism for these differences? To answer this question, we must carefully analyse the processes taking place at the plasma-liquid interfaces from which the H2O2 is formed during the plasma-liquid interactions.
Figure 2. H2O2 yields at different experimental conditions.

Plasma-liquid interactions were performed with liquids of (a) initial conductivities of 1.60, 1440, 4800 and 10500 μS cm−1, (b) NaCl and NaOH with the same initial conductivity of 4800 μS cm−1, (c) NaCl with the initial conductivity of 4800 μS cm−1 at discharge currents of 30 mA, 40 mA and 50 mA, and (d) NaCl with an initial conductivity of 4800 μS cm−1 (The liquid acts as cathode or anode).
Water molecule transfer processes at the plasma-liquid interface
When the liquid acts as cathode, a cathode voltage fall (VC) will be built between the plasma and the liquid surface due to the space charge accumulation36,37,38,39,40. The VC will be located at a limited distance called cathode sheath, and for an atmospheric pressure discharge plasma, the thickness of the cathode sheath is smaller than 100 μm when the discharge current is more than 5 mA36. In our case, the discharge current is 30 mA, it is reasonable to take the sheath thickness as 100 μm. Table 1 presents the VC for liquids with different initial conductivities (see SI for the details of the VC estimation). The following analysis will demonstrate that this cathode region near the liquid surface is very important to the water molecules transfer at the plasma-liquid interface.
Table 1. Cathode voltage falls (V C ) for liquids with different initial conductivities. NaCl was used to adjust the solution conductivity.
| Time (min) | VC (V) | |||
|---|---|---|---|---|
| 1.60 μS cm−1 | 1440 μS cm−1 | 4800 μS cm−1 | 10500 μS cm−1 | |
| 0 | N/A[a] | 581 | 504 | 512 |
| 20 | 663 | 559 | 494 | 505 |
| 40 | 590 | 544 | 507 | 510 |
| 60 | 558 | 526 | 493 | 506 |
[a] Data are unavailable because the discharge is unstable in the very beginning for the liquid with an initial conductivity of 1.60 μS cm−1.
Figure 3 depicts three processes contributing to the water transfer from the liquid phase to the gaseous plasma, and the qualitative characteristics of the voltage potential (V) and the electric field (E) in the plasma-liquid system (ref. 41). Firstly, positive ions in the plasma passing the cathode sheath will be accelerated by the VC, and the constituents of liquid will be sputtered into the gaseous phase by the accelerated energetic ions. This sputtering process has been widely used in low pressure plasma for material fabrication41. Secondly, the VC measured in our case is in the magnitude of ~500 V (see Table 1) which forms an electric field in the order of 100 kV cm−1 near the liquid surface (see latter analysis for the electric field estimation). This electric field is high enough to pull out the hydrated negative ions (carrying water molecules) from the liquid surface and transfer them to the gaseous plasma36,42,43,44, which is similar to the field emission at a solid surface. Thirdly, there is evaporation at the liquid surface caused by plasma and Joule heating. Obviously, all these three processes can transfer water molecules from the liquid phase into the gaseous plasma, and then the number of water molecules entering the plasma phase is influenced by the above three processes. Water molecules in the gaseous plasma can react with plasma species to form OH, and then H2O2 is formed by the combination of OH. Finally, H2O2 dissolves into the liquid to form an aqueous solution. Therefore, H2O2 production will increase with the number of water molecules entering the plasma phase which is strongly dependent on the above mentioned processes.
Figure 3. Characteristics of the plasma-liquid interface.

(a) Three main processes occurring at the plasma-liquid interface as the liquid is cathode, and (b) the qualitative characteristics of the voltage potential (V) and electric field (E) in the plasma-liquid system (ref. 41). z is the coordinate along the plasma-liquid direction and at the liquid surface z = 0, VC is the cathode voltage fall, dC is the thickness of the cathode sheath, and EC is the electric field at the liquid surface.
