

Graphical abstract

Keywords: QSAR method, Gamma aminobutyric acid aminotransferase, Molecular docking, Density functional theory, Anticonvulsant, Genetic function algorithm
Abstract
Quantitative structure-activity relationship and molecular docking studies were carried out on a series of quinazolinonyl analogues as anticonvulsant inhibitors. Density Functional Theory (DFT) quantum chemical calculation method was used to find the optimized geometry of the anticonvulsants inhibitors. Four types of molecular descriptors were used to derive a quantitative relation between anticonvulsant activity and structural properties. The relevant molecular descriptors were selected by Genetic Function Algorithm (GFA). The best model was validated and found to be statistically significant with squared correlation coefficient (R2) of 0.934, adjusted squared correlation coefficient (R2adj) value of 0.912, Leave one out (LOO) cross validation coefficient (Q2) value of 0.8695 and the external validation (R2pred) of 0.72. Docking analysis revealed that the best compound with the docking scores of −9.5 kcal/mol formed hydrophobic interaction and H-bonding with amino acid residues of gamma aminobutyric acid aminotransferase (GABAAT). This research has shown that the binding affinity generated was found to be better than the commercially sold anti-epilepsy drug, vigabatrin. Also, it was found to be better than the one reported by other researcher. Our QSAR model and molecular docking results corroborate with each other and propose the directions for the design of new inhibitors with better activity against GABAAT. The present study will help in rational drug design and synthesis of new selective GABAAT inhibitors with predetermined affinity and activity and provides valuable information for the understanding of interactions between GABAAT and the anticonvulsants inhibitors.
Introduction
Epilepsy is a perpetual and regularly dynamic issue described by the occasional and erratic event of epileptic seizures, which are brought on by an anomalous release of cerebral neurons [1]. It is a standout among the most widely recognized neurological issue that influences around 70 million individuals around the world [2]. Epilepsy causes seizure to occur and these seizures can cause a variety of symptoms depending on the areas of the brain affected. Symptoms can vary from mild to severe and can include complete or partial loss of consciousness, loss of speech, uncontrollable motor behavior, and unusual sensory experiences [3]. Gamma aminobutyric acid aminotransferase (GABAAT) is a validated target for anti-epileptic drugs because its selective inhibition raises GABA concentration in brain which has an antiepileptic effect [4]. There is a proceeding with an interest for new anticonvulsant agents, as it has not been conceivable to control each sort of seizure with the right now accessible antiepileptic drugs. Additionally, the present treatment of epilepsy, with advanced antiepileptic medications, is connected with measurement related symptoms, unending lethality, and teratogenic impacts [5], [6]. Therefore, developing a new antiepileptic drug with approved therapeutic properties is an important challenge for medicinal chemists.
Quantitative Structure-Activity Relationships (QSAR) are mathematical frameworks which interface molecular structures of compounds with their natural activities in a quantitative way [7]. The main success of the QSAR method is the possibility to estimate the properties of new chemical compounds without the need to synthesize and test them. This analysis represents an attempt to relate structural descriptors of compounds to their physicochemical properties and biological activities. This is broadly utilized for the prediction of physicochemical properties in the chemical, pharmaceutical, and environmental spheres [8]. Moreover, the QSAR strategies can save resources and accelerate the process of developing new molecules for use as drugs, materials, and additives or for whatever purposes [9]. Molecular docking is a computational method used to determine the binding compatibility of the active site residues to specific groups and to reveal the strength of interaction [10], [11]. Molecular docking is a very popular and useful tool used in the drug discovery arena to investigate the binding of small molecules (ligands) to macromolecule (receptor) [12], [13], [14]. The objective of this research was to develop various QSAR models using Genetic Function Algorithm (GFA) method and to predict the GABAAT inhibitory activity of the compounds. We also docked the compounds against GABAAT protein (10HV) with bound ligand (quinazolinonyl analogues).
Material and methods
Data sets used
24 Molecules of quinazolinonyl derivatives used as anticonvulsant activity were selected from the literature and used for the present study [15]. The anticonvulsant activities of the molecules measured as ED50 (μM) were expressed as logarithmic scale as pED50 (pED50 = log1/ED50) was used as dependent variable, consequently correlating the data linearly with the independent variable/ descriptors. The observed structures and the biological activities of these compounds are presented in Table 1.
Table 1.
Biological activities of training and test set derivatives.
| Comp. number | Compound | pED50 | Pred.Pred.pED50ED50 | Residual |
|---|---|---|---|---|
| a | 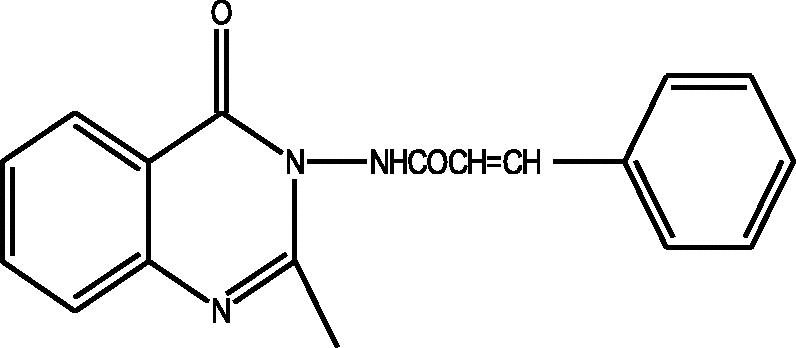 |
1.69 | 1.67 | 0.02 |
| a | 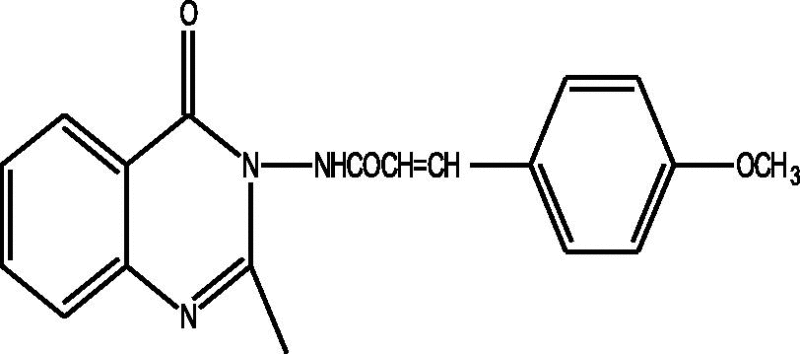 |
1.77 | 1.77 | 0.00 |
| b | 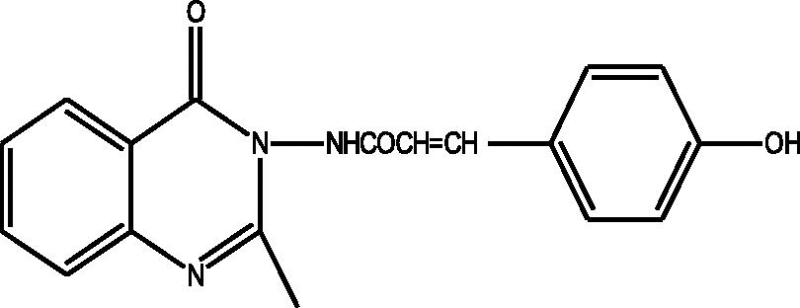 |
1.69 | 1.68 | 0.01 |
| a | 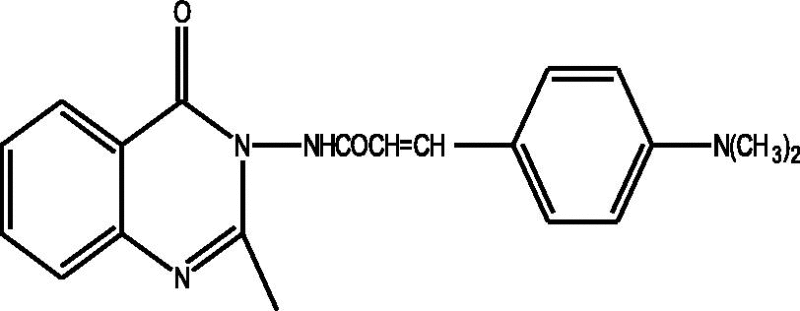 |
1.69 | 1.67 | 0.02 |
| a | 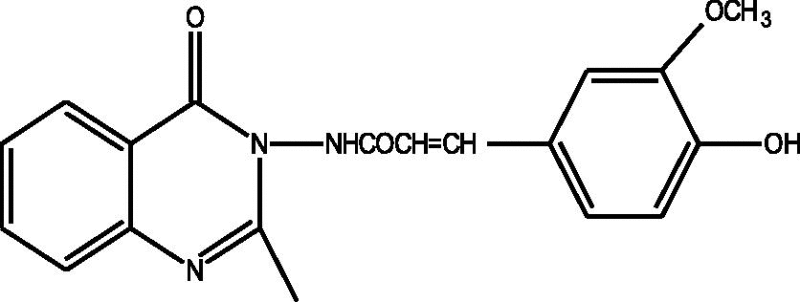 |
1.77 | 1.78 | −0.01 |
| b | 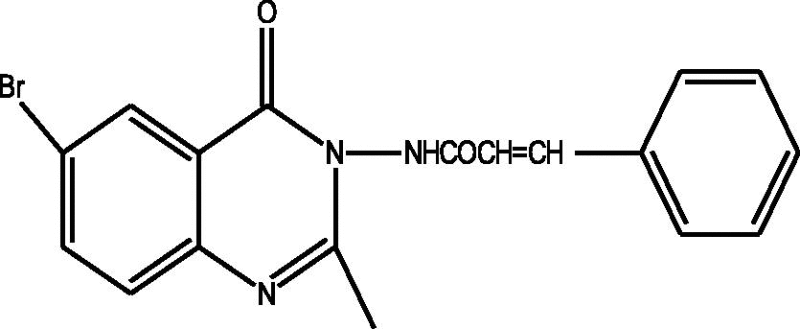 |
1.77 | 1.76 | 0.01 |
| a | 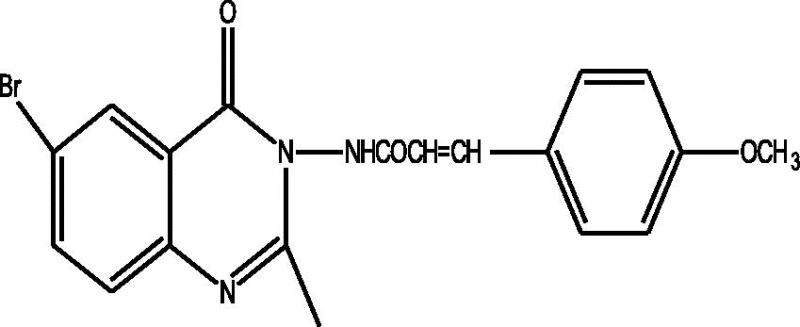 |
1.84 | 1.83 | 0.01 |
| a | 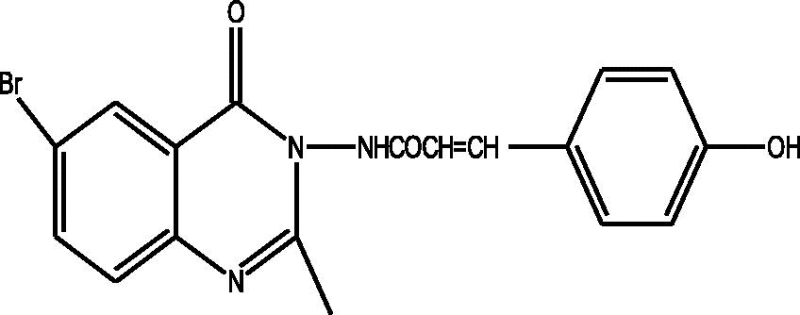 |
1.77 | 1.77 | 0.00 |
| b | 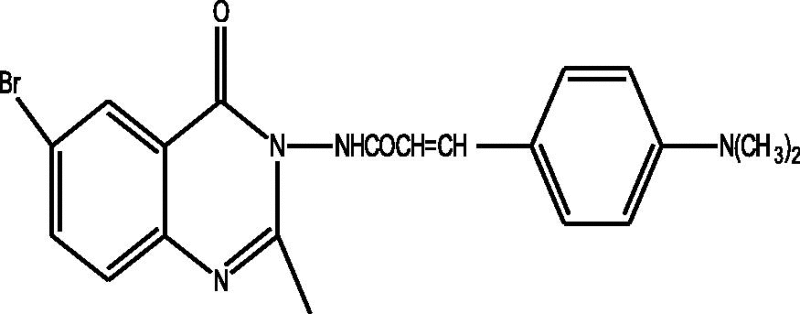 |
1.77 | 1.76 | 0.01 |
| a | 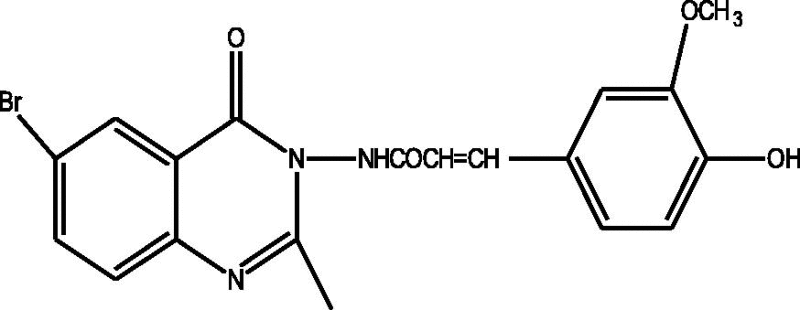 |
1.69 | 1.72 | 0.03 |
| a | 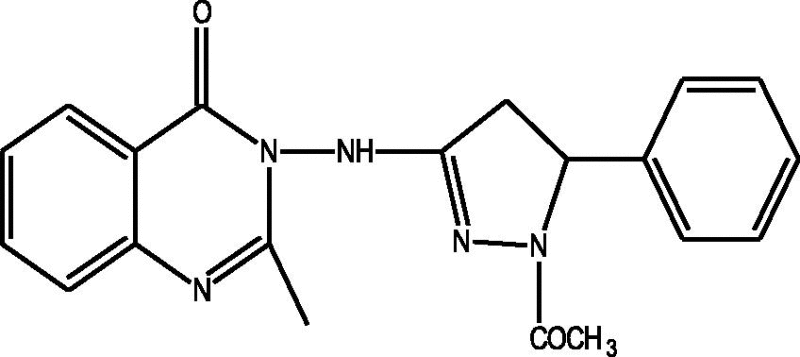 |
1.77 | 1.73 | 0.04 |
| b | 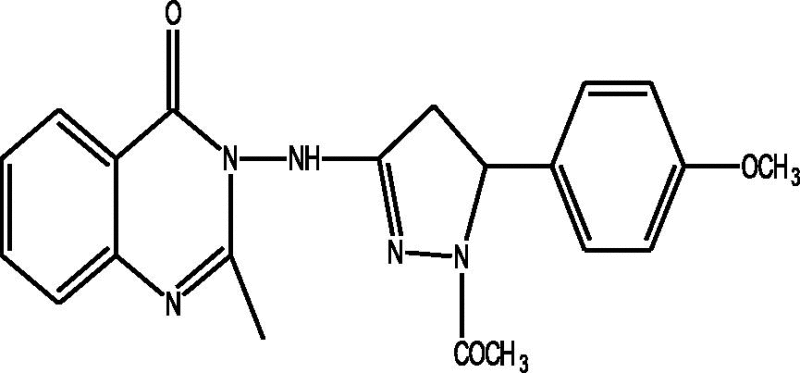 |
1.90 | 1.83 | 0.07 |
| a | 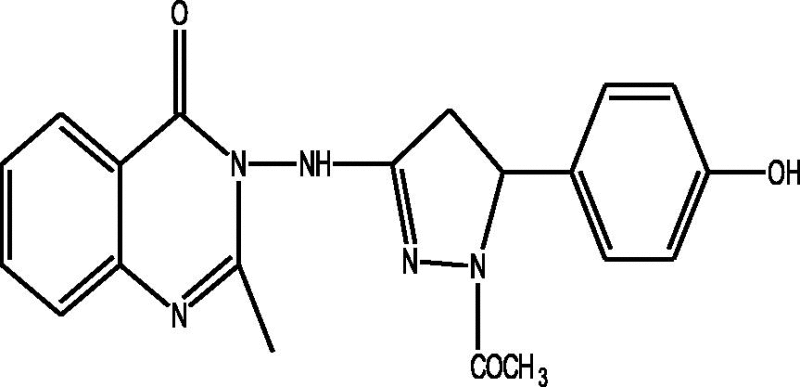 |
1.77 | 1.77 | 0.00 |
| a | 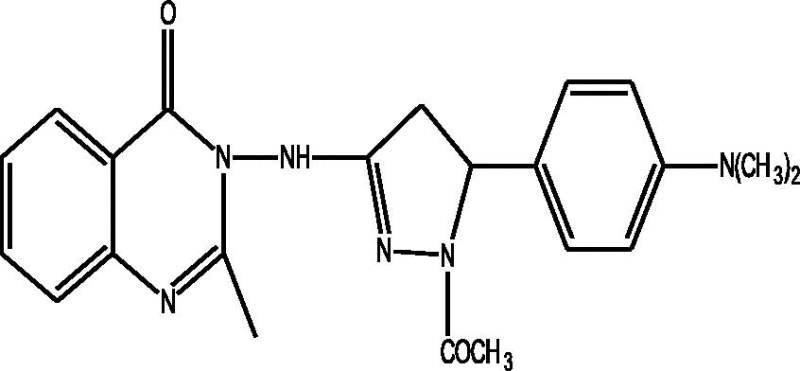 |
1.77 | 1.77 | 0.00 |
| b | 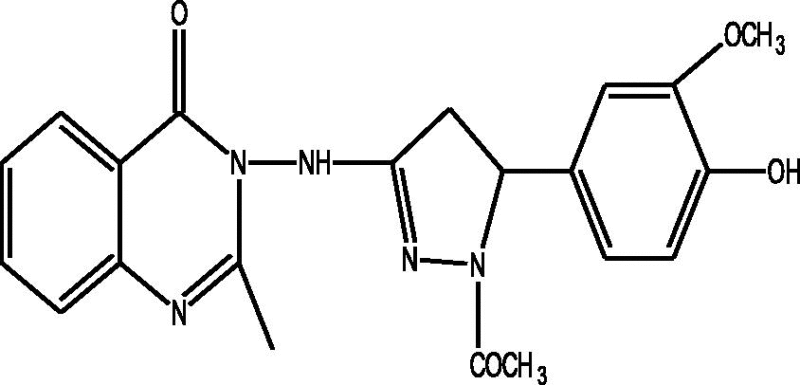 |
1.84 | 1.81 | 0.03 |
| a | 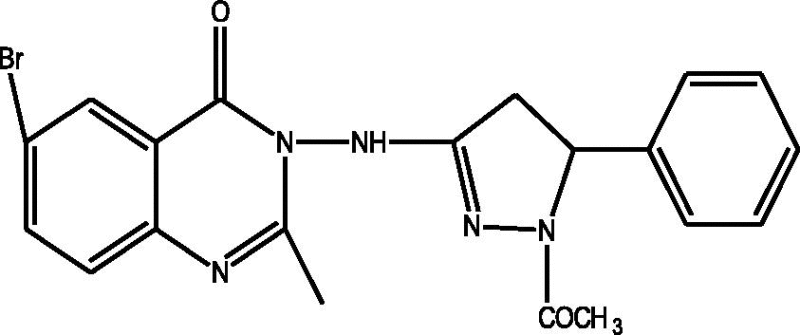 |
1.84 | 1.83 | 0.01 |
| a |  |
1.95 | 1.95 | 0.00 |
| a |  |
1.90 | 1.90 | 0.00 |
| b |  |
1.84 | 1.88 | −0.04 |
| a |  |
1.90 | 1.91 | −0.01 |
| a | 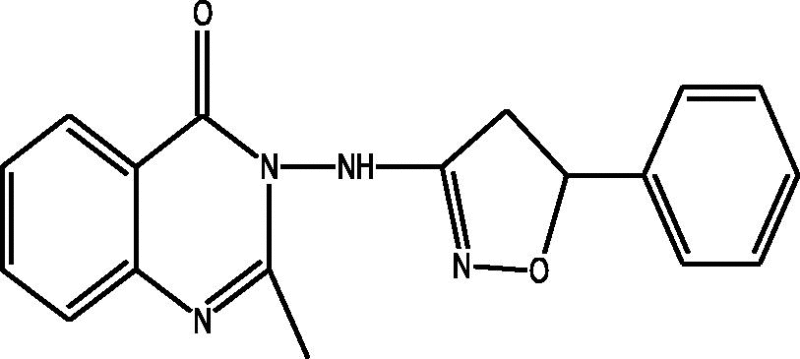 |
1.69 | 1.73 | −0.04 |
| a | 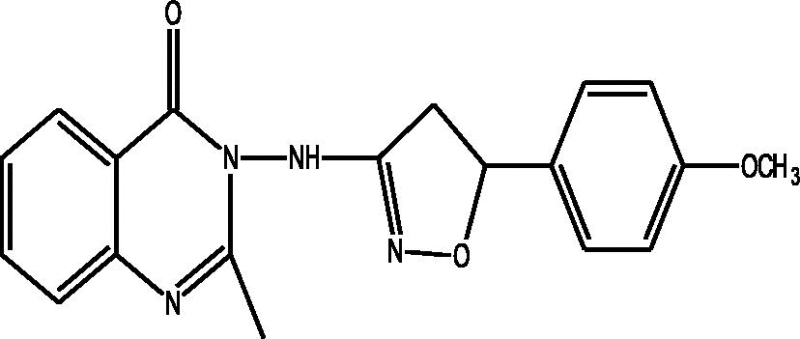 |
1.90 | 1.88 | 0.02 |
| b | 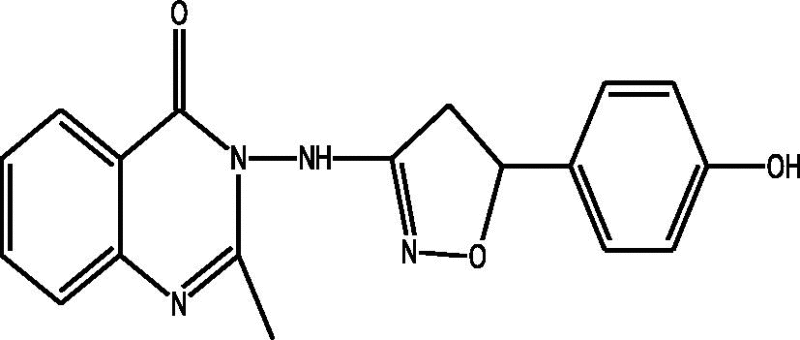 |
1.84 | 1.82 | 0.02 |
| a | 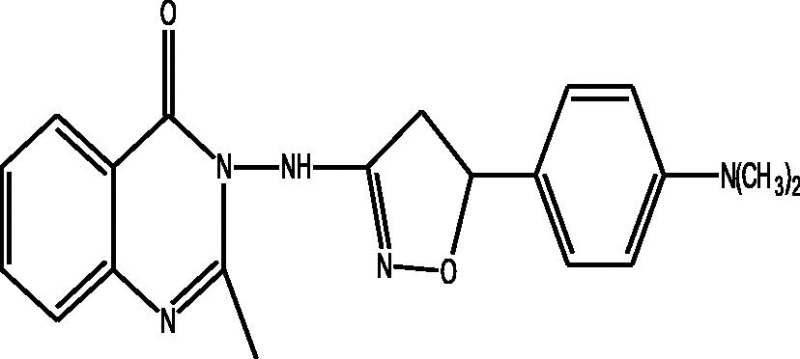 |
1.77 | 1.80 | −0.03 |
Training set.
Test set.
Molecular modeling
All molecular modeling studies were done utilizing Spartan’14 version 1.1.2 [16] and PaDEL Descriptor version 2.18 [17] running on Toshiba Satellite, Dual-core processor window 8.0 operating system. The molecular structures of the compounds were drawn in the graphic user interface of the software. 2D application tool was used to build the structures and exported in 3D format. All 3D structures were geometrically optimized by minimizing energy. Calculation of the structural electronic and other descriptors of all the 24 quinazolinonyl derivatives was conducted by means of density functional theory (DFT) using the B3LYP method and 6-31G∗ basis set. The lowest energy structure was used for each molecule to calculate their physicochemical properties. The optimized structures that were from the Spartan’14 version 1.1.2 quantum chemistry package [16] were saved in sdf format, and transferred to PaDEL-Descriptor version 2.18 tool kits [17] where the calculation of 1D, 2D and 3D descriptors took place.
Computational method
In order to obtain validated QSAR models, the descriptors (1D-3D) generated from the PaDEL version 2.18 tool kits [17] were divided into training and test sets. The training set was used to generate the model, while the test set was used for the external validation of the model [18]. The correlation between activity values of the molecules against GABAAT and the calculated descriptors was obtained through correlation analysis using the material studio software version 8. Pearson’s correlation matrix was used as a qualitative model, in order to select the suitable descriptors for regression analysis. The generated descriptors from the PaDEL version 2.18 tool kits [17] were subjected to regression analysis with the experimentally determined activities as the dependent variable and the selected descriptors as the independent variables using Genetic Function Algorithm (GFA) method in material studio software version. The number of descriptors in the regression equation was 4, and Population and Generation were set to 600 and 600, respectively. The number of top equations returned was 4. Mutation probability was 0.1, and the smoothing parameter was 0.5. The models were scored based on Friedman’s Lack of Fit (LOF). In GFA algorithm, an individual or model was represented as one-dimensional string of bits. It was a distinctive characteristic of GFA that it could create a population of models rather than a single model. GFA algorithm, selecting the basic functions genetically, developed better models than those made using stepwise regression methods. And then, the models were estimated using the LOF, which was measured using a slight variation of the original Friedman formula, so that the best fitness score can be received. The revised formula of LOF [19] is as follows:
| (1) |
where SSE is the sum of squares of errors, c is the number of terms in the model, other than the constant term, d is an user-defined smoothing parameter, p is the total number of descriptors contained in all model terms (ignoring the constant term) and M is the number of samples in the training set. Unlike the commonly used least squares measure, the LOF measure cannot always be reduced by adding more terms to the regression model. While the new term may reduce the SSE, it also increases the values of c and p, which tend to increase the LOF score. Thus, adding a new term may reduce the SSE, but actually increases the LOF score. By limiting the tendency to simply add more terms, the LOF measure resists over fitting better than the SSE measure (Materials Studio 8.0 Manual).
Quality assurance of the model
The reliability and predictive ability of the developed QSAR models were evaluated by internal and external validation parameters.
Internal and external validations
The internal and external validation parameters were compared with the minimum recommended value for the evaluation of the quantitative QSAR model [20] as shown in Table 2. The square of the correlation coefficient (R2) describes the fraction of the total variation attributed to the model. The closer the value of R2 is to 1.0, the better the regression equation explains the Y variable. R2 is the most commonly used internal validation indicator and is expressed as follows:
| (2) |
where Yobs, Ypred, and Ytraining are the experimental property, the predicted property and the mean experimental property of the samples in the training set, respectively [20]. Adjusted R2 (R2adj) value varies directly with the increase in number of repressors i.e. descriptors; thus, R2 cannot be an useful measure for the goodness of model fitness. Therefore, R2 is adjusted for the number of explanatory variables in the model. The adjusted R2 is defined as follows:
| (3) |
where
Table 2.
General minimum recommended value for the evaluation of the quantitative QSAR model.
| Symbol | Name | Value |
|---|---|---|
| R2 | Coefficient of determination | ⩾0.6 |
| P(95%) | Confidence interval at 95% confidence level | <0.05 |
| Q2 | Cross validation coefficient | ⩾0.5 |
| R2 - Q2 | Difference between R2 and Q2 | ⩽0.3 |
| Next. test set | Minimum number of external test set | ⩾5 |
| R2ext | Coefficient of determination for external test set | ⩾0.6 |
n is the number of training compounds.
p = number of independent variables in the model [21].
The leave one out cross validation coefficient (Q2) is given by the following:
| (4) |
where Yp and Y represent the predicted and observed activity respectively of the training set and Ym the mean activity value of the training set [22].
Applicability domain
The applicability domain (AD) of the generated models was assessed in order to specify the scope of their proposed models by defining the mathematical model limitations with respect to its structural domain and response space.
Docking study
Docking materials
Docking preparation and energy (kcal/mol) calculations of active anticonvulsant compounds and GABAAT receptor were performed by MGL tool and AutoDock Vina of PyRx virtual screening software [23]. Autogrid precalculation of the docking anticonvulsant compounds was performed by Autodock Vina of Pyrx by describing the target GABAAT protein. The energy grid was performed based on Lamarckian genetic algorithm [24]. Ligplot, discovery studio 3.5 and PyMol visualization software were used to perform the virtual analysis of docking site.
Preparation of the target receptor
The 3D structure of GABAAT receptor (1OHV) was obtained from the protein data bank in PDB format. All Heteroatomic molecules were excluded from the file using Discovery Studio 3.5 software. GABAAT receptor structure was minimized, protonated and saved in PDBQT file format in all polar residues. Fig. 1(a and b) shows the prepared three dimensional structure of GABAAT (10HV).
Fig. 1.

(a) Structure of GABAAT (10HV), (b) Structure of GABAAT(10HV) Preparation of compounds for docking.
Preparation of the ligands
The 24 synthesized compounds of quinazolinonyl derivatives (Table 1) were selected from the literature and used as ligands [15]. Chemdraw software was used to draw the 2D structures of these compounds and was then converted to 3D structures, optimized and saved in pdb file format by Spartan’14 version 1.1.2 [16]. The compounds were converted to PDBQT format by Autodock 4.2 software. The 3D structures of the prepared ligands are shown in Fig. 2.
Fig. 2.

3D structures of the prepared ligands.
Structure validation
Native ligands present in the protein structure were removed. In order to check the confirmation, root mean square deviation (RMSD) value was calculated between the original structure and the ligand deleted structure [25], [26].
Analysis of binding
The docking software binding sites were designed such that the entire ligand binding area was included within the GRID. An Autodock tool was used to select the ligand binding area of macromolecule. Docking analysis of GABAAT with the ligands was carried out using Autodock Vina. Macromolecule (GABAAT) was kept as rigid while ligand molecules were kept as flexible throughout the docking studies.
Results and discussion
QSAR studies
All the four developed QSAR models were recorded out of which the best model (model 1) was identified and reported due to the statistical significance. The name and symbol of the descriptors used in the QSAR optimization model are shown in Table 3 below. Table 4 gives the result of Validation of the Genetic Function Algorithm (GFA) of model 1 that was generated from material studio. Minimum recommended value of validation Parameters for a generally acceptable QSAR model [20] was in agreement with the model 1 parameters. Based on this analysis, Model 1 was selected and reported as the best QSAR model.
Table 3.
List of some physiochemical descriptors used for the best model.
| S/NO | Symbol | Names of descriptors | Class |
|---|---|---|---|
| 1 | ETA_Eta_L | Local index Eta_local | 2D |
| 2 | XLogP | XLOgP | 2D |
| 3 | PPSA-3 | Charge weighted partial positive surface area | 3D |
| 4 | RNCG | Relative negative charge – most negative charge/total negative charge | 3D |
Table 4.
Validation of the genetic function approximation from material studio.
| Eq. (1) | Eq. (2) | Eq. (3) | Eq. (4) | |
|---|---|---|---|---|
| Friedman LOF | 0.002815 | 0.002876 | 0.002912 | 0.003107 |
| R-squared, R2 | 0.934053 | 0.932637 | 0.931777 | 0.92723 |
| Adjusted R-squared, | 0.912071 | 0.910182 | 0.909036 | 0.902973 |
| Cross validated R-squared, | 0.869587 | 0.832929 | 0.806221 | 0.87158 |
| Significant regression | Yes | Yes | Yes | Yes |
| Significance-of-regression F-value | 42.49129 | 41.53468 | 40.97325 | 38.22585 |
| Critical SOR F-value (95%) | 3.306215 | 3.306215 | 3.306215 | 3.306215 |
| Replicate points | 0 | 0 | 0 | 0 |
| Computed experimental error | 0 | 0 | 0 | 0 |
| Lack-of-fit points | 12 | 12 | 12 | 12 |
| Min expt. error for non-significant LOF (95%) | 0.018506 | 0.018704 | 0.018823 | 0.01944 |
Model 1
pED50 = 0.114383001 ∗ ETA_Eta_L + 0.190098515 ∗ XLogP + 0.028759587 ∗ PPSA-3 + 4.201924750 ∗ RNCG − 0.690224604, N = 17, 0.72028, , = 0.912071, = 0.869587, , Min expt. error for non-significant LOF (95%) = 0.018506.
Model 2
pED50 = 0.279901890 ∗ VP-6 + 0.188955711 ∗ XLogP + 0.033018384 ∗ PPSA-3 + 3.694884401 ∗ RNCG − 0.404755657, N = 17, = 0.62704, = 0.932637, = 0.910182, = 0.832929, , Min expt. error for non-significant LOF (95%) = 0.018704.
Model 3
pED50 = 0.148446854 ∗ VP-4 + 0.190534973 ∗ XLogP + 0.032884549 ∗ PPSA-3 + 4.028075797 ∗ RNCG − 0.595730073, N = 17, , , = 0.909036, , , Min expt. error for non-significant LOF (95%) = 0.018823.
Model 4
pED50 = 0.109267006 ∗ SP-6 + 0.197509169 ∗ XLogP + 0.029112087 ∗ PPSA-3 + 4.163767660 ∗ RNCG − 0.562852003, N = 17, = 0.69, = 0.92723, = 0.902973, , , Min expt. error for non-significant LOF (95%) = 0.01944.
The result from the Correlation matrix (Table 5) shows clearly that the correlation coefficients between each pair of descriptors are very low, and this means that there exist no significant inter correlation among the descriptors used in development of the model. Suppl. Fig. 1 gives the plot of predicted activities of both training and test sets against observed activities; the reliability of the model (best QSAR model) was further confirmed as the GFA derived R2 value was in agreement with R2 value of 0.93 recorded in this graph.
Table 5.
Pearson’s correlation matrix for descriptors used in QSAR model for the activities of anticonvulsant molecules.
| ETA_Eta_L | XLogP | PPSA-3 | RNCG | |
|---|---|---|---|---|
| ETA_Eta_L | 1 | |||
| XLogP | 0.17959 | 1 | ||
| PPSA-3 | 0.1924 | −0.25267 | 1 | |
| RNCG | −0.57017 | −0.35028 | −0.54108 | 1 |
The Williams plot, the plot of the standardized residuals against the leverage (suppl. Fig. 2), was used to visualize the applicability domain (AD) [27]. Leverage indicates a compound’s distance from the centroid of X. The leverage of a compound in the original variable space is defined as follows:
| (5) |
The danger leverage (h∗) is defined as follows:
| (6) |
where N is the number of training compounds, and p is the number of predictor variables. Where is the descriptor vector of the considered compound and X is the descriptor matrix derived from the training set descriptor values. In suppl. Fig. 3, it is obvious that all compounds in the test set fall inside the domain of the model (the danger leverage limit is 0.88). All the training and test sets are good leverages since none of the chemical compounds go beyond the danger value, so they can be regarded as good prediction for the model.
Molecular docking studies
Molecular docking studies were carried out between the targets (GABAAT) and the inhibitors. All the compounds were found to strongly inhibit by completely occupying the active sites in the target protein (GABAAT). All inhibitors showed low energy values (high docking scores) than the binding energies of vigabatrin (-4.4 kcal/mol), the standard antiepileptic drug. For target protein, binding energy values range from -6.0 to -9.5 kcal/mol. In Table 6, most of the inhibitors were found to involve in both the hydrophobic interactions and hydrogen bonding with the receptor (GABAAT). In addition, ligand number 13a with binding energies of -9.5 kcal/mol showed better binding energies than other co-ligands.
Table 6.
GABAAT active site residues involved in docking interactions with the inhibitors and docking scores.
| Ligand(s) | Receptor | Binding Affinity (kcal/mol) | Hydrophobic interaction | Hydrogen bonding | Hydrogen bond length (Å) |
|---|---|---|---|---|---|
| GABAAT | −6.0 | Pro91,Glu50,Gln92, Ser95,Val94,Pro82, Val85, | Arg53 | 2.80 | |
| GABAAT | −8.1 | Ile72,Glu270,Tyr69, Tyr348,Ile351, Asn423,Ser427,Arg430, Ile426, | His44,Gly438 | 3.05,3.04 | |
| GABAAT | −8.0 | Gly438,Tyr69, Glu270,Phe351,Ile105, Ile72,Tyr348,His44, Ser427 | Asn423 | 3.19 | |
| GABAAT | −8.3 | Phe351,Ile72,Glu270, Tyr348,Asn423, Arg430,Ser427,Ile426, Tyr69 | His44,Gly438 | 3.02,3.05 | |
| GABAAT | −8.0 | His44,Tyr348,Ile105, Ile72,Phe351,Glu270,Tyr69,Gly438, Ser427 | Asn423 | 3.13 | |
| GABAAT | −7.0 | Ile72,His206, Arg430,Ser427,Tyr348 | |||
| GABAAT | −7.9 | Ile72,His206, Arg430,Ser427,Tyr348 | |||
| GABAAT | −8.1 | Ile72,His206, Arg430,Ser427,Tyr348 | |||
| GABAAT | −7.2 | Ala381,Gly409,Leu388, Gly407,Leu227,Asn234, Glu238,Val231,Leu223, Ser277 | Arg208 | 2.90,3.18 | |
| GABAAT | −8.2 | Asn423,Arg423,Tyr69, Ile72,Tyr345,Ser427, phe351 | Arg192,Act500 | 2.87,2.92 | |
| GABAAT | −7.0 | His275,Ser277,Leu227, Tyr225,Gly407,Arg406, Ala276,Arg408 | Asp278,Asp279 | 3.05,2.07 | |
| GABAAT | −8.6 | Gly438,His44,Ile426, Arg430,Lys203,His206, Glu270,Cys439, Arg422,Tyr348,Ile72, Tyr69 | Gly440 | 2.79 | |
| GABAAT | −9.5 | Cys439,Asn423,Arg422, His44,Arg430,Leu436, Ile426,Tyr438,Ile72, Tyr69,His206,Gly438, Lys203,Glu270 | Gly440 | 3.04 | |
| GABAAT | −8.8 | Lys203,Gly438,Cys439, Tyr69,Ile72, Phe351,Ile105,Ala42, His44,Glu41,Asn423, Glu419, | Glu270 | 3.24 | |
| GABAAT | −9.4 | Ile426, Arg430, Arg422, Tyr348, His44, Ile72, Ile105, Glu270, His206, Lys203, Cys439 | Tyr69, Gly440 | 3.04,3.05 | |
| GABAAT | −8.8 | Arg422, Arg430,His44, Tyr69,Gly438,Tyr348, His206,Ile105 | Asn423 | 3.04 | |
| GABAAT | −9.0 | Ser277,Leu223,Asn234, Leu227,Arg408,Leu388, Gly407,Asp278 | |||
| GABAAT | −7.1 | Ser277,Leu223,Asn234, Leu227,Arg408,Leu388, Gly407,Asp278 | |||
| GABAAT | −8.9 | Gly438,His44,Ile426, Asn423,Lys203,Glu419, Ile205,His206,Tyr348, Arg422 | |||
| GABAAT | −9.1 | Arg430,Ile426,His44, Ile72,Ile105,Tyr69, Tyr348,Glu270,His206, Lys203,Cys439 | Gly440 | 3.07 | |
| GABAAT | −9.1 | Phe351,Ile72,Arg422, Cys439,Gly438,Glu419, Ile205,Lys203,Ile105, Tyr348,Tyr69 | Gly440 | 2.99 | |
| GABAAT | −8.5 | Ile105,Phe351,Ile72, Tyr348,Tyr69,Ile426, Asn423,Ser427 | His44,Arg430,Gly438 | 2.83,3.16,3.13 | |
| GABAAT | −8.7 | Tyr270,Phe351,Ile105, Ile72,His44, Tyr69,Lys203,Pro347, Ala346, Ile205,Tyr348 | |||
| GABAAT | −9.2 | Arg422,Tyr69, Ile105,Ile72,Phe351, Tyr348,Glu270, Ile205,Lys203,Glu419 | Gly440 | 3.06 |
Binding mode of inhibitors
Table 6 shows the docking scores, hydrogen bond length (in angstrom) and interacting residues involved in the docking of inhibitors (ligands) at the active site of GABAAT. Fig. 3 shows the best first-three docking results. Ligand number 24a shows that Arg422, Tyr69, Ile105, Ile72, Phe351, Tyr348, and Glu270 residues of target are involved in hydrophobic interactions. In addition, it also forms hydrogen bonds (3.06 Å) with Gly440. Strong inhibitor binding is also reflected by the frequency of hydrogen bonds as shown in Table 4. Compound 15b made two hydrogen bonds (3.04 Å and 3.05 Å) with two residues Tyr69 and Gly440, while hydrophobic interactions are observed with Ile426, Arg430, Arg422, Tyr348, His44, Ile72, Ile105, Glu270, Act500, His206, Lys203, and Cys439. Compound 13a (compound with the best binding score of -9.5 kcal/mol) forms a hydrogen bond with Gly440 (3.04 Å), and hydrophobic interactions with Cys439, Asn423, Arg422, His44, Arg430, Leu436, Ile426, Tyr438, Ile72, Tyr69, His206, Gly438, Lys203, and Glu270.
Fig. 3.

Three-dimensional docked GABAAT - Ligands Complex. (A) Interactions between GABAAT and Ligand 13a. (B) Interactions between GABAAT and Ligand 15b. (C) Interactions between GABAAT and Ligand 24b. Ligand:H-bond interactions, green dashed lines: Hydrophobic interactions, red dashed line.
Conclusions
It has been clearly demonstrated that the approach utilized in this study was successful in finding novel GABAAT inhibitors from the data set developed by computational methods. The model generated from various physicochemical descriptors corresponds to the essential structural features of quinazolinonyl analogues and found to have significant correlation coefficient of determination (R2) of 0.934 with GABAAT inhibiting activity. Substituted quinazolinonyl analogues showed good interactions with GABAAT protein. Compound (13a), in particular, showed high binding affinity with docking score of -9.5 kcal/mol against GABAAT in docking analysis and predicted pED50 value of 1.77 in QSAR analysis. The ligand was docked deeply within the binding pocket region forming a hydrogen bond with Gly440 (3.04 Å), and hydrophobic interactions with Cys439, Asn423, Arg422, His44, Arg430, Leu436, Ile426, Tyr438, Ile72, Tyr69, His206, Gly438, Lys203, and Glu270. From the docking analysis, we realized that the binding scores generated were found to be better than the one proposed by other researcher [28].
Furthermore, all the quinazolinonyl analogues were found to be docked to GABAAT better than the standard anti-epilepsy drug (vigabatrin). The physicochemical descriptors used in QSAR analysis (model 1) in this study were important parameters to consider in improving the potency of these substituted quinazolinonyl analogues as inhibitors of GABAAT. Our QSAR model (high correlation coefficient of determination R2 of 0.934) and molecular docking results (high binding affinity with docking score of −9.5 kcal/mol) corroborate with each other and propose the directions for the design of new inhibitors with better activity toward GABAAT. This study will help in rational drug design and synthesis of new selective GABAAT inhibitors with predetermined affinity and activity and provides valuable information for the understanding of interactions between GABAAT and the novel compounds and might pave the way toward discovery of novel GABAAT inhibitors.
Conflict of Interest
No conflict of interest.
Funding
The authors received no direct funding for this research.
Compliance with Ethics Requirements
This article does not contain any studies with human or animal subjects.
Footnotes
Peer review under responsibility of Cairo University.

Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.jare.2016.10.004.
Appendix A. Supplementary material
References
- 1.Loscher W. New visions in the pharmacology of anticonvulsant. Eur J Pharm. 1998;342:1–13. doi: 10.1016/s0014-2999(97)01514-8. [DOI] [PubMed] [Google Scholar]
- 2.Guerrini R. Epilepsy in children. Seminar at Department of child Neurology and Psychiatry, University of Pisa and IRCCS Foundazionestella Maris. 2006: 367: 499–52.
- 3.Atshunler L.L. Depression, anxiety and temporal lobe epilepsy. Laterality of focus and Symptoms. Arch Neurol. 1990;47(3):284–288. doi: 10.1001/archneur.1990.00530030050016. [DOI] [PubMed] [Google Scholar]
- 4.Storici P., Capitani G., Baise D.D., Moser M., John R.A., Jansonius J.N. Crystal structure of GABA aminotransferase, a target for antiepileptic drug therapy. Biochemistry. 1999;38:8628–8634. doi: 10.1021/bi990478j. [DOI] [PubMed] [Google Scholar]
- 5.Wilk I.J. Chemical aspects of anticonvulsant drugs. J Chem Educ. 1957;34:199. [Google Scholar]
- 6.Mattson R.H. Efficacy and adverse effects of established and new antiepileptic drugs. Epilepsia. 1995;36:S13–26. doi: 10.1111/j.1528-1157.1995.tb05995.x. [DOI] [PubMed] [Google Scholar]
- 7.Hansch C., Leo A., Hoekman D.H. Exploring QSAR. Am Chem Soc.; Washington, DC, USA: 1995. (Fundamentals and Application in Chemistry and Biology). [Google Scholar]
- 8.Wong Kai Y., Andrew G., Mercader L.M., Saavedra B.H., Gustavo P., Romanelli P.R. QSARAnalysis on tacrine-related acetylcholinesterase inhibitors. J Biomed Sci. 2014;21:84. doi: 10.1186/s12929-014-0084-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mohammad H.F., Zohreh A. In-silico prediction of rgs4 inhibitory activity of sometiadiazolidinone. Int J Med Pharm. Dec. 2013;1 [Google Scholar]
- 10.Sudha K.N., Shakira M., Prasanthi P., Sarika N., Kumar C.N., Babu P.A. Virtual screening fornovel COX-2 inhibitors using the ZINC database. Bioinformation. 2008;2:325–329. doi: 10.6026/97320630002325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abraham D.J. 6th ed. John Wiley and sons; New York: 2003. Burger’s med chem drug discov. [Google Scholar]
- 12.Lill MA, Danielson ML. Computer aided drug design platform using PyMOL. J Comput Aided Mol Des. 25: 13–19. [DOI] [PubMed]
- 13.Barril X., Morley S.D. Unveiling the full potential of flexible receptor docking using multiple crystallographic structures. J Med Chem. 2005;48:4432–4443. doi: 10.1021/jm048972v. [DOI] [PubMed] [Google Scholar]
- 14.Hawkins P.C.D., Skillman A.G., Nicholls A. Comparison of shape matching and docking as virtual screening tools. J Med Chem. 2007;50:74–82. doi: 10.1021/jm0603365. [DOI] [PubMed] [Google Scholar]
- 15.Archana S.V.K., Chandra R., Kumar A. Synthesis of potential quinazolinonyl pyrazolines and quinazolinyl isoxazolines as anticonvulsant agents. Indian J Chem. 2002;41B:2371–2375. [Google Scholar]
- 16.Anonymous. Wavefunction. Inc., Spartan’14, version 1.1.2. Irvine, California, USA; 2013.
- 17.Yap C.W. PaDEL-descriptor: open source software to calculate molecular descriptors and ingerprints. J Comput Chem. 2011;32(7):1466–1474. doi: 10.1002/jcc.21707. [DOI] [PubMed] [Google Scholar]
- 18.Kennard R.W., Stone L.A. Computer aided design of experiments. Technometrics. 1969;11(1):137–148. [Google Scholar]
- 19.Khaled K.F. Modeling corrosion inhibition of iron in acid medium by genetic function approximation method: a QSAR model. Corro Sci. 2011;53(11):3457–3465. [Google Scholar]
- 20.Ravinchandran Rajak V., Jain H., Sivadasan A., Varghese S., Kishore-Agrawal C.P. R Int J Drug Des Discov. 2011;2:511–519. [Google Scholar]
- 21.Brand.on-Vaughn, Orr KA, Comprehensive R archive network (CRAN): http:// CRAN.Rproject.org. retrieved; 2015.
- 22.Jalali-Heravi M.J., Kyani A. Use of computer-assisted methods for the modeling of theretention time of a variety of volatile organic compounds: A PCA-MLR-ANN approach. J Chem Inf Model. 2004;44:1328–1335. doi: 10.1021/ci0342270. [DOI] [PubMed] [Google Scholar]
- 23.Trott O., Olson A.J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kumar D.B., Kumar P.V., Bhubaneswaran S.P., Mitra A. Advanced drug designing softwares and their application in medical research. Int J Pharm Sci. 2010;2:16–18. [Google Scholar]
- 25.Arumugam M., Muthuswamy U., Kuppusamy A., Thirumalaisamy S., Vardharajan S., Puliyath J. Computational drug discovery of potential phosphodiesterase inhibitors using in silico studies. Pac J Trop Discov. 2012:S822–S826. [Google Scholar]
- 26.Daisy P., Nivedha R.P., Bakiya R.H. In silico drug designing approach for biotin protein Ligase of Mycobacterium tuberculosis. Asian J Pharm Clin Res. 2013;6(l1):103–107. [Google Scholar]
- 27.Netzeva T.I., Worth A.P., Aldenberg T., Benigini R., Cronin M.T.D., Gramatica P. Lab Anim. 2005;33:155–173. doi: 10.1177/026119290503300209. [DOI] [PubMed] [Google Scholar]
- 28.Iftikhar H., Batool S., Deep A., Narasimhan B., Sharma P.C., Malhotra M. In silico analysis of the inhibitory activities of GABA derivatives on 4-aminobutyrate transaminase. Arab J Chem. 2013 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.