

Abstract
Tristetraprolin (TTP) and TIS11d are two human RNA-binding proteins that belong to the CCCH-type tandem zinc finger family. In the RNA-free state, TIS11d coordinates a zinc ion in each of its two fingers, while TTP coordinates a single zinc ion with the N-terminal zinc finger. We have previously identified three residues, located in the C-terminal half of a short α-helix in the second zinc finger, that control how structured the RNA-binding domain is in these two proteins: Y151, L152, Q153 in TTP and H201, T202, I203 in TIS11d. Here, we have used molecular dynamics, NMR spectroscopy and other biochemical methods to investigate the role of these three residues in the stability of the RNA-binding domain. We found that the intra-helical hydrogen bond formed by the T202 hydroxyl group in the C-terminal zinc finger of TIS11d is necessary to allow for π – π stacking between the side chains of a conserved phenylalanine and the zinc coordinating histidine. We demonstrated that the lack of this hydrogen bond in TTP is responsible for the reduced zinc affinity of the C-terminal zinc finger.
1 Introduction
Tristetraprolin (TTP) is the prototype of the family of CCCH-type zinc finger proteins. In the cell, TTP production is induced by extracellular stimuli such as insulin, polypeptide growth factor, phorbol esters and mitogens. By controlling the activation of many genes, TTP plays an important role in modulating the inflammatory response1. TTP binds to AU-rich elements located in the 3′ untranslated region of many mRNAs, including tumor necrosis factor-α, granulocyte-macrophage colony-stimulating factor, and interleukin-22–4. The binding of TTP promotes deadenylation and consequent degradation of these transcripts, thus decreasing the production of these proteins3,5,6. The RNA-binding domain of TTP is a 70-amino acid fragment that contains the tandem zinc finger domain. The integrity of both zinc fingers is necessary for binding RNA, as any mutation of the CCCH Zn2+-coordinating residues (henceforth defined as Cys1, Cys2, Cys3 and His4) abolishes binding7.
There are two other mammalian members in this protein family with TTP-like activity: TIS11b and TIS11d8. The RNA-binding domains of these three proteins are highly homologous: the primary sequences of TTP and TIS11d are 71% identical (Fig. S1) while those of TIS11d and TIS11b are 91% identical.
The solution structure of the RNA-binding domain of TIS11d bound to the RNA oligonucleotide 5′-UUAUUUAUU-3′ has been solved using NMR spectroscopy9. This structure is a novel fold characterized by few secondary structural elements, with the two zinc finger domains, having nearly identical conformations, separated by an extended eighteen-residue linker (Fig. S1).
Molecular dynamics (MD) simulations of TIS11d free and bound to RNA have shown that a major structural reorganization occurs in TIS11d when it dissociates from RNA10,11. This reorganization primarily alters the structure of the linker residues, thereby dramatically changing the relative orientations of the two zinc fingers, yet leaving the internal structure of the zinc fingers essentially unperturbed. This structural transition involves the burial of hydrophobic surface area that would otherwise (in the absence of RNA) have an energetically unfavorable exposure to the solvent. Given the high sequence identity of the RNA binding domains of TTP and TIS11d (Fig. S1), these two proteins were expected to have similar structures. Indeed, in the RNA-bound state the secondary structure of TTP, predicted using the backbone and Cβ chemical shifts using the δ2D12 and SPARTA+13 programs, is the same as that of TIS11d14. In the absence of RNA, however, only the N-terminal zinc finger (ZF1) of TTP adopts a stable fold while the C-terminal zinc finger (ZF2) does not stably bind Zn2+ 14–16. The effect of a fully folded RNA-binding domain on the cellular activity of TTP has been determined using a luciferase reporter assay, where luciferase was placed under the control of the tumor necrosis factor-α 3′ untranslated region14. Lower reporter activity was observed when the partially disordered RNA-binding domain of TTP was replaced with the fully structured domain of TIS11d, indicating that increased structure is associated with higher RNA-degradation activity14. This result showed that folding of the RNA-binding domain is tightly coupled with the activity of TTP and TIS11d in the cell.
We have previously shown14 that the stability of ZF2 is determined by the identity of three residues, located in the C-terminal half of the α-helix of ZF2: Y151, L152, Q153 in TTP and H201, T202, I203 in TIS11d. Here, we investigate the following question: how do the residues located at the C-terminal half of the α-helix determine the affinity of ZF2 for Zn2+ and consequently the folding and stability of ZF2? To address this question we simulated TIS11d and a homology model of TTP in solution using molecular dynamics. Analysis of the resulting trajectories points to specific key interactions that stabilize the structure of ZF2 in TIS11d but that are absent in TTP. To validate the findings from simulation, we tested the role of these specific interactions using mutagenesis, NMR and CD spectroscopy experiments.
2 Methods
2.1 TTP and TIS11d RNA-binding domain homology model building and preparation
The unknown structure of the ligand-free RNA-binding domain of TTP (residues 101 to 170) was generated starting from the lowest energy NMR structure of TIS11d (pdb entry: 1RGO) bound to ARE (5′-UUAUUUAUU-3′) using the SWISS-MODEL Server17–19. The resulting structure was solvated using VMD 1.9.220 in an orthorhombic water box (50×66×52 Å3). Six Cl− ions were added to the system to neutralize the charge. Similarly, to obtain the structure of TIS11d in the apo state, the lowest energy NMR structure of TIS11d (residues 151 to 220) was solvated, after removal of the RNA molecule, in an orthorhombic water box (60×75×66 Å3) containing a single Cl− ion to enforce charge neutrality. The size of the water box used to solvate TIS11d needed to be larger than that used for TTP. This choice was to avoid the interaction of the protein with its image in the surrounding boxes due to an infrequently observed extension of the linker region. This rare linker extension was never observed for TTP. For the purpose of detecting a finite-size effect in the TTP simulations, two additional trajectories of 30 ns were collected for TTP solvated in the same size water box that was used for TIS11d (60×75×66 Å3). As expected, the size of the box did not affect the structure and dynamics of TTP in solution. The mutation of threonine 202 into leucine (T202L) was introduced in the sequence of wild type TIS11d using the Mutator plugin (v. 1.3) of VMD20.