The number density of water molecules (n) entering the plasma phase can be expressed as
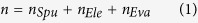 |
where
nSpu: the number density of water molecules entering the plasma phase due to the sputtering, nSpu is related to the cathode voltage fall (VC), discharge current (Id), liquid surface tension etc. and it can be described as nSpu = kspuId(Ei)1/241, kspu is the sputtering coeffieient, and Ei is the energy of incident ion. The positive ion in the plasma obtains its energy by passing the plasma cathode sheath, and therefore the obtained energy is proportional to the Vc. Thus, nSpu can be expressed as nSpu = kspuId(VC)1/2.
nEle: the number density of water molecules entering the plasma phase due to the electric field induced hydrated ion emission, and nEle is an increasing function of the ion concentration in the liquid phase (CIon) and the electric field near the liquid surface (EC) but depends on these parameters in a complicated way44. We can express it as nEle = f(VC, CIon). In addition, the electric field near the liquid surface (EC) can be estimated as follows. In the cathode sheath, E = EC(1 − z/dC), and V = VC(z/dC)(2 − z/dC), where E is the electric field in the cathode sheath, z is the distance away from the liquid surface, dC is the thickness of cathode sheath, V is the voltage potential in the cathode sheath, and VC is the voltage potential near the liquid surface41. If we integrate E to get V, we can find that EC = 2VC/dC. From the results and analysis in the main paper, we know that the VC in liquid cathode is in the magnitude of ~500 V, and dC is taken as 100 μm, and then EC is estimated to be in the order of 100 kV cm−1.
nEva: the number density of water molecules entering the plasma phase due to evaporation. Based on the thermodynamics law, nEva can be expressed as nEva = n0 exp(−W/kT), where n0 is the number density of water molecules in the liquid phase, W is the heat of evaporization, k is the Boltzmann constant and T is the liquid temperature.
Therefore, n can be expressed as
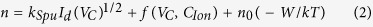 |
Besides depending on Id for nSpu, and on CIon for nEva, both nSpu and nEle depend on the cathode voltage fall (VC), and nEva depends on the solution temperature.
H2O2 energy yields
Based on the discharge voltages shown in Fig. S2, we estimated the average power consumed in the plasma-liquid interactions, and then the H2O2 generation energy yields were calculated for liquids with different initial conductivities and the results are summarized in Table 2. The H2O2 energy yields are larger than most of the H2O2 energy yields generated from discharge plasmas which have been summarized in ref. 28. The maximum energy yield appears for liquid with an initial conductivity of 10500 μS cm−1, although the liquid with an initial conductivity of 1.6 μS cm−1 produces the largest generation rate. The energy yield can be expressed as
Table 2. Consumed power, energy yield and the generation rate of hydrogen peroxide generation for liquids with different initial conductivities.
| Initial conductivities (μS cm−1) | Power (10−3 kW) | Energy Yield (mg kWh−1) | Generation Rate (mg h−1) |
|---|---|---|---|
| 1.60 | 38.1 | 1089 | 41.48 |
| 1440 | 24.5 | 866 | 21.22 |
| 4800 | 23.1 | 1354 | 31.28 |
| 10500 | 22.8 | 1741 | 39.78 |
NaCl was used to adjust the solution conductivity.
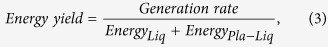 |
where EnergyLiq and EnergyPla-Liq are energy consumed in the liquid (mostly taking a form of heating the liquid cylinder) and energy consumed in the plasma-liquid interactions, respectively, and only the latter contributes to the H2O2 production. Therefore, EnergyLiq can affect the energy yield of the H2O2 formation. The largest generation rate for the liquid with an initial conductivity of 1.6 μS cm−1 can be attributed to the high resistance of the liquid cylinder between the plasma and the graphite electrode. The Joule heating consumes a large portion of the supplied power in the liquid with the initial conductivity of 1.6 μS cm−1, but in this case, a larger cathode voltage fall is close to the liquid surface as shown in Table 1, and therefore, it produces a large H2O2 generation rate with a lower energy yield if compared with the case using liquid with the initial conductivity of 10500 μS cm−1. In the plasma-induced H2O2 generation, to select the liquid with a high or low initial conductivity is dependent on one’s aim: to obtain high energy yield or high generation rate.
