


Abstract
In vivo single molecule tracking has recently developed into a powerful technique for measuring and understanding the transient interactions of transcription factors (TF) with their chromatin response elements. However, this method still lacks a solid foundation for distinguishing between specific and non-specific interactions. To address this issue, we took advantage of the power of molecular genetics of yeast. Yeast TF Ace1p has only five specific sites in the genome and thus serves as a benchmark to distinguish specific from non-specific binding. Here, we show that the estimated residence time of the short-residence molecules is essentially the same for Hht1p, Ace1p and Hsf1p, equaling 0.12–0.32 s. These three DNA-binding proteins are very different in their structure, function and intracellular concentration. This suggests that (i) short-residence molecules are bound to DNA non-specifically, and (ii) that non-specific binding shares common characteristics between vastly different DNA-bound proteins and thus may have a common underlying mechanism. We develop new and robust procedure for evaluation of adverse effects of labeling, and new quantitative analysis procedures that significantly improve residence time measurements by accounting for fluorophore blinking. Our results provide a framework for the reliable performance and analysis of single molecule TF experiments in yeast.
INTRODUCTION
Binding of certain transcription factors (TF) to their target DNA sequences is highly dynamic, with residence time on the scale of seconds, first observed in mammalian cells by McNally et al. (1) and in yeast by Karpova et al. (2), reviewed in (3). The role that this rapid exchange plays in transcription remains hotly debated. Is this noise, or is it transcriptionally productive? Estimates of TF binding to specific sites may clarify the function of this fast exchange and also provide a deeper understanding of the molecular mechanisms of transcription initiation. Morisaki et al. (4) applied single molecule tracking (SMT) to two mammalian TFs, p53 and glucocorticoid receptor, and demonstrated that TF molecules that participate in transcription bind transiently to polymerase foci. This important observation supports the hypothesis that transient binding is transcriptionally productive.
SMT provides direct information about molecular dynamics (5). Other microscopy methods, such as fluorescence recovery after photobleaching (FRAP) and fluorescence correlation spectroscopy (FCS), are based on population analysis and mathematical modeling that adds additional difficulties in data analysis. In addition, FRAP and FCS are difficult to implement on small specific targets such as transcription factories or gene loci. Thus, the powerful technique of SMT is the method of choice for studies of transcription factor dynamics. However, this method still lacks a solid foundation for distinguishing between specific and non-specific interactions of transcription factors with chromatin. SMT provides a set of residence times for a given protein, but then criteria have to be defined to decide which residence times reflect specific or non-specific binding, where specific binding refers to binding of the transcription factor at specific response elements and non-specific binding refers to binding at all other DNA sites (6). Distinguishing between those two types of binding requires a benchmark for non-specific binding.
Development of this benchmark requires proteins that are conditionally incapable of DNA binding while preserving non-specific DNA binding. Such experiments are easier to perform in Saccharomyces cerevisiae, a unicellular eukaryotic model system. In this paper we took advantage of the well-studied yeast transcription factor Ace1p. With Ace1p we can distinguish specific from non-specific binding because Ace1p has only five specific sites in the genome (7); moreover, Ace1p may bind to these sites only upon associating with Cu2+ (8). Thus, in the absence of copper Ace1p should only bind to DNA non-specifically and therefore Ace1p provides a perfect benchmark for non-specific binding.
To track TF in yeast we had to overcome two specific technical problems. A first key technical problem is a lack of robust procedures to determine if the labeling strategies used to mark the transcription factor are innocuous and therefore to ensure that SMT estimates are correct. The current state of the art in SMT is to use organic dyes that enter cells and then bind covalently to genetically encoded HaloTag- or SNAP proteins (9,10). These bright and stable organic dyes enable SMT for extended periods (11) and were extensively used in mammalian cells. However, labeling of yeast cells is problematic due to the low retention of the dyes, and thus no SMT experiments were yet reported for yeast. Organic dyes have been introduced into yeast for the staining of cellular structures but not for SMT by either electroporation (12) or triple disruption of transporters (13,14). It was reported that triple disruption of ATP-binding cassette multidrug resistance (ABC-MDR) transporters encoded by PDR5, SNQ2 and YOR1 (15) substantially reduces the cell viability. Electroporation is also a harsh treatment that may lead to unintended physiological effects. Currently there are no tests sensitive enough to assay subtle effects of the labeling procedure on transcription. Therefore, it is important to develop such assays and to apply them to development of benign procedure for organic dye incorporation into yeast cells. The second technical problem is to the small size of the yeast nucleus, which is ∼10-fold smaller in diameter than mammalian cells, and therefore the imaged area is ∼100-fold smaller. The significantly smaller volume reduces the total number of easily separable tracks that can be obtained from a single yeast nucleus.
To weed out potential artifacts of different organic dye incorporation procedures we developed a biological control based on Ace1p. After treatment with CuSO4, Ace1p undergoes a slow cycle of binding that peaks at 5–10 min after copper addition (2). Yeast strains carry 10–15X tandem copies of CUP1 (16). This clustering of multiple Ace1p-3xGFP molecules in the tandem array amplifies the fluorescent signal and allows microscopic observation of TF binding. We tested whether different organic dye incorporation procedures perturb the Ace1p slow cycle in two different ways: first, by checking for binding of Ace1p to CUP1 promoters in the absence of copper, and second by monitoring the dynamics of the slow cycle of copper-induced TF binding. We found that electroporation procedure in yeast is detrimental to slow cycle, indicating that this commonly used procedure is problematic and cannot be applied to studies of yeast transcription. We overcame this problem by disrupting only one ABC-MDR transporter PDR5. We show here that disruption of this transporter allows organic dyes to be retained long enough to form covalent links to HaloTag. Importantly, we also show that cells of this single mutant (pdr5Δ) exhibit completely normal binding of Ace1p to the promoters of CUP1. To address the yeast nuclear size limitations, we optimized label density and track visualization in yeast cells. Finally, we developed a new computational correction procedure that can account for the reversible and irreversible photobleaching, which otherwise leads to an artifactual reduction in the length of some tracks and therefore to underestimation of residence time.