2.2 Simulation Protocol
The solvated proteins, described above, were energy-minimized and equilibrated using the NAMD 2.10 molecular modeling package21 and the CHARMM27 force field22. The force field was modified following Sakharov and Lim23 in order to include polarization and charge transfer effects for the Zn2+ ions and the side chain atoms of the zinc coordinating residues. Simulations including a fixed charge non-bonded model of Zn2+ were not able to accurately reproduce the tetrahedral coordination geometry for the zinc ions in TIS11d10. In order to further assess the accuracy of the Sakharov and Lim polarizable charge transfer model for these CCCH-family tandem zinc fingers, five molecular configurations of ZF2 from TTP, representing diverse coordination states for the Zn2+ cation, were selected and examined using electronic structure calculations and a series of levels of theory. The charges transferred in the model of Sakharov and Lim reproduce accurate quantum chemical charges reasonably well and substantially improve upon the accuracy of the fixed charges from CHARMM2722 (see Fig. S2). Prior to equilibration, all systems were subjected to energy minimization in three stages, with restraints sequentially removed: first, all heavy atoms were constrained; next, only Cα atoms were constrained and finally, minimization was done without constraints. The systems were subsequently subjected to step-wise heating during constant volume MD with restraints applied to Cα atoms, followed by 10 ps of unconstrained constant-NPT molecular dynamics equilibration at 1 atm and 298 K. Trajectories were subsequently collected from constant-NPT MD simulations at 1 atm and 298 K. Temperature and pressure were maintained using Langevin dynamics (damping coefficient: 5 ps−1) and the Nosé-Hoover Langevin piston method, respectively. The equations of motion were integrated using the SHAKE constraint algorithm in order to use a 2 fs time step24. Non-bonded interactions were calculated at every time step with a cut-off distance of 12 Å and a switching distance of 10 Å. The particle mesh Ewald method was used to treat electrostatic interactions with periodic boundary conditions25,26. Three trajectories of TTP were run, each for a total of 100 ns, with the last 80 ns used for data collection and analysis. Two of the three trajectories of TTP, where the Zn2+ ion remains bound to ZF2, were extended for a further 50 ns, to a total of 150 ns. Loss of Zn2+ from ZF2 was not observed in the extended trajectories. Six trajectories of TIS11d were run, each for a total of 100 ns, with the last 80 ns used for data collection and analysis. Six trajectories were run for the T202L mutant of TIS11d, each for a total of 100 ns, with the last 80 ns considered for data collection and analysis. Although the thermodynamic values for each trajectory of TTP and TIS11d were equilibrated in the initial configurations, on average it took about 20 ns to reach structural equilibration. Conformations were judged equilibrated as quantified by root mean square displacement (RMSD) from the original structure. The structure of the zinc fingers is well maintained for the entire duration of the simulations, while the linker region remains flexible, as shown by the average RMSD calculated for the full protein and the zinc fingers, see Figs. S3, S4, S5, S6 and Table S1. Trajectories were analyzed using VMD 1.9.220 and molecular configurations were visualized using STRIDE27 and Tachyon28.
2.3 Protein Expression
The RNA-binding domain of human TIS11d (residues 152–220) and TTP (residues 102 to 170) were synthesized by Genescript and cloned into a modified pet28 vector with a SUMO tag between BamH1 restriction site. The F200A, T202L and C212S mutations of TIS11d and the L152T mutation of TTP were generated via Quikchange mutagenesis. TIS11d wild type, F200A, T202L and C212S mutants and TTP wild type and L152T mutant were expressed within BL21(DE3) E. coli competent cells. Isotopic labeling with 15N was performed by growing the cells in M9 containing 1 g of 15NH4Cl per liter. The cells were grown at 37°C to an OD600 of 0.8 and then induced for 4 hours with 1 mM Isopropyl β-D-1 thiogalactopyranoside (IPTG) and 0.1 mM ZnSO4 at the same temperature. Harvested cells were lysed using a cell disruptor in 50 mL buffer containing 50mM Tris HCl, pH 8.0, 50 mM NaCl, and 1 EDTA free cOmplete™ protease inhibitor tablet (Roche). Lysates were centrifuged at 19500 RPM for 1 hour at 4°C and passed through a 20 mL pre-packed PrepEase His Tagged resin (Affymetrix), washed with 5 column volumes of 50 mM Tris HCl, pH 8.0, 50 mM NaCl, 5 mM imidazole, and eluted with 50 mM Tris HCl, pH 8.0, 50 mM NaCl, 350 mM imidazole. The SUMO tag was cleaved off with ULP1. The cleavage reaction was performed for 2 hours at room temperature, using a ULP1-to-protein ratio of 1:10. The protein was then passed through a 5 mL HiTRAP Q and SP column (GE Healthcare Life Sciences) pre-equilibrated with a buffer containing 50 mM Tris HCl, pH 8.0, 50 mM NaCl. Purified protein solution was buffer exchanged into 10 mM Tris, pH 6.2, 20 mM KCl, 2 mM DTT, 0.1 mM ZnSO4 by dialysis and concentrated using a 3 KDa Centriprep concentrator (Millipore).
2.4 CD spectroscopy
Far-UV circular dichroism (CD) spectra were recorded for TIS11d wild type, F200A and T202L mutants and for TTP wild type and L152T mutant in 50 mM HEPES, pH 7.0, 20 mM KCl and 1 mM TCEP using a Jasco-810 spectropolarimeter (Jasco Inc., Easton, MD). Curves were monitored from 200–260 nm in a 0.1 cm path length quartz cuvette using a scan rate of 20 nm min−1 and a response time of 8 s. The sample temperature for all CD measurements was maintained at 293 K.
2.5 NMR spectroscopy
Folding of TIS11d wild type, F200A and T202L mutants and TTP wild type and L152T mutant was monitored via NMR spectroscopy. 15N-1H heteronuclear single quantum coherence (HSQC) spectra were collected at 298 K on a Varian Inova spectrometer operating at 600 MHz equipped with a triple-resonance cold probe. Data processing was performed using Sparky29 and NMRPipe30 software.