Discussion
Based on the expression of n, the water moleclues transfer from the liquid phase to the gaseous plasma is largely determined by the sputtering and the electric field induced hydrated ion emission in the case of liquid cathode since the evaporation for all cases are estimated to be similar [Fig. S6(a)]. The results in Table 1 indicate that the VC decreases with increasing time for liquids with initial conductivities of 1.60 μS cm−1 and 1440 μS cm−1, while it almost keeps constant for liquids with initial conductivities of 4800 μS cm−1 and 10500 μS cm−1. Therefore, from the expression of n and the data in Table. 1, one can deduce that n for liquids with initial conductivities of 1.60 μS cm−1 and 1440 μS cm−1 will decrease with increasing time, while it is almost constant for the liquids with initial conductivities of 4800 μS cm−1 and 10500 μS cm−1. Therefore, the H2O2 production rate decreases as time increases for liquids with low initial conductivity, while it is almost constant for liquids with high initial conductivity. Because the evaporation for all cases are estimated to be similar [see Fig. S6(a)], n is determined by Id, VC, and CIon. For the case with a constant Id, n is related to VC and CIon. Although CIon is low in case of the liquid with low conductivity (1.6 μS cm−1), n can be still large since the high VC (see Table 1) in this case. Therefore, there is a high H2O2 production rate for the liquid with a low conductivity (1.6 μS cm−1). In addition, the estimated VC are almost the same for discharge currents of 30 mA, 40 mA and 50 mA (~500 V, not shown). When the discharge current increases, the ion flux (related to Id) near the liquid surface is enhanced and thus water transfer from the liquid phase to the gaseous plasma is increased. Consequently, the H2O2 yield increases with increasing discharge current as shown in Fig. 2(c). The estimated VC for NaOH solution with an initial conductivity of 4800 μS cm−1 is ~30 V higher than that for the NaCl solution with the same conductivity. Based on the above anlysis, one must expect that the NaOH solution should produce more H2O2. However, the H2O2 yield for NaOH solution is almost zero as shown in Fig. 2(b). The reason might be as follows. H2O2 is a weak acid and it can react with OH− to form HO2− in concentrated NaOH solution (H2O2 + OH− → HO2− + H2O)45,46,47. Therefore, the produced H2O2 was consumed by reacting with NaOH, resulting in a very low H2O2 yield.
When liquid acts as anode, the cathode voltage fall is formed on the tungsten steel electrode, and therefore there is no sputtering and electric field induced ion emission at the liquid surface, and evaporation is the only way to transfer water molecules from liquid phase into the gasesous plasma. Compared with the case of liquid cathode, n is insignificantly small in the case of liquid anode, resulting in a low H2O2 yield as shown in Fig. 2 (b). Comprison of OH opitical emission intensity in cases of liquid cathode and anode also confirms this conclusion (Fig. S9).
In summary, using water as the only consumed material, H2O2 is directly synthesized by plasma-water interations and the H2O2 production rate can reach 1200 μmol/h when the liquid cathode is purified water or NaCl solution with an initial conductivity of 10500 μS cm−1. The H2O2 production rate strongly depends on the plasma-liquid interactions at the liquid surface: sputtering, high electric field induced hydrated ion emission, and evaporation. Liquid cathode performs much better than liquid anode in producing H2O2 by plasma-water interations. In addition, the synthesized H2O2 can be consumed if the liquid contains some constituent able to react with H2O2 such as NaOH.
Methods
Measurement of the H2O2 yield
Because H2O2 can react with titanium sulfate. in strong acid to form H2TiO4 (Ti4+ + H2O2 + 2H2O → H2TiO4 + 4 H+) and the absorption intensity of the yellow-coloured H2TiO4 in 410 nm is proportional to the reacted H2O2 concentration15,48,49,50. We can use it to determine the synthesized H2O2 concentration. 7.5 ml [Ti(SO4)2,120 g/l) was added to 250 ml H2SO4 (1.5 M) to obtain the test solution of titanium sulfate. We used H2O2 with standard concentrations to obtain the proportionality between the absorption intensity of H2O2 at 410 nm, and the results are presented in Fig. S8(a). Once the proportionality is obtained, the H2O2 yield is estimated by the following equation:
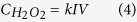 |
where k is the proportionality obtained by linearly fitting Fig. S8(b), I is the absorption intensity of synthesized H2O2 at 410 nm, and V is the solution volume (in our case, 400 ml).
Measurement of the pH value, conductivity, and temperature of the liquid
The pH value and temperature of the solution were measured by a pH detector with a temperature sensor (Yesmylab SX620), and the solution conductivity was measured by a conductivity detector (Yesmylab SX650).