In this paper we have characterized non-specific binding of Ace1p and two other DNA-binding proteins, histone H3 (HHT1) and heat shock factor (HSF1). Estimates for non-specific residence times of these very different DNA-binding proteins are surprisingly similar. This result helps to define a characteristic residence time for non-specific binding, and also suggests that those non-specific interactions occur in very similar ways perhaps reflecting predominantly simple electrostatic interactions. In sum, our results provide a framework for the reliable performance and analysis of SMT of transcription factors in yeast.
MATERIALS AND METHODS
Strains and plasmids, media and growth conditions
Saccharomyces cerevisiae strains of this study listed in Supplementary Table S1 are derivatives of the haploid strains isogenic to S288C: BY4742, BY4741 from the Saccharomyces deletion project (Research Genetics/Invitrogen, Huntsville, AL, USA). Standard methods were used for yeast growth, yeast transformation and genomic DNA isolation (17) (See Supplementary data for details).
Fluorescent labeling of HaloTag fusion proteins
A total of 5 mM stock solutions for HaloTag ligands were prepared in DMSO. The electroporation procedure for labeling of HaloTag proteins in yeast has been described in (12). A Genepulser (BioRad, Hercules, CA, USA) was used at 1000 V, 800 Ω, 25 μF settings for the electroporation. Cells were imaged either 5 min later or after 2 h recovery in CSM at 30°C. To label HaloTag fusion proteins (Hht1p or Ace1p or Hsf1p) in cell culture, strains were grown overnight in 3 ml CSM (or CSM+SC, as stated for appropriate experiments) to saturation in 14 ml polypropylene round-bottom tubes (Falcon, Mexico C.P., Cat. No.: 352059). Fifty microliters of this culture was inoculated into 3 ml of fresh medium and grown for another 4 h. Cells were pelleted and resuspended in the same tube in 1 ml of fresh medium with appropriate concentration of the HaloTag ligand. Cells were grown further for another 30 min at 30°C. Cells were washed twice with 3 ml of the growth medium and resuspended in 25 μl of CSM for imaging. We used lower concentration of HaloTag ligands for SMT and higher concentration of HaloTag ligands for nuclear staining (Supplementary Table S3). To stain the nuclei, DAPI was added to the live culture at 25 μg/ml from 5 mg/ml stock in DMSO. For the successful DAPI staining of live yeast cells concentration of DMSO had to be maintained at 1%. TMR staining and DAPI staining were performed simultaneously for 30 min.
Epifluorescence and HILO microscopy
Cells were concentrated and mounted for imaging as described in (2). In short, 5 μl of the concentrated yeast cell suspension was placed into LabTek II chamber (Nalge Nunc Intl., Rochester, NY, Cat. No.: 155379) and subsequently covered by a 10 mm × 10 mm CSM or CSM+SC agarose slab. For the induction of the CUP1 arrays, 100 μM CuSO4 was added to agarose slabs. For epifluorescence microscopy, the DeltaVision Elite microscopy system (GE Healthcare Bio-Sciences, Pittsburgh, PA, USA) with 100X, 1.4 NA, oil-immersion objective (UPLSAPO100XO, Olympus Scientific Solutions, Waltham, MA, USA) was used. To observe the difference in staining efficiency between pdr5Δ Hht1p-HaloTag and PDR5 Hht1p-HaloTag strains, live cells were stained with 10–15 nM TMR and DAPI as described above; then washed twice with 0.1 M KPi buffer (pH 7) and mounted on the LabTek II chamber with KPi agarose pads for imaging. Single focal plane (SFP) images were acquired in TMR and DAPI channels, TMR images were deconvolved. Particles were counted from 83–105 nuclei. To visualize dynamics of Ace1p-3xGFP at the CUP1 array (Figure 4A, B and C), 11 z-sections with 0.4 μm step size were acquired with 4 min time lapse for 48 min total. These images were deconvolved and projected onto a single plane using SoftWoRx software (GE Healthcare Bio-Sciences, Pittsburgh, PA, USA). Cells containing visible arrays were counted from these projections. For SMT, SFP time lapse movies were acquired on a custom-built Highly Inclined Laminated Optical (HILO) sheet, (18)) imaging system (see Supplementary Methods for details). All the lasers were set at 2 mW output power, and images were acquired as fast as possible with constant laser exposure. Image size was cropped to 256 × 256 pixels, which resulted in a camera readout time of ∼15 ms, corresponding to a frame rate of 66.7 Hz (frames per second), with total time acquisition of 4.5 s per 300 frames. To avoid loss of temporal resolution in two-color experiments of single molecules and saturated nuclei, the entire image sequence in the single molecule channel (647 nm) was collected first, after which the nuclear images were collected (561 nm). The 561 nm laser line was used to illuminate TMR, JF549 and JF585 whereas the 647 nm laser line was used to illuminate JF635 and JF646. In heat-shock experiments, cells were mounted onto pre-warmed (39°C) Lab-Tek II coverglass chambers and covered by pre-warmed (39°C) CSM+SC agarose slabs. The stage of the HILO microscope was preheated to 37°C using a stage-top incubation system (H501-NG, Okolab USA, Inc., Burlingame, CA, USA). For all samples, imaging from the same agarose pad was limited to the first 15 min to obtain consistent results and to record the molecular movements under active metabolic conditions.