3 Results and Discussion
3.1 ZF2 in TTP does not maintain Zn2+-coordination in the MD simulations
MD simulations of TIS11d in the free state show that the linker region of TIS11d (Fig. S1) is flexible in solution, while the structures of both ZF1 and ZF2 are maintained throughout all trajectories, in agreement with the experimental spectroscopic data9–11,14. The homology model of the RNA-binding domain of TTP used in this study was based upon the solution structure of TIS11d. For this reason, the initial structure of TTP used in the MD simulations has both zinc fingers folded and coordinating Zn2+ ions. Experimental evidence indicates, however, that only ZF1 of TTP can stably bind Zn2+ in the RNA-free state14–16. Consistent with this evidence, one of the three MD trajectories of TTP shows the loss of Zn2+-coordination at ZF2, Fig. 1, through a series of events that are described in detail below. The remaining two trajectories exhibit the earliest events that promote this loss (Figs. S5 and S6). To characterize the Zn2+-coordination of each finger, the six angles and the four distances between the Zn2+ cation and the Zn2+-coordinating atoms (S from the cysteine residues, and Nε from the histidine) were monitored as a function of time, Fig. 1.
Figure 1.
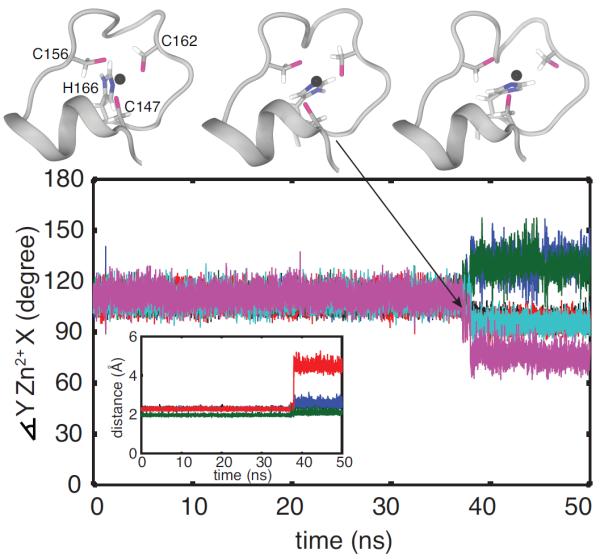
Unfolding of ZF2 during MD simulation of TTP. Conformations of the unfolding C-terminal zinc finger of TTP, sampled from an MD trajectory, are shown at top and correspond to t =37.130 ns (top, left), 37.145 ns (top, center) and 38.420 ns (top, right). Below, the geometry of the zinc coordination in the C-terminal zinc finger of TTP is monitored. In the main figure, the angles between the zinc ion and the zinc coordinating atoms are shown for the first 50 ns: ∡SC147-Zn2+-SC156 in black, ∡SC147-Zn2+-SC162 in blue, in red, ∡SC156-Zn2+-SC162 in green, in cyan and in magenta. In the inset, the distances between the zinc ion and the zinc coordinating atoms are shown: SC147-Zn2+ in black, SC156-Zn2+ in blue, SC162-Zn2+ in red and in green.
To determine the order of events that lead to the loss of Zn2+-coordination at ZF2, the structure, intramolecular fluctuations and overall dynamics were analyzed from the trajectory where this loss was exhibited, as described in the following sections.
3.2 The Zn2+-coordinating histidine in ZF2 of TTP samples two conformations
Comparison of the trajectories collected for TTP and TIS11d reveals important differences in the structures of ZF2 in the two proteins. In particular, analysis of the dihedral angles χ1 (defined by the following atoms: N, Cα, Cβ, Cγ) and χ2 (defined by the following atoms: Cα, Cβ, Cγ and Nδ1) for the Zn2+-coordinating residues shows that in TTP the Zn2+-coordinating histidine (i.e. His4) in ZF2 (H166 in TTP) samples two different states characterized by different orientations of the aromatic side chain: χ2 = 180° and χ2 = 90° (see Fig. 2B). The His4 residues in ZF1 of TTP and in both ZFs of TIS11d mostly sample the conformation characterized by χ2 = 180°, and visit the second conformation, characterized by χ2 = 90°, very infrequently, with ≈ 1% probability (see Fig. 2A,C,D).
Figure 2.
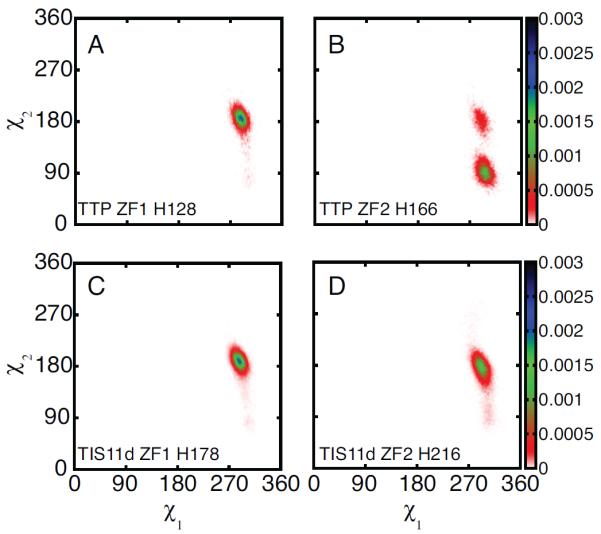
His4 of ZF2 populates two rotameric conformations in TTP but not in TIS11d. Probability density distribution of the dihedral angles χ1 and χ2 for the side chains of H128 (A) and H166 (B) of TTP and H178 (C) and H216 (D) of TIS11d. Data are taken from the 100 ns MD trajectory of TTP where loss of Zn2+-coordination was observed and from the six 100 ns MD trajectories of TIS11d. The color bars show the values of the probability density calculated for χ1 and χ2 as the number of counts normalized by the total number of observations and by the area of each bin.
Rotation of the imidazole ring of H166, Fig. 3A, leads to steric hindrance with the side chain of C162, the third Zn2+-coordinating residue in ZF2, Fig. 3B. As a consequence, C162 separates from both H166 and the Zn2+ cation, thereby disrupting proper Zn2+-coordination, Fig. 3C. Thus, one of the two orientations of the H166 side chain, χ2 = 90°, is incompatible with Zn2+-binding.
Figure 3.

Side chain rotation of H166 causes a steric clash with C162. The χ2 dihedral angle of H166 (A), van der Waals interaction energy between residues C162 and H166 (B) and SC162-Zn2+ distance (C) are shown as functions of time. Data are shown for the first half of the unfolding MD trajectory of TTP (100 ns in total).