Electrical characterization of the discharge plasma
The voltage between the tungsten steel and the graphite electrodes was measured by a high voltage (H.V.) probe (Tektronix P6015A) and the current was achieved from dividing the voltage across a 10-Ω resistor which was in series connected with the graphite electrode.
Additional Information
How to cite this article: Liu, J. et al. Direct synthesis of hydrogen peroxide from plasma-water interactions. Sci. Rep. 6, 38454; doi: 10.1038/srep38454 (2016).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
This work was partially supported by the National Natural Science Foundation of China (Grant Nos.: 11405144, 11275261, 11304132, 61376068), the Fundamental Research Funds for the Central Universities (Grant Nos: 20720150022, 20720150083), the Natural Science Foundation of Fujian Province (2014J01025), and the Natural Science Foundation of Guangdong Province (Grant No. 2015A030313005). One of the authors Q. X. gives thanks for the funding supports by National Natural Science Foundation of China (Grant No: 11305273), and the Fundamental Research Funds for the Central Universities (Grant No: 0213005202054).
Footnotes
The authors declare no competing financial interests.
Author Contributions Q.C. designed the experiments and supervised the project. J.L. and B.H. conducted the experiments, all the data analysis and interpretation guided by Q.C. and H.L. J.L., Q.X., G.Y., X.Z., S.Y. and Q.H.L. provided valuable discussions on the purpose and focus of the study. All authors discussed the results and Q.C. wrote the manuscript.
References
- Hill C. A Vertical Empire: the History of the UK Rocket and Space Programme, 1950–1971. (World Scientific, 2001).
- Campos‐Martin J. M., Blanco‐Brieva G. & Fierro J. L. Hydrogen peroxide synthesis: an outlook beyond the anthraquinone process. Angew. Chem. Int. Ed. 45, 6962–6984 (2006). [DOI] [PubMed] [Google Scholar]
- Centi G., Perathoner S. & Abate S. in Modern Heterogeneous Oxidation Catalysis: Design, Reactions and Characterization 253–287 (2009). [Google Scholar]
- Landon P., Collier P. J., Papworth A. J., Kiely C. J. & Hutchings G. J. Direct formation of hydrogen peroxide from H2/O2 using a gold catalyst. Chem. Comm., 2058–2059 (2002). [DOI] [PubMed] [Google Scholar]
- Nomura Y. et al. Nanocolloidal Pd‐Au as Catalyst for the Direct Synthesis of Hydrogen Peroxide from H2 and O2. ChemSusChem 1, 619–621 (2008). [DOI] [PubMed] [Google Scholar]
- Edwards J. K. et al. The Direct Synthesis of Hydrogen Peroxide Using Platinum‐Promoted Gold–Palladium Catalysts. Angew. Chem. Int. Ed. 53, 2381–2384 (2014). [DOI] [PubMed] [Google Scholar]
- Murayama T. & Yamanaka I. Electrosynthesis of neutral H2O2 solution from O2 and water at a mixed carbon cathode using an exposed solid-polymer-electrolyte electrolysis cell. J. Phys. Chem. C 115, 5792–5799 (2011). [Google Scholar]
- Yamanaka I., Onizawa T., Takenaka S. & Otsuka K. Direct and Continuous Production of Hydrogen Peroxide with 93% Selectivity Using a Fuel‐Cell System. Angew. Chem. Int. Ed. 42, 3653–3655 (2003). [DOI] [PubMed] [Google Scholar]
- Murayama T. & Yamanaka I. Neutral H2O2 Synthesis by Electrolysis of O2 and Water. ECS Trans. 25, 19–24 (2010). [DOI] [PubMed] [Google Scholar]
- Yamanaka I. & Murayama T. Neutral H2O2 synthesis by electrolysis of water and O2. Angew. Chem. Int. Ed. 47, 1900–1902 (2008). [DOI] [PubMed] [Google Scholar]
- Liu Y., Quan X., Fan X., Wang H. & Chen S. High‐Yield Electrosynthesis of Hydrogen Peroxide from Oxygen Reduction by Hierarchically Porous Carbon. Angew. Chem. Int. Ed. 54, 6837–6841 (2015). [DOI] [PubMed] [Google Scholar]
- Cai R., Hashimoto K., Fujishima A. & Kubota Y. Conversion of photogenerated superoxide anion into hydrogen peroxide in TiO 2 suspension system. J. Elecctroanal. Chem. 326, 345–350 (1992). [Google Scholar]
- Kormann C., Bahnemann D. W. & Hoffmann M. R. Photocatalytic production of hydrogen peroxides and organic peroxides in aqueous suspensions of titanium dioxide, zinc oxide, and desert sand. Environ. Sci. Technol. 22, 798–806 (1988). [DOI] [PubMed] [Google Scholar]
- Cai R., Kubota Y. & Fujishima A. Effect of copper ions on the formation of hydrogen peroxide from photocatalytic titanium dioxide particles. J. Catal. 219, 214–218 (2003). [Google Scholar]
- Satterfield C. N. & Bonnell A. H. Interferences in titanium sulfate method for hydrogen peroxide. Anal. Chem. 27, 1174–1175 (1955). [Google Scholar]
- Shiraishi Y. et al. Sunlight‐Driven Hydrogen Peroxide Production from Water and Molecular Oxygen by Metal‐Free Photocatalysts. Angew. Chem. Int. Ed. 126, 13672–13677 (2014). [DOI] [PubMed] [Google Scholar]
- Venugopalan M. & Jones R. Chemistry of dissociated water vapor and related systems. Chem. Rev. 66, 133–160 (1966). [Google Scholar]
- Morinaga K. The Reaction of Hydrogen and Oxygen through a Silent Electric Discharge. I. The Formation of Hydrogen Peroxide. Bull. Chem. Soc. Jpn. 35, 345–348 (1962). [Google Scholar]
- Yi Y. et al. Safe direct synthesis of high purity H2O2 through a H2/O2 plasma reaction. Angew. Chem. Int. Ed. 52, 8446–8449 (2013). [DOI] [PubMed] [Google Scholar]
- Liu Z. et al. Physicochemical processes in the indirect interaction between surface air plasma and deionized water. J. Phys. D: Appl. Phys. 48, 495201 (2015). [Google Scholar]
- Chen Q., Li J. & Li Y. A review of plasma–liquid interactions for nanomaterial synthesis. J. Phys. D: Appl. Phys. 48, 424005 (2015). [Google Scholar]
- Mariotti D., Patel J., Švrček V. & Maguire P. Plasma–liquid interactions at atmospheric pressure for nanomaterials synthesis and surface engineering. Plasma Process Polym. 9, 1074–1085 (2012). [Google Scholar]
- Akolkara R. & Sankarana R. M. Charge transfer processes at the interface between plasmas and liquids. J. Vac. Sci. Technol. A 31, 050811 (2013). [Google Scholar]
- Richmonds C. et al. Electron-Transfer Reactions at the Plasma–Liquid Interface. J. Am. Chem. Soc. 133, 17582–17585 (2011). [DOI] [PubMed] [Google Scholar]
- Rumbach P., Bartels D. M., Sankaran R. M. & Go D. B. The solvation of electrons by an atmospheric-pressure plasma. Nat. Comm. 6, 8248 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruggeman P. & Schram D. C. On OH production in water containing atmospheric pressure plasmas. Plasma Sources Sci. Technol. 19, 045025 (2010). [Google Scholar]
- Bobkova E., Shikova T., Grinevich V. & Rybkin V. Mechanism of hydrogen peroxide formation in electrolytic-cathode atmospheric-pressure direct-current discharge. High Energy Chem. 46, 56–59 (2012). [Google Scholar]
- Locke B. R. & Shih K.-Y. Review of the methods to form hydrogen peroxide in electrical discharge plasma with liquid water. Plasma Sources Sci. Technol. 20, 034006 (2011). [Google Scholar]
- Lukeš P. Water treatment by pulsed streamer corona discharge (2001). [Google Scholar]
- Burlica R. & Locke B. R. Pulsed plasma gliding-arc discharges with water spray. IEEE Trans. Ind. Appl. 44, 482–489 (2008). [Google Scholar]
- De Baerdemaeker F., Šimek M., Člupek M., Lukeš P. & Leys C. Hydrogen peroxide production in capillary underwater discharges. Czechoslovak J. Phys. 56, B1132–B1139 (2006). [Google Scholar]