Figure 4.

Development of correction procedures for the blinking in tracking data. (A and B) Extracted distributions of unbound, short- and long-lived binding events for simulations of molecules without blinking, and (A) without, or (B) with photobleaching. (C–F) Extracted distributions of unbound, short- and long-lived binding events for simulation with blinking and photobleaching where tracking is performed by filling in gaps with a maximum length of (C) 1 frame, (D) 4 frames, (E) 13 frames and (F) 37 frames. For all distributions, the average residence times of the short and long binding events are included next to the appropriate slice with the 95% confidence interval. The 1-frame and 4-frame gap setting data were adequately fit with a single-component exponential, while the remaining data sets required a two-component exponential based on the results of an F-test comparing the 2 fits. (G) Cumulative distribution of gap lengths for simulations (black, solid), Hht1p-HaloTag (blue), Ace1p-HaloTag (red) and Hsf1p-HaloTag (yellow). The dashed line at a fraction of 0.75 is the cut-off value used to determine the number of frames within a gap to fill for each tracked protein. (H) Comparison of gap length distributions for Hht1p-HaloTag in fixed (black) and live (red) cells. The longer gaps present in live cells are believed to be due to dissociation/re-binding events.
SMT analysis
For each experimental condition, we obtained ∼250 tracks which were sufficient to generate a reasonably smooth cumulative histogram. We tracked the single molecules and analyzed the data with TrackRecord software (19) based on MATLAB (freely available at https://sourceforge.net/projects/single-molecule-tracking). TrackRecord identifies particle positions based on their peak intensities and fitting to a 2D Gaussian function. Tracks are generated using a nearest-neighbors approach that we prefer over other algorithms due to its use of parameters that are more readily interpretable by biologists and to its low computational demands. This software permits us to assay (i) the fraction of molecules that are freely diffusing and those that are immobilized due to binding (ii) to discern long-residence molecules within the fraction of immobile molecules (iii) to calculate residence times for long-residence and short-residence binding events. We introduced the following improvements to this software: (i) the ability to select multiple regions if interest (ROI) that allows tracking in only the nuclei of all cells located in the field of view; (ii) additional noise removal by Wiener and morphological top-hat filters to reduce speckle noise and uneven illumination, respectively; (iii) display and use of particle kymographs to simplify the manual correction of erroneous automatically generated tracks. We used kymographs to manually bridge the gaps generated by blinking of the ligands.
To extract binding times, the cumulative distribution of residence times (‘survival plot’) is fit with least-squares to both a one- and two-component exponential decay. Because of the chromatin motion, it is required to set a threshold on the allowable motion for a bound particle, called rmax. Tracking performed on chromatin-bound nucleosomal histone showed that 99% of single molecules of Hht1p had a frame-to-frame displacement of <0.22 μm at 66.7 Hz frame rate. It is also important to specify a minimum number of frames, Nmin that a particle must jump less than rmax in order to remove slowly diffusing free molecules that contaminate the survival plot. The probability that a free molecule will be considered bound depends on its diffusion coefficient, D, the frame rate, Δt and the values of rmax and Nmin (5).
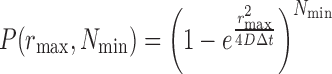 |
To achieve P(rmax,Nmin) < 0.01 for free molecules with a diffusion coefficient of 0.5 μm2/s, and rmax = 0.22 μm, Nmin is set to 21. A statistical F-test between the two fits was used to determine whether the two-component exponential significantly improves the fitting.
Simulations of photobleaching and blinking
Simulated images of single particles were generated using custom software developed in MATLAB. Three-dimensional positions of the particles were randomly generated from a uniform distribution within a sphere with the specified radius, and these positions were convolved with a Gaussian to estimate the point spread function at the particles depth, which could be adjusted with parameters for the objective N.A., refractive index of the imaging medium, wavelength and pixel size of the camera. At each exposure (texp), there is the possibility that each molecule can either irreversibly photobleach or reversibly photoswitch to the dark state. Modeling of photobleaching is governed by a 2-component exponential decay, with the 2 rates (kB1 and kB2) and the proportionality (FB1) between them adjustable. The time-dependent photobleaching curve is generated based on the user-supplied parameters, and at each time step, the number of molecules that should bleach is calculated. This number of randomly selected molecules is then switched off at this time point. Photoswitching is governed by power-law statistics (20) with an off rate constant (moff) that is also adjustable. When a particle has reversibly gone dark, it will recover with a probability that is also modeled with power law with an adjustable rate constant (mon). Each ‘on’ and ‘off’ period for every molecule is randomly selected from a power law distribution with the indicated exponents.