3.3 The stacking of histidine and phenylalanine aromatic rings stabilizes Zn2+-binding in ZF2
The interaction of H166 with F150, in TTP, and of H216 with F200, in TIS11d, stabilizes the His4 side chain in the Zn2+-binding conformation, characterized by χ2 = 180°. For TIS11d, both ZF1 and ZF2 exhibit stacking of aromatic residues throughout our simulations. The aromatic side chain of His4 stacks with that of a conserved phenylalanine located in the α-helix, three residues after the first Zn2+-coordinating cysteine, Cys1+3 (see Fig. 4 and Fig. S7). For TTP however, while this aromatic stacking is consistently observed in ZF1 (see Fig. S7), it is observed in ZF2 only in the first part of the trajectory that precedes to the loss of Zn2+-coordination (see Fig. 4). When this stacking interaction between H166 and F150 is lost, H166 preferentially samples the rotameric configuration characterized by χ2 = 90° (Fig. 5), resulting in the loss of Zn2+-coordination from ZF2.
Figure 4.
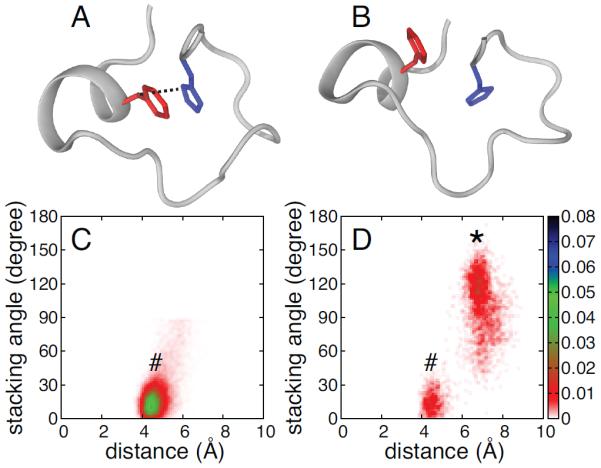
Aromatic side chain stacking of phenylalanine and histidine in ZF2. Representative ZF2 structures for TTP showing F150 (red) and H166 (blue) side chains stacked (A) and not stacked (B). Probability density distributions of the stacking angle and distance between the aromatic rings are shown for F200 and H216 in TIS11d (C) and for F150 and H166 in TTP (D). In the figure, conformations where the phenylalanine and histidine side chains are stacked (as shown in A) and not stacked (as shown in B) are represented by a # or a *, respectively. The stacking angle was calculated as the angle between the normals of the two aromatic rings (the planes for the side chains are defined by atoms Cδ2, Cε1 and Nε2 for histidine and Cζ, Cε2, Cδ2 for phenylalanine). The distance between the aromatic rings was calculated as the distance between the centers of mass for the heavy atoms of the two side chains. The color bars show the values of the probability density calculated for the stacking angle and distance as the number of counts normalized by the total number of observations and by the area of each bin. Configurations and distributions were extracted from the unfolding MD trajectory for TTP (100 ns) and from six 100 ns MD trajectories for TIS11d.
Figure 5.
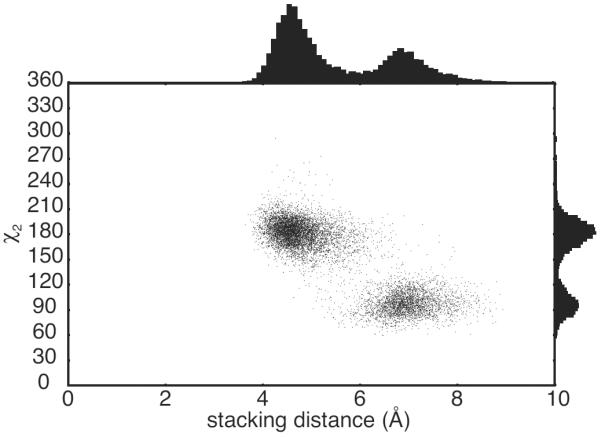
H166 in TTP populates the rotameric conformation with χ2 = 90° upon loss of stacking with F150. Scatter plot of the stacking distance between the aromatic ring of F150 and H166 of TTP and the χ2 dihedral angle of H166. Probability distributions of the stacking distance and χ2 dihedral are shown on the axes. The distance between the aromatic rings was calculated as the distance between the centers of mass for the heavy atoms of the two side chains. Data were extracted from the three MD trajectory for TTP.
3.4 The stacking interaction between histidine and phenylalanine occurs if the α-helix axis is bent
As described above, the phenylalanine residue that interacts with His4 is located in the α-helix that separates Cys1 and Cys2 in each zinc finger. Structural alignment of the α-helices of ZF2 of TIS11d and TTP reveals a difference in their conformations (see Fig. 6) that is also evident from the difference in the backbone dihedral angles of the three residues located in the C-terminal half of the α-helix (see Fig. S8). In TIS11d, the α-helix axis is bent due to the presence of a hydrogen bond between the side chain hydroxyl of T202 (the fifth residue in the α-helix, Cys1+5 position) and the backbone acyl group of R198 (Cys1+1 position). This hydrogen bond is present in all trajectories collected for TIS11d. The equivalent hydrogen bond in ZF1, between the Oγ of S165, Cys1+6, and the acyl oxygen of P161 in TIS11d and between the corresponding residues S115 and T111 in TTP, is always observed in all the trajectories collected for TIS11d and TTP. A hydrogen bond between the Oγ atom of a serine or threonine residue with the acyl oxygen of the i − 3 or i − 4 residue has been previously observed to induce a bend in the α-helix axis of ≈ 3 − 4°, particularly in transmembrane α-helices31, 32. Because of the lack of the corresponding hydrogen bond in TTP (between the side chain of Cys1+5 and the backbone of Cys1+1), the bend in the α-helix is not stabilized as it is in TIS11d. Fig. 6 shows the straightening of the α-helix axis occurring in TTP at ≈ 37 ns. This rearrangement of the α-helix results in a displacement of the phenylalanine side chain located at the middle of the helix, Cys1+3. In this new position, the phenylalanine side chain is not positioned to stack against the imidazole ring of H166 as well as the corresponding stacking interaction in ZF2 of TIS11d (see Figs. 4 and 6). This helix reorganization is the first step in the zinc finger destabilization that eventually leads to the loss of Zn2+-coordination in TTP.
Figure 6.