- Lukes P., Appleton A. T. & Locke B. R. Hydrogen peroxide and ozone formation in hybrid gas-liquid electrical discharge reactors. IEEE Trans. Ind. Appl. 40, 60–67 (2004). [Google Scholar]
- Lukes P., Dolezalova E., Sisrova I. & Clupek M. Aqueous-phase chemistry and bactericidal effects from an air discharge plasma in contact with water: evidence for the formation of peroxynitrite through a pseudo-second-order post-discharge reaction of H2O2 and HNO2. Plasma Sources Sci. Technol. 23, 015019 (2014). [Google Scholar]
- Hsieh K. C., Wang H. & Locke B. R. Analysis of Electrical Discharge Plasma in a Gas‐Liquid Flow Reactor Using Optical Emission Spectroscopy and the Formation of Hydrogen Peroxide. Plasma Process Polym (2016). [Google Scholar]
- Thagard S. M., Takashima K. & Mizuno A. Chemistry of the positive and negative electrical discharges formed in liquid water and above a gas–liquid surface. Plasma Chem. Plasma Process 29, 455–473 (2009). [Google Scholar]
- Bruggeman P. et al. Dc excited glow discharges in atmospheric pressure air in pin-to-water electrode systems. J. Phys. D: Appl. Phys. 41, 215201 (2008). [Google Scholar]
- Chen Q., Kaneko T. & Hatakeyama R. Reductants in gold nanoparticle synthesis using gas–liquid interfacial discharge plasmas. Appl. Phys. Express 5, 086201 (2012). [Google Scholar]
- Cserfalvi T. & Mezei P. Direct solution analysis by glow discharge: electrolyte-cathode discharge spectrometry. J. Anal. At. Spectrom. 9, 345–349 (1994). [Google Scholar]
- Chen Q., Kaneko T. & Hatakeyama R. Rapid synthesis of water-soluble gold nanoparticles with control of size and assembly using gas–liquid interfacial discharge plasma. Chem. Phy. Lett. 521, 113–117 (2012). [Google Scholar]
- Kaneko T., Chen Q., Harada T. & Hatakeyama R. Structural and reactive kinetics in gas–liquid interfacial plasmas. Plasma Sources Sci. Technol. 20, 034014 (2011). [Google Scholar]
- Lieberman M. A. & Lichtenberg A. J. Principles of Plasma Discharges and Materials Processing. (John Wiley & Sons, 2005). [Google Scholar]
- Kebarle P. A brief overview of the present status of the mechanisms involved in electrospray mass spectrometry. J. Mass Spectrom. 35, 804–817 (2000). [DOI] [PubMed] [Google Scholar]
- Thomson B. & Iribarne J. Field induced ion evaporation from liquid surfaces at atmospheric pressure. J. Chem. Phys 71, 4451–4463 (1979). [Google Scholar]
- Kebarle P. & Tang L. From ions in solution to ions in the gas phase-the mechanism of electrospray mass spectrometry. Anal. Chem. 65, 972A–986A (1993). [Google Scholar]
- Nicoll W. & Smith A. Stability of dilute alkaline solutions of hydrogen peroxide. Ind. Eng. Chem. 47, 2548–2554 (1955). [Google Scholar]
- Gurman M., Shcherbak L. & Rasskazov A. Gold and arsenic recovery from calcinates of rebellious pyrite–arsenopyrite concentrates. J. Min. Sci 51, 586–590 (2015). [Google Scholar]
- Chlistunoff J. & Simonin J.-P. Ionic association of hydroperoxide anion HO2-in the binding mean spherical approximation. Spectroscopic study of hydrogen peroxide in concentrated sodium hydroxide solutions. J. Phys. Chem. A 110, 13868–13876 (2006). [DOI] [PubMed] [Google Scholar]
- Alshammari Y. & Hellgardt K. Partial oxidation of n-hexadecane through decomposition of hydrogen peroxide in supercritical water. Chem. Eng. Res. Des 93, 565–575 (2015). [Google Scholar]
- Dai X. J. et al. Efficient and Selectable Production of Reactive Species Using a Nanosecond Pulsed Discharge in Gas Bubbles in Liquid. Plasma Process Polym. 306–310 (2015). [Google Scholar]
- Eisenberg G. Colorimetric Determination of Hydrogen Peroxide. Ind. Eng. Chem. 15, 327–328, doi: 10.1021/i560117a011 (1943). [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.