Molecules are allowed to move randomly within the volume with a mean squared displacement (MSD) dictated by the set diffusion coefficient, D: MSD = 6DΔt, where Δt is the time between frames. Molecular motion is reflected when the volume boundary is encountered. The molecules also undergo binding and dissociation events, which are governed by user-supplied parameters. The time that each molecule spends diffusing before it binds is randomly selected from an exponential distribution with mean, 1/kbind, while the length of time that the molecule is bound is selected from one of two exponential distributions with means 1/kd1 or 1/ kd2, to model both short and long binding events.
RESULTS
Optimization of HaloTag labeling of yeast proteins for single molecule tracking
Incorporation of TMR in cell culture of pdr5Δ Hht1p-HaloTag. The first challenge of SMT in yeast to overcome is inefficient organic dye incorporation. We optimized the labeling procedure by disrupting the gene encoding a transporter involved in resistance to xenobiotic compounds, PDR5 (21). By disrupting just one of the three multidrug transporters we expected to obtain more efficient retention of the ligand in yeast cells with the least effect on physiology. We compared TMR staining efficiency in wild-type and pdr5Δ strains homozygous for in situ C-terminal fusion of HaloTag to Hht1p (histone H3). This protein is largely immobile because of its incorporation into the nucleosomes and thus it is suitable for prolonged observation of the same molecules in the same focal plane. We treated live cells of pdr5Δ Hht1p-HaloTag strain and PDR5 Hht1p-HaloTag strain with TMR and counter stained the nuclei with DAPI (as described in Materials and Methods). After staining with 10–15 nM of TMR we observed bright particles in the nuclei of Hht1p-HaloTag strains and these particles were co-localized with the nuclear DAPI staining. We could not observe any particles either in the absence of TMR or in the absence of Hht1p-HaloTag (Figure 1A). Thus, the particles correspond to individual molecules of Hht1p-HaloTag labeled with TMR. The average number of particles was 2.14 ± 0.21 in pdr5Δ strain and 0.13 ± 0.04 in PDR5 strain (Figure 1B). Thus, pdr5Δ disruption increases the efficiency of dye incorporation for the HaloTag by a factor of 20.
Figure 1.

Disruption of PDR5 increases efficiency of TMR staining of Hht1p-HaloTag cells. (A) Live cells of WT (YTK1479), WT Hht1p-HaloTag (YTK1490), pdr5Δ Hht1p-HaloTag (YTK1491) and pdr5Δ (YTK1492) were treated with 15 nM TMR/0 nM TMR and DAPI. Single focal plane (SFP) images were acquired on DeltaVision Elite microscope; TMR images were deconvolved. All images in the TMR channel are scaled equally. Yellow circles represent nuclei with TMR particles, whereas green circles represent nuclei without TMR particles. Scale bar = 2 μm. (B) Particles from the TMR channel were counted for 10 nM TMR/0 nM TMR. Center lines show the median values; box limits indicate the 25th and 75th percentiles, outliers are represented by dots; crosses represent sample means. N ranges from 83 to 105.
Tracking single molecules and correcting the tracks in small nuclei. A critical aspect for successful SMT is the labeling density. Excessive labeling of individual molecules interferes with the tracking algorithm and with manual observation of the tracks. Higher particle densities make it impossible to follow each particle as the tracks overlap and cross each other. In a mammalian cell nucleus, the limit for successful tracking is ∼30 molecules per cell (4,19). The area of the yeast nucleus is ∼100-fold smaller than that of a mammalian nucleus. To establish a limit of labeling for yeast, we performed tracking of Hht1p-HaloTag fusion stained with JF646, using a previously developed semi-automated tracking software (19). Briefly, the software takes images such as shown in Figure 2A, and filters out noise (Figure 2B) to identify single molecules. Tracking is performed within user-defined ROIs (Figure 2C and D) delineating the nucleus to confine analysis to molecules inside the nuclear compartment (see Materials and Methods for more details). Even at low labeling densities there is a possibility that two (or more) labeled molecules will be close together, and due to chromatin motion, even if both molecules are bound their tracks will likely cross. Reversible photoswitching of organic dyes (‘blinking’) may lead to artificial shortening of auto-detected tracks. It is therefore necessary to correct the mis-assignment that happened in the automatic tracking due to crossing tracks, and due to blinking. To improve manual quality control and thus to maximize correct detection of the tracks, we used kymographs, rather than explored the two spatial dimensions at each time-point. Kymographs display only one of the spatial dimensions versus time, with the second spatial dimension being maximum projected (Figure 2E and F). Kymographs allowed us to observe the entire length of the tracks, and add/correct many track-points at once, rather than adding/correcting the points in a track one frame at a time.
Figure 2.

Overview of the steps involved in kymograph-based tracking of single molecules of Hht1p-HaloTag labeled with JF646. Imaging is done on HILO microscope. (A) Projection of the intensity summed over all 300 frames of a representative movie to visualize all particles in the 647 nm channel. (B) Same projection as in (A) after noise reduction with the band-pass filtering module of the software. (C) Summed intensity projection of the same field-of-view as in (A) in the 561 nm channel to visualize nuclei. Red outlines indicate user-identified ROIs in which particle tracking is performed. (D) Overlay of 647 nm (red) and 561 nm (green) channels demonstrating particles residing in the nuclei. (E and F) Kymographs XT, (E) Y-projections, and YT, (F) X-projection of all 300 frames from the movie shown in (B) after filtering. Both kymographs are used for manual correction of tracks because apparent track overlap in one dimension resulting from particles in different nuclei are typically well-separated in the other dimension. Colored arrows indicate the corresponding particles shown in the summed image displayed in (B). Examples of (G and H) correctly autodetected and (I and J) manually edited tracks shown as kymographs. (G) The software successfully identifies tracks that are continuous or contain gaps of a single frame. Red, green and blue spots indicate three separate tracks from this image. (H) The algorithm also identifies freely diffusing molecules, which are characterized by a bright spot in a single frame with dimmer (usually not autodetected) spots in the preceding and/or following frames. (I) If a track contains gaps of several frames, the user must manually join the autodetected track segments. (J) Over-crowding makes it challenging to follow a single particle, and so these types of tracks are typically thrown out. See text for our recommendations on when to merge track segments as in (I), or leave them separated as in (G).