The hydrogen bond between T202 and R198 stabilizes a bend in the axis of the α-helix. Top: Structures of the C-terminal zinc finger of TTP (red) and TIS11d (cyan). The orientations of the side chain of F150 (TTP, red) and F200 (TIS11d, cyan) are shown on the left. The hydrogen bond between and OR198, depicted as black dashed line, is shown on the right. Oxygen atoms are depicted in red, nitrogen in blue and hydrogen in white. Bottom: The root-mean-square deviation of the backbone of the α-helix in TTP (residues 147 to 153) is shown as a function of time. The dashed line indicates the change in the α-helix conformation that causes a displacement of the F150 side chain to a position where it does not stack against H166. Data are shown for the first half of the unfolding MD trajectory of TTP (100 ns in total)
3.5 MD simulations of the TIS11d T202L mutant show decreased stacking between the side chains of H216 and F200
Comparison of the MD simulations of TTP and TIS11d highlighted the role of the hydrogen bond between the backbone acyl oxygen of R198 and the side chain of T202 of TIS11d in stabilizing the bend in the α-helix. This bent conformation of the α-helix places the aromatic ring of F200 in a position suitable for stacking against H216, thus maintaining H216 in the rotameric state that allows Zn2+-binding. To further assess this mechanism of stabilization of ZF2, we collected six 100 ns MD trajectories of a mutant of TIS11d where T202 was mutated into leucine, the equivalent residue found in TTP. Due to the absence of a hydroxyl group on the side chain of the leucine, this mutant cannot form the hydrogen bond. For this reason the α-helix located in ZF2 is not stabilized in a bent conformation, and the stacking interaction between F200 and H216 is not as stable as that in the wild type protein. This relative instability results in H216 sampling both rotameric side chain conformations, one that allows ZF2 to stably coordinate Zn2+ (χ2 = 180°) and one that does not (χ2 = 90°), as shown in Figs. S9 and S10. The conformation that is incompatible with Zn2+-binding is sampled by H216 with higher probability (9.6%) in the mutant than in the wild type protein (4.7%), due to the lack of a stacking interaction with F200 (Figs. S9 and S10).
3.6 Mutation of threonine 202 to leucine is sufficient to destabilize the structure of ZF2 in TIS11d
Our simulations of TTP and TIS11d indicate that the structural difference observed between ZF2 of TTP and that of TIS11d arises from the different amino acid compositions of the α-helix, in agreement with reported experimental observations14. In particular, our simulations reveal that the conformation of the α-helix is different in the two proteins. In TIS11d, the hydrogen bond between the side chain of T202 and the backbone of R198 causes a bend in the α-helix axis, thereby positioning the aromatic ring of F200 to stack against the imidazole ring of H216. Stacking of the aromatic side chains of F200 and H216 stabilizes H216 in the rotameric conformation (χ2 = 180°) that stabilizes Zn2+-binding in ZF2 (see Fig. 3). In TTP, the homologous residue to T202 in TIS11d is L152, whose side chain cannot form a hydrogen bond with the backbone of residue 148. For this reason, the α-helix axis is not bent in TTP as it is in TIS11d, resulting in a lower probability for the F150 side chain of stacking against that of H166 (compare Figs. 4 A and B). Thus, F150 does not stabilize the H166 side chain in the conformation with χ2 = 180° (Fig. 5), that maintains Zn2+-coordination in ZF2 (Fig. 3).
To validate the role of T202 in stabilizing the structure of ZF2 based on our MD results, we mutated threonine 202 to leucine in TIS11d and used NMR and CD spectroscopy to characterize the structure of the mutant protein. We found that in T202L mutant of TIS11d, ZF2 is less structured than in the wild type protein, Figs. 7 A and B. Cross-peaks from ZF1 and ZF2 are present in the 15N-1H HSQC spectrum of the T202L mutant TIS11d, however cross-peaks corresponding to the residues in the linker and in the α-helix of ZF2 are broadened beyond detection, Fig. 7A. Cross-peaks from ZF2 show the largest chemical shift differences from the wild type and have lower intensities than cross-peaks from ZF1, Fig. S11. These results suggest that ZF2 is more flexible and that the structure of ZF2 is affected by the single point mutation. The CD spectrum of the RNA-binding domain of TIS11d T202L indicates that this protein is less structured than the wild type TIS11d, and more similar to TTP, Fig. 7B.
Figure 7.

Structural studies of T202L and F200A mutant of TIS11d. 15N-1H HSQC spectra (A) of TIS11d wild type (black), T202L (blue) and F200A (red) mutants of TIS11d. On top, a schematic representation of the RNA-binding domain depicts the ZFs as rectangles and the linker region as a line. The circles indicate residues along the primary sequence with a cross-peak in the 15N-1H HSQC spectrum. Black box indicates the α-helix in ZF2. The F200A mutant protein is less stable than the wild type protein as indicated by the presence of degradation peaks in the region between 7.8 and 8.2 ppm in the 1H dimension and 125 and 130 ppm in the 15N dimension. Circular dichroism spectra (B) of TIS11d wild type (black), T202L (blue) and F200A (red) mutants of TIS11d and TTP (gray).
Zn2+ titrations of TIS11d T202L monitored by NMR spectroscopy show that only ZF1 stably binds the metal cation. The titration endpoint occurred at a [Zn2+]/[TIS11d T202L] ratio of 1:1, and further addition of Zn2+ resulted in no changes in the cross-peaks intensity or in their position, Figs. S12 and S13. Cross peaks from ZF2 have low signal-to-noise ratios, indicating that a small fraction of ZF2 is folded, and that ZF2 binds Zn2+ with lower affinity than ZF1.
In agreement with what is observed for TTP, addition of RNA stabilizes the structure of ZF2 in TIS11d T202L, as indicated by the presence of cross-peaks from ZF2 in the 15N-1H HSQC spectrum of the T202L mutant TIS11d that were missing in the free state, Fig. S14. All together these results indicate that mutation of threonine 202 to leucine decreases the affinity of ZF2 for Zn2+ and destabilizes its structure.
3.7 Mutation of leucine 152 to threonine is sufficient to stabilize the structure of ZF2 in TTP
Following the same rationale discussed in the previous section that leads to the mutation of threonine 202 to leucine in TIS11d, we made the equivalent mutation to TTP, L152T. The introduction of a threonine in the middle of the α-helix allows for the formation of a hydrogen bond with the acyl oxygen of the forth preceding residue, as observed in TIS11d. Such a hydrogen bond introduces a bend in the α-helix that supports the stacking of phenylalanine located on the opposite side of the helix with the Zn2+-coordinating histidine. For this reason, we expected ZF2 of this mutant to have higher affinity for Zn2+ than the wild type protein. As shown by NMR and CD spectroscopy, this mutant stably binds Zn2+ with both zinc fingers (Fig. 8). The 15N-1H HSQC spectrum of TTP L152T contains cross-peaks from both ZF1 and ZF2 (Fig. 8A) and the CD spectrum closely resembles that of TIS11d (Fig. 8B). Combined these results indicate that both zinc fingers are properly folded in this mutant protein.
Figure 8.

Structural studies of L152T mutant of TTP. 15N-1H HSQC spectra (A) of TTP (gray) and L152T mutant of TTP (magenta). Circular dichroism spectra (B) of TIS11d wild type (black) and TTP wild type (gray) and L152T mutant (magenta).
Taken together TIS11d T202L mutant and the corresponding TTP L152T mutant confirm the role of the intra-helical hydrogen bond formed by the side chain of the threonine in stabilizing the π − π interaction, essential for the stability of ZF2.
3.8 ZF2 is unfolded in the F200A mutant of TIS11d
To validate the importance in TIS11d of the aromatic stacking interaction between F200, located in the middle of the α-helix, and H216 in stabilizing the structure of the ZF2, we mutated F200 to alanine. The 15N-1H HSQC spectrum of the F200A mutant TIS11d is missing all cross-peaks from ZF2, indicating that ZF2 samples multiple states and, as a consequence, cross-peaks are broadened beyond detection (see Fig. 7A). Cross-peaks from ZF2 are observed in the 15N-1H HSQC spectrum of wild type TIS11d, where both ZFs are folded, but are missing in the 15N-1H HSQC spectrum of TTP, where only ZF1 is folded. Missing cross-peaks from ZF2 are an indication of lack of ZF2 structure. To prove it, we compared the spectrum of F200A with that of a mutant protein of TIS11d where ZF2 is known to be unfolded, C212S14 (Fig. S15). In TIS11d C212S, Cys3 in ZF2, is mutated to serine thus, by removal of one of the essential Zn2+-coordinating residues, zinc binding is abrogated and the structure of ZF2 destabilized14.
We used CD spectroscopy to characterize the structure of the mutant protein. Fig. 7B shows that the RNA-binding domain of F200A is less structured than that of wild type TIS11d, and more similar to that of TTP. The shift in minimum of the CD spectrum is due to the loss of the aromatic interactions in the F200A mutant. These experimental data demonstrate that F200 is essential in stabilizing the structure of ZF2. It is important to note that F200 is located on the protein surface, it is partially solvent exposed and there are no hydrophobic side chains within a 5 Å radius (see Fig. S16). The destabilization of ZF2 observed in the F200A mutant, therefore, is not due to the disruption of a hydrophobic core within the protein but only to the disruption of the aromatic stacking with H216.
While RNA-binding by TTP is sufficient to stabilize the structure of ZF2 and bind a Zn2+ cation14, RNA-binding is not sufficient to stabilize the structure of ZF2 in the F200A mutant of TIS11d. The 15N-1H HSQC spectrum of F200A shows minor changes upon addition of RNA: small shifts are present in the position of the peaks from ZF1 and no additional peaks appear from ZF2 residues (see Fig. S17). The addition of RNA, therefore, does not stabilize the structure of ZF2 and this mutant protein does not bind RNA with high affinity, Kd < 1 μM, as confirmed by a fluorescence polarization binding assay. These data show that a phenylalanine located three residues after the first Zn2+-coordinating cysteine (i.e. Cys1), in the middle of the α-helix in TIS11d, is essential for stabilizing the structure of the zinc finger and for increasing the binding affinity of the zinc finger for Zn2+.
3.9 Sequence alignment
Analysis of the primary sequences of 14,851 zinc finger domains from the tandem zinc finger CCCH family obtained from PFAM (PF00642)33 shows that the aromatic character of third residue after the first Zn2+-coordinating cysteine (Cys1+3) is highly conserved (see Fig. 9A). In addition, we have shown that a bend in the α-helix axis facilitates the stacking of the phenylalanine at the Cys1+3 position with His4. This bend is stabilized by the formation of a hydrogen bond between the Oγ of either a threonine or a serine residue, located either at Cys1+5 or Cys1+6, and the acyl oxygen of the preceding fourth residue. Primary sequence analysis of the tandem zinc finger CCCH family32 shows that a residue containing an Oγ, although not fully conserved, is present with high frequency at either Cys1+5 or Cys1+6 (see Fig. 9B), with threonine being the most abundant amino acid (46%). A charged or polar residue is often observed at either of these positions with Arg, Lys and Glu having the highest probabilities after threonine. The probability of finding an Asp is much lower than that of Glu, suggesting that the side chain length of the charged amino acid is important.
Figure 9.

Sequence analysis of the tandem zinc finger CCCH family (PFAM ID: PF00642). A: Probability of finding an aromatic or not aromatic residue at position Cys1+3. B: Probability of finding a given amino acid at position Cys1+5 or Cys1+6.
Primary sequence analysis indicates that the stacking of an aromatic side chain with the Zn2+-coordinating histidine is the strategy adopted by a large majority (94.2%) of CCCH-type zinc finger domains to coordinate Zn2+ with high affinity (see Fig. 9A). Stabilization of the stacking interactions of the aromatic side chains, however, is likely achieved in different ways by different zinc fingers in this family. We have shown that in TIS11d, both ZF1 and ZF2 use an intra-helical hydrogen bond to stabilize the stacking interactions between the phenylalanine in the middle of the α-helix and His4. In TTP, however, only ZF1 adopts this strategy to stabilize its structure, while ZF2 binds Zn2+ with low affinity in the absence of RNA and is partially unstructured in solution. Our previous studies indicate that TTP has recently evolved to modulate its activity through its folded stability14. The CCCH-type tandem zinc finger domain contains few secondary structural elements and thus has a relatively low thermodynamic stability. A single point mutation, therefore, can easily destabilize the fold and shift the equilibrium toward a disordered zinc finger state. For this reason, we expect proteins from this family to have evolved to use an unfolded-to-folded transition to regulate their activity in the cell.
4 Conclusions
In this work, we investigated the interactions occurring in the RNA-binding domains of TTP and TIS11d that stabilize their coordination of zinc ions. Using molecular dynamics we were able to observe the loss of structure of the C-terminal zinc finger of TTP and characterize the events that underlie this loss. We found that, in the C-terminal zinc finger of TTP, the zinc coordinating histidine, H166, populates two rotameric states. The rotamers correspond to the χ2 angle of the side chain centered at 180° or at 90°, respectively: the first conformation allows the correct tetrahedral geometry between the three cysteines, the histidine and the zinc ion; the latter causes a steric clash between the side chain of H166 and C162 that results in the disruption of the zinc binding site. We found that when the rotameric state of H166 has χ2 = 180°, a π − π interaction is present between the side chains of H166 and F150; when χ2 = 90°, no such interaction is present. When the stacking between the two aromatic moieties is formed, the side chain of histidine residue is kept in the χ2 = 180° conformation. In TTP, this stacking interaction is only marginally stable, however.
In TIS11d, the rotamer of H216 with χ2 = 180° is stabilized by the π − π interaction with the side chain of F200. As in TTP, the phenylalanine is in the center of the short α-helix spanning six residues and starting at the first cysteine. An intra-helix hydrogen bond between the hydroxyl group in the side chain of T202 and the acyl oxygen of R198 restrains the α-helix in a conformation that allows the side chain of F200 to stack against the imidazole ring of H216. Although TTP and TIS11d share the majority of their primary sequence in the RNA-binding domain, the residues that form the α-helix are not conserved. The residue corresponding to T202 in TIS11d is L152 in TTP. Leucine side chains are unable to form hydrogen bonds and hence in TTP the phenylalanine is not kept in proximity of the imidazole ring of H156 in a conformation that can stack against the side chain of H156. MD simulations of TIS11d T202L mutant support the importance of this hydrogen bond in stabilizing the Zn2+-coordination: in the mutant protein, which cannot form the hydrogen bond, the H216 rotamer with χ2 = 90° is more populated than in the wild type TIS11d.
The mechanism of stabilization of ZF2 that we proposed based on the MD simulations was experimentally validated using mutagenesis. First, we constructed two mutant proteins, TIS11d T202L and TTP L152T, to probe the role of the hydrogen bond in stabilizing the π − π interaction observed between the side chains of the Zn2+-coordinating histidine and the conserved phenilalanine in the middle of the α-helix. We have shown that a single point mutation of threonine 202 to leucine in TIS11d is sufficient to decrease the affinity for Zn2+ of the C-terminal zinc finger and destabilize the structure, while the corresponding mutation of leucine 152 to threonine in TTP is sufficient to increase zinc binding affinity of ZF2. Second, we mutated the conserved phenylalanine 200 to alanine in TIS11d, to verify that its stacking against the Zn2+-coordinating histidine is essential for the folding of ZF2. Indeed, this mutant is partially unstructured and ZF2 is unable to fold even upon addition of RNA. These results unequivocally verify the critical residues and interactions identified using MD that are necessary to increase Zn2+-binding affinity and stabilize the fold of ZF2.
The sequence alignment of 14,851 CCCH-type zinc finger domains shows that the residue three positions away from the first cysteine (i.e. Cys1+3) is likely to be aromatic (Phe, Trp, Tyr or His) with a probability > 94%. In addition, experimental studies of the F200A mutant of TIS11d confirm that, in the absence of this aromatic residue, the C-terminal zinc finger is unable to stably coordinate Zn2+. These results suggest that most CCCH-type zinc finger proteins employ π − π interactions to stabilize the Zn2+-coordinating histidine in a rotameric state that is compatible with the tetrahedral geometry of the Zn2+-binding site. The strategy adopted to maintain the stacking of the aromatic side chain with the Zn2+-coordinating histidine is not conserved, however. Previous studies had shown that the extent of disorder of the RNA-binding domain affects the activity of the protein in the cell14. Of the 14,851 CCCH-type zinc finger sequences that we examined, roughly half of them support the formation of hydrogen bonds using the Oγ from residues located either at Cys1+5 or Cys1+6: the remaining zinc fingers may use alternative mechanisms to stabilize the coordination of Zn2+. Through this apparent variety of Zn2+-coordination stabilizing mechanisms, evolution can modulate the thermodynamic stability for this class of zinc fingers and, ultimately, regulate their biological activity.
Supplementary Material
Acknowledgement
The authors thank Asli Ertekin for helpful discussions and C. Robert Matthews for sharing equipment.
Funding Sources This work was supported by National Institutes of Health Grant GM098763.
Footnotes
Author Contributions DT, LMD, TWW and FM designed the experiments and simulations. DT carried out the MD simulations, analysis of the data and prepared and performed all the NMR and CD experiments of the TIS11d T202L and TTP L152T mutants. LMD prepared the samples and performed all the NMR and CD experiments of TTP and TIS11d wild type, F200A and C212S. TWW performed the DFT calculations. DT, TWW and FM wrote the paper.
Supporting Information Available A figure showing the sequence alignment of the RNA-binding domains of TIS11d and TTP, with the structure of TIS11d and TTP. A Figure comparing the charges from the CHARMM27 force field with those from the polarizable charge-transfer model of Sakharov and Lim along with the natural bond orbital (NBO) charges corresponding to a series of electronic structure calculations. A table showing the average values of the RMSD for the protein and ZF1 and ZF2 of TIS11d and TTP. A figure showing the RMSD as a function of time for TIS11d and its ZF1 and ZF2. A figure showing the Zn2+-coordinating angles of TIS11d ZF1 and ZF2 and TTP ZF1 as a function of time. Two figures showing the Zn2+-coordinating angles and the stacking distance between F150 and H166 of TTP as a function of time for the two trajectories of TTP where Zn2+-coordination is not lost. A figure showing the probability distribution for the stacking angle and distance of histidine and phenylalanine in ZF1 of TTP and TIS11d. A figure showing the distribution of ϕ and ψ angles for residues 151, 152 and 153 of TTP and 201, 202 and 203 of TIS11d. A figure showing the probability density distribution of χ2 dihedral angle of His4 and the conserved phenylalanine for the ZFs of TIS11d wild type and T202L mutant. A figure showing the χ2 dihedral angle of H216 of TIS11d T202L mutant as a function of time and as scatter plot versus the stacking distance with F200. A figure showing the cross-peak shifts in the 15N-1H HSQC spectrum of TIS11d T202L relative to TIS11d wild type. A figure showing the 15N-1H HSQC spectra corresponding to the zinc titration of TIS11d T202L. A figure showing the cross-peak intensities of 15N-1H HSQC spectrum of TIS11d T202L as a function of zinc in solution. A figure showing the 15N-1H HSQC spectra of TIS11d T202L free and bound to 5′-UUUAUUUAUUUU-3′. A figure showing the 15N-1H HSQC spectra of TIS11d F200A and TIS11d C212S. A figure showing the solution structure of TIS11d and the location of hydrophobic side chains and F200, H216. A figure showing the 15N-1H HSQC spectra of TIS11d and F200A mutant of TIS11d free and bound to 5′-UUUAUUUAUUUU-3′.
This material is available free of charge via the Internet at http://pubs.acs.org/.
References
- (1).Blackshear PJ. Biochem. Soc. Trans. 2002;30:945–952. doi: 10.1042/bst0300945. [DOI] [PubMed] [Google Scholar]
- (2).Carrick DM, Lai WS, Blackshear PJ. Arthritis Res. Ther. 2004;6:248–264. doi: 10.1186/ar1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Carballo E, Lai WS, Blackshear PJ. Science. 1998;281:1001–1005. doi: 10.1126/science.281.5379.1001. [DOI] [PubMed] [Google Scholar]
- (4).Ogilvie RL, Abelson M, Hau HH, Vlasova I, Bohjanen PR, Blackshear PJ. J. Immunol. 2005;174:953–961. doi: 10.4049/jimmunol.174.2.953. [DOI] [PubMed] [Google Scholar]
- (5).Lai WS, Carballo E, Strum JR, Kennington EA, Phillips RS, Blackshear PJ. Molecular and Cellular Biology. 1999;19:4311–4323. doi: 10.1128/mcb.19.6.4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Chen CY, Gherzi R, Ong SE, Chan EL, Raijmakers R, Pruijn GJ, Stoecklin G, Moroni C, Mann M, Karin M. Cell. 2001;107:451–464. doi: 10.1016/s0092-8674(01)00578-5. [DOI] [PubMed] [Google Scholar]
- (7).Lai WS, Kennington EA, Blackshear PJ. J. Biol. Chem. 2002;277:9606–9613. doi: 10.1074/jbc.M110395200. [DOI] [PubMed] [Google Scholar]
- (8).Lai WS, Carballo E, Thorn JM, Kennington EA, Blackshear PJ. J. Biol. Chem. 2000;275:17827–17837. doi: 10.1074/jbc.M001696200. [DOI] [PubMed] [Google Scholar]
- (9).Hudson BP, Martinez-Yamout MA, Dyson HJ, Wright PE. Nat Struct Mol Biol. 2004;11:257–264. doi: 10.1038/nsmb738. [DOI] [PubMed] [Google Scholar]
- (10).Morgan BR, Massi F. Protein Sci. 2010;19:1222–1234. doi: 10.1002/pro.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Morgan BR, Deveau LM, Massi F. Biophys. J. 2015;108:1503–1515. doi: 10.1016/j.bpj.2015.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Camilloni C, De Simone A, Vranken WF, Vendruscolo M. Biochemistry. 2012;51:2224–2231. doi: 10.1021/bi3001825. [DOI] [PubMed] [Google Scholar]
- (13).Cornilescu G, Delaglio F, Bax A. J. Biomol. NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- (14).Deveau LM, Massi F. ACS Chem. Biol. 2016 doi: 10.1021/acschembio.5b00639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Brewer BY, Ballin JD, Fialcowitz-White EJ, Blackshear PJ, Wilson GM. Biochemistry. 2006;45:13807–13817. doi: 10.1021/bi061320j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Blackshear PJ, Lai WS, Kennington EA, Brewer G, Wilson GM, Guan X, Zhou P. J. Biol. Chem. 2003;278:19947–19955. doi: 10.1074/jbc.M301290200. [DOI] [PubMed] [Google Scholar]
- (17).Bordoli L, Kiefer F, Arnold K, Benkert P, Battey J, Schwede T. Nat Protoc. 2009;4:1–13. doi: 10.1038/nprot.2008.197. [DOI] [PubMed] [Google Scholar]
- (18).Arnold K, Bordoli L, Kopp J, Schwede T. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- (19).Schwede T, Kopp J, Guex N, Peitsch MC. Nucleic Acids Res. 2003;31:3381–3385. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Humphrey W, Dalke A, Schulten K. Journal of Molecular Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- (21).Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, Schulten K. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, Karplus M. J Phys Chem B J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- (23).Sakharov DV, Lim C. J. Am. Chem. Soc. 2004;127:4921–4929. doi: 10.1021/ja0429115. [DOI] [PubMed] [Google Scholar]
- (24).Ryckaert JP, Ciccotti G, Berendsen HJC. J. Comput. Phys. 1997;23:327–341. [Google Scholar]
- (25).Darden T, York DM, Pedersen LG. J. Chem. Phys. 1993;96:10089–10092. [Google Scholar]
- (26).Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. J. Chem. Phys. 1995;103:8577–8593. [Google Scholar]
- (27).Frishman D, Argos P. Proteins: structure, function and genetics. 1995;23:566–579. doi: 10.1002/prot.340230412. [DOI] [PubMed] [Google Scholar]
- (28).Stone J. M.Sc. thesis. Computer Science Department, University of Missouri-Rolla; 1998. An Efficient Library for Parallel Ray Tracing and Animation. [Google Scholar]
- (29).Goddard TD, Kneller DG. University of California; San Francisco: [Google Scholar]
- (30).Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- (31).Ballesteros JA, Deupi X, Olivella M, Haaksma E. Biophys. J. 2000;79:2754–2760. doi: 10.1016/S0006-3495(00)76514-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Gray TM, Matthews BW. J. Mol. Biol. 1984;175:75–81. doi: 10.1016/0022-2836(84)90446-7. [DOI] [PubMed] [Google Scholar]
- (33).Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, Sonnhammer ELL, Tate J, Punta M. Nucleic Acids Research. 2014;42:D222–30. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.