Examples of auto-detected bound molecules and diffusing molecules, and examples of the tracks that were manually joined or rejected are presented in Figure 2G–J. The decision to manually join tracks is made based on their x–y coordinates, and on whether any residual fluorescence may be seen in the frames between the identified tracks indicating track continuity. For example, in Figure 2G the blue, red and green track segments are slightly displaced from one another, and the intensity in between them is low. In contrast, the five track segments (blue, red, green, magenta and cyan) in Figure 2I are roughly at the same position, and the intensity, although below the threshold that was set to locate particles, appears to show a single continuous track. Of course, the threshold could be lowered to capture the interstitial regions, however, this would also increase the number of false-positive particle detections in the background signal. Manual track correction is very labor-intensive, typically taking an entire day for an experiment consisting of ∼200 tracks from ∼10 movies. We found that using kymographs allowed us to do the same number of corrections in half the time. Even with the help of kymographs we have found that the number of labeled molecules in a yeast nucleus should not exceed three. Although this means that each cell does not provide much data, the lack of statistics can be compensated by simply observing more cells.
Development of the test for the physiological effects of organic dye incorporation
Assay for the physiological effects of labeling procedure. Although neither staining in culture, facilitated by pdr5Δ, nor fusions of the proteins to HaloTag have an impact on growth rate or resistance to copper (Supplementary Figure S1A and B), disruption of the transporter may exhibit subtle specific effects on transcription by introducing ionic imbalance. To test for such effects, we designed an assay based on the visualization of binding of the TA Ace1p to the CUP1 promoter. In unperturbed cells, Ace1p binds to the promoter of the CUP1 locus only in presence of copper (8). After a treatment with 100 μM of CuSO4, Ace1p undergoes a slow cycle of binding that peaks at 5–10 min after copper addition (2). We tested whether either pdr5Δ or ligand staining induces binding of Ace1p to CUP1 promoters in the absence of copper, or impedes the slow cycle of copper-induced TA binding by counting the fractions of Ace1p-3xGFP cells exhibited green spots over time. No effect was observed for both, and this indicates that neither pdr5Δ disruption nor TMR staining by itself result in a measurable difference in Ace1p binding to CUP1 promoters (Figure 3A). By RT-PCR we observed similar dynamics of CUP1 transcription for wild-type Ace1p and Ace1p-HaloTag; thus HaloTag fusion to Ace1p does not affect Ace1p function (Supplementary Figure S1C).
Figure 3.

Disruption of PDR5 does not alter the normal physiology of the cell in contrast to electroporation. (A) Dynamics of Ace1p-3xGFP occupancy on CUP1 array were assayed by counting the number of cells with visible CUP1 arrays observed over time with 4-min time intervals in PDR5 Ace1p-3xGFP (YTK1479), and pdr5Δ Ace1p-3xGFP (YTK1404). N > 100. Error bars represent standard error of the proportion (s.e.p). Arrows point to visible arrays in the representative diploid nucleus of PDR5 Ace1p-3xGFP. Scale bar = 2 μm. (B) PDR5 Ace1p-3xGFP (YTK1479) cells were observed on DeltaVision Elite microscope before and 5 min after electroporation (EP). Error bars represent the standard error of the mean (s.e.m.). Scale bar = 2 μm. (C) PDR5 Ace1p-3xGFP (YTK1479) were allowed to recover for 2 h before being treated with 100 μM CuSO4 to observe dynamics of Ace1p binding to the CUP1 promoters. Cells treated with TMR by electroporation (EP+TMR) were compared with Non-electroporated cells (No EP), treated and not treated with TMR. Error bars represent s.e.p. N ranges from 29 to 100.
Adverse effects of electroporation. Electroporation (EP) is an efficient method of introducing HaloTag ligands into the yeast cell (12). However, the effects of this procedure on transcription may be adverse. We tested TMR staining procedure by EP on the diploid carrying Ace1p-3xGFP. Logarithmically growing cell culture was split in three; one culture was stained with 1 μM TMR by EP (EP+TMR), another culture was treated with TMR in culture as described above (NoEP+TMR), and the third culture was left untreated (NoEP+NoTMR). We applied the Ace1p visualization assay to all three cultures. First, all three cultures were observed immediately after staining. No arrays were visible in controls, as expected, if no Cu2+ was added to the culture. However, in EP+TMR culture arrays were observed in 20% of cells, indicating that EP induces Ace1p binding to CUP1 promoters even in absence of Cu2+ (Figure 3B). EP further severely affected cell morphology, as observed microscopically (Figure 3B). Furthermore, in plate assays we found >50% reduction in the cell viability after electroporation (Supplementary Figure S2). When the cultures were allowed to recover for 2 h in CSM and then tested again, arrays were no longer visible in absence of Cu2+ in EP+TMR, as well as in controls. However, the response to Cu2+ induction was severely impaired in ER+TMR cells, while normal dynamics were observed in controls tested at the same time (Figure 3C). Thus, EP exhibits a long-term inhibitory effect on the dynamics of Cu2+-induced slow cycle of Ace1p binding. Therefore, electroporation is a harsh procedure, adversely affecting cell morphology, viability and TA binding, and it is not suitable for the staining of the HaloTag fusion proteins if physiological functions such as transcription initiation are to be investigated. Conversely, pdr5Δ disruption does not affect the transcription and the growth rate. In addition, pdr5Δ cells require a simpler procedure for staining the HaloTag proteins with the HaloTag ligands. Thus, pdr5Δ staining enhancement is preferable to electroporation for the labeling of HaloTag proteins in yeast.
Development of compensation procedure for the reversible photoswitching of HaloTag ligands
In order to obtain reliable estimates for the diffusion coefficient and residence times, we had to overcome artifacts that originate from photo-physical characteristics of the fluorescent dyes. Irreversible photobleaching shortens the visible tracks. Reversible photoswitching introduces gaps dividing a single track into several shorter ones. Thus, both types of photobleaching lead to underestimation of the real track lengths. The original TrackRecord software contained corrections for both photoswitching and photobleaching (19). Photobleaching was corrected by counting the number of detected particles in each frame of an experiment, and fitting the time-course to a 2-component exponential. The experimental survival distribution was then divided by this photobleaching curve to account for the fact that a disappearing molecule could be due to either irreversible photobleaching or dissociation. Photoswitching (blinking) was corrected by filling in gaps that appeared in otherwise continuous tracks. The allowed length of a gap was limited to a single frame to avoid the possibility of a dissociation event followed by the binding of a second molecule at the same site. However, as kymographs improved visualization of the tracks, we were able to measure the gap lengths more accurately. When analyzing immobile molecules of H3 histone in live cells we often found gaps of up to 10 frames where a particle would disappear and then reappear in the same spot. Thus, the maximum gap length could potentially be increased to 10 frames (150 ms) for Hht1p-HaloTag stained with JF646. However, most TF are more mobile than H3 and other dyes may have different blinking and photobleaching characteristics.
We therefore used simulations to assess how the extracted residence time depends on performance of the photobleaching and blinking corrections. Parameters for the simulations were chosen using previously observed experimental values, reproducing a population consisting of freely diffusing, long-residence and short-residence particles. The particles were allowed to randomly bind with a rate of kB = 0.1 s−1, and randomly dissociate with one of two dissociation constants, k1 = 0.6 s−1 and k2 = 0.1 s−1, corresponding to residence times of 1.67 and 10 s, respectively, with 90% of dissociation events occurring at k1. With these parameters we expect to have 82% 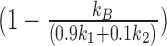 of molecules freely diffusing, 16% bound with the short residence time, and 2% bound with the long residence time. For all simulations of live cells, particles were set to move randomly in a 3D volume that approximated the size of a diploid yeast nucleus, 4.2 μm3 with a diffusion coefficient of D = 0.2 μm2/s. 200 particles were simulated in 200 cells (1 particle per cell). We analyzed these simulated images with the same analysis workflow as used for experimental data.
of molecules freely diffusing, 16% bound with the short residence time, and 2% bound with the long residence time. For all simulations of live cells, particles were set to move randomly in a 3D volume that approximated the size of a diploid yeast nucleus, 4.2 μm3 with a diffusion coefficient of D = 0.2 μm2/s. 200 particles were simulated in 200 cells (1 particle per cell). We analyzed these simulated images with the same analysis workflow as used for experimental data.
As a base-line measurement, a first set of simulations (S1) was run with molecules that were always in their ‘on’ state, i.e. exhibited no photobleaching or blinking. The fractions of the three populations with the residence times extracted through TrackRecord are shown in the pie-chart of Figure 4A, and closely match the expected values above.
For the second set of simulations (S2), we added photobleaching (with an exponential rate constant of 0.9 s−1), but no blinking, and left all other parameters as for S1. The distribution of residence times found for photobleaching molecules after automated tracking, and photobleaching correction is shown in Figure 4B, again giving a close match to expected values. Therefore, the correction for photobleaching recapitulates the results from simulation with no photobleaching.
Next, to characterize the blinking correction of TrackRecord, we conducted a third set of simulations (S3) with photobleaching as above but adding fluorophore blinking with a power law exponent of −1.5 (see Materials and Methods, (20)). After analysis with TrackRecord, with the single-frame gaps bridged, and without performing the manual correction, we get, not surprisingly, a stark difference in the fitting results as shown in Figure 4C. In this case, the survival distribution can be adequately fit with a single-component exponential. We therefore examined the possibility of improving the automated tracking by simply increasing the allowable number of frames that a particle can be absent.
To determine the proper limit for gap filling, in the fourth set of simulations (S4) we used the same photobleaching and blinking properties as in S3 but simulated immobile molecules by setting the diffusion coefficient to 0. For immobile molecules the gaps are generated only by blinking, not by dissociation. The number of missing frames was determined in each track by automatically identifying time-points where the particle is not found. The cumulative distribution of gap lengths for the simulated fixed cells is shown in the graph of Figure 4G (‘Simulation’). Of note is that only 20% of the gaps consisted of a single frame, while 47% were 4 frames or fewer, 78% were at most 8 frames and 90% are 13 frames or fewer.
We then determined the minimal number of frames to bridge that would allow us to closely match the residence time for diffusing molecules obtained in the simulations of S1. We tracked the particles in the S3 simulation, setting the gap parameter to 4, 8 or 13 frames. The extracted parameters for each case are shown in Figure 4D–F. Although the parameters from S1 were never matched precisely, 8- and 13-frame gap settings generated better matches to values obtained in the S1 than the original setting of 1 frame. We therefore chose to use 8 frames, corresponding to the 75th percentile as the maximum gap setting for diffusing molecules. Manual correction of the tracks returns a closer match to the S1 parameters (data not shown), however, the manual correction step added 10 h to the analysis in addition to the 2 h required for automated tracking.
Because the simulations indicated that we obtained the smallest errors when we close at least ∼75% of gaps, it was important to determine the distribution of gap lengths in real fixed cells. We first asked if fixation affects the distribution of gap lengths by checking the observed gaps in JF646 in Hht1p-HaloTag cells (Figure 4H). The gaps observed in live cells were longer than those in fixed cells. The difference in gap length could potentially be caused by changes in photophysics of the fluorophores due to environmental effects, such as a change in pH, resulting from fixation. Tracks in live cells could also exhibit molecule interchange (where one molecule leaves a binding site, and another molecule binds to the same site), which would not happen in fixed cells. The gap length resulting from this interchange would depend on the concentration of labeled molecules, with a higher concentration decreasing the amount of time the site remains vacant. Because we label only a small number of molecules in each cell, the gap length from this phenomenon would likely be longer than from fluorophore blinking. We chose to use the lower gap limit established from fixed molecules to reduce the risk of falsely linking unrelated tracks, although it may result in an under-estimation of residence times.
We found that the distribution of gap lengths differed for Hht1p, Ace1p and Hsf1p in fixed cells (Figure 4G, colored curves), which could be due to slight changes in the fluorophores’ local environment because of where it is bound on the folded protein. The value corresponding to the 75th percentile of gap lengths in fixed cells was 4 frames for Hht1p, 4 frames for Ace1p and 6 for Hsf1p. However, to maintain consistent parameters when analyzing the different data sets, we chose to use the shortest gap value of 4 frames for all subsequent data analysis. Increasing the gap setting from 1 frame to 4 frames eliminated the need for manual corrections.
To verify that increasing the allowable gap-length improved the tracking, and resulting residence time analysis, we compared results using manually corrected tracks from 251 cells expressing Hht1p-HaloTag to the automated 1-frame gap setting, and to the 4-frame gap setting. Using the 1-frame gap setting, we found 66% of the 72 000 total particles (0.5/cell/timepoint) were bound, while 25% had a short residence time of 0.25 ± 0.01 s, and 8% had a long residence time of 1.2 ± 0.07 s (Figure 5A). After manually correcting this data, the fraction of freely diffusing molecules dropped to 50%, and the fraction of short, and long binding events increased to 33% and 17%, respectively (Figure 5B). The extracted residence times did not change dramatically, and were 0.23 ± 0.006 s for short binding events, and 1.1 ± 0.03 s for long binding events. With the 4-frame gap setting, the extracted numbers are much closer to the manually corrected tracks than with the 1-frame setting (Figure 5C). With the longer gaps filled, we obtain 40% of freely diffusing molecules, and 40% short binding events with an average residence time of 0.27 ± 0.005 s, and 20% long binding events with a residence time average of 1.7 ± 0.03 s. Thus, the use of the 4-frame gap setting allows us to extract parameters that closely match those obtained with manual tracking without the need to spend many hours manually processing images and risk introducing operator bias.
Figure 5.

DNA binding is protein- and environment-specific, (A–C) Raw survival distribution (black dots), the best fitting 2-component exponential decay (red line), and extracted distributions of diffusing, short and long binding events (pie chart) in Hht1p-HaloTag after (A) automated tracking filling in 1-frame gaps, (B) manual correction of the automated tracking in (A), and (C) automated tracking filling in gaps up to 4 frames. (D–F) Raw survival (black dots), best fit (red line), and distribution of binding events (pie chart) from automated tracking while filling in 4-frame gaps for (D) Ace1p-HaloTag, (E) Hsf1p-HaloTag in heat shock conditions, and (F) Hsf1p-HaloTag in absence of heat shock. For all distributions, the average residence times of the short and long binding events are included next to the appropriate slice with their 95% confidence interval. All distributions required a two-component exponential fit based on an F-test comparison between the one- and two-component fitting.
Single molecule tracking of DNA-binding proteins reveals similarities in characteristics of short-residence binding
TFs are capable of non-specific (and transient) binding (6). To evaluate transient binding and to estimate its parameters, we evaluated the dynamics of three different DNA-binding proteins: TF encoded by ACE1 gene, heat shock transcription factor, encoded by HSF1 and nucleosomal protein histone H3 encoded by HHT1. Homozygous diploids for HHT1-HaloTag, HSF1-HaloTag and ACE1-HaloTag were constructed in pdr5Δ background. The strains were treated with 0.005 nM and 0.05 nM JF646, respectively, to get the label density of 2–3 molecules per nucleus. To detect the nuclear boundaries, the nucleus was counterstained with JF549 as described in Materials and Methods and Supplementary Table S3. Imaging for SMT was conducted for 600 frames at 66.7 Hz (9 s total). This fast frame rate was selected as only imaging as fast as possible allowed us to extract any tracks from the Ace1p-HaloTag cells. However, at such a fast frame rate, we expected to be limited to measuring maximum residence times of 1–2 s (∼100 images) after which time the majority of fluorophores would be photobleached. Tracking was performed using TrackRecord with automatic linking closing of maximum 4 frames as discussed above. Bound track segments were identified as those that had at least 21 frames where the frame-to-frame displacement did not exceed 220 nm, based on tracking performed on chromatin-bound nucleosomal histone. These parameters reduce the probability that a freely diffusing molecule will be classified as bound to <1% (see Materials and Methods).
Ace1p in absence of Cu2+ may be used as a benchmark for non-specific binding since under these experimental conditions Ace1p binds to DNA only non-specifically. Ace1p requires Cu2+ for the conformational change leading to specific binding (8). In absence of externally added Cu2+, no specific binding occurs, as confirmed by absence of visible arrays in of Ace1p-3xGFP cells (Figure 3C and (2)). In addition, Ace1p may bind specifically only to five sites in the whole genome (CUP1, CRS5, FET3, FTR1 and SOD1) (7). Furthermore, we previously confirmed that Ace1p is capable of non-specific binding by FRAP (22). Thus, a predominant majority of Ace1p binding that we may observe in SMT experiments must be non-specific. For Ace1p-HaloTag, we performed automated tracking in 257 cells. Of the 60 300 total particles (0.4/cell/timepoint), 80% were freely diffusing, while 17% were bound with a short residence time comparable to the short residence time observed in Hht1p-HaloTag (0.13 ± 0.003 s), and 3% were bound with a longer residence time of 1.3 ± 0.06 s (Figure 5D).
We compared Ace1p estimates for non-specific binding to those of the heat shock factor Hsf1p. We performed tracking on Hsf1p-HaloTag at physiological temperature and under heat shock conditions (HS). Search for Hsf1p binding sites yielded ∼1200 binding sites for Hsf1p, called heat shock elements (HSEs), localized in promoters and in open reading frames (23). Those sites are of three types (‘perfect’, ‘gapped’ and ‘stepped’) that differ by sequence and by the binding affinity of Hsf1p (24). Different HSEs may be present in the same promoter, and some of them may be constitutively occupied. For the promoter of HSF82 it has been shown that Hsf1p may bind constitutively at least to some HSE (25). However, by whole-genome analysis, it has been shown that Hsf1p binds to the great majority of the HSEs only after HS (26). Based on high number of HSEs and on HS induced binding of Hsf1p to most of those HSE, we expected a dramatic difference in Hsf1p mobility between HS and control conditions. We also expected to observe a fraction of constitutively bound molecules of Hsf1p even in absence of HS.
Upon the induction of heat shock, Hsf1p behaves more like Hht1p, binding strongly to its target sequences. From the analysis of the residence times of 101 500 particles in 226 cells (0.75/cell/timepoint), we obtained 48% freely diffusing molecules, 38% bound with an average residence time of 0.32 ± 0.008 s, and 14% with a residence time averaging 2.2 ± 0.06 s (Figure 5E). Although this residence time appears to be longer than the long residence time observed in histones (1.7 ± 0.03 s, Figure 5C) further studies are required to determine if upon heat shock, the binding of Hsf1p is indeed stronger than Hht1p. In absence of HS we obtained data on 72 700 particles in 226 Hsf1p-HaloTag cells (0.5/cell/timepoint). The residence time analysis of these particles revealed that 68% were freely diffusing, 25% showed short binding events with an average residence time of 0.21 ± 0.008 s, and 6% showed longer binding events with an average residence time of 1.1 ± 0.07 s (Figure 5F). Thus in absence of HS, the distribution of three fractions of molecules of Hsf1p (not-bound and bound with short and long residence time) is similar to Ace1p (No Cu2+) and dramatically different from Hht1p. These data indicate that in control cells Hsf1p is predominantly bound non-specifically, similarly to Ace1p.
Surprisingly, the residence times for the short-residence molecules is very similar for all proteins/conditions tested. To ensure that the computation does not impose an artificial ceiling on residence time for the short-residence fraction we performed simulations for the sensitivity of curve fitting to residence time values up to 1 s (Supplementary Figure S3). Simulations demonstrate that the fitting algorithm is sensitive to residence times of the ‘short-residence’ fraction at least 5 times higher than those detected in our experiments.
DISCUSSION
Biological significance of SMT estimates of residence time
SMT provides important biophysical characterization for the mammalian nuclear proteins. Notably SMT allowed direct measurement of the dwell time of the TF on transcriptional factories and therefore shed light on the long-standing question of the role for the dynamics of transcriptional complexes. However, important criteria are still not defined to accurately distinguish sequence-specific binding, non-specific binding and diffusion. The tracking algorithm is set to always generate the parameters for the three populations of the molecules: diffusing, bound with short residence time and bound with long residence time. How can we decide on the biological significance of short and long residence times? Do they correspond to specific and non-specific binding, or do they correspond to two different types of specific binding?
Yeast is a model eukaryotic organism that allows development of a simple benchmark for non-specific binding. DNA-binding TF may exhibit not only sequence-specific binding but also sequence-non-specific binding that may reflect a searching mechanism for the specific sites (6). The best control for the nonspecific TF binding would be not a freely diffusing protein, but a TF that binds specific sites conditionally, reflecting the ability of TF to bind both directly and indirectly by interacting with co-activators. Yeast TF Ace1p achieves the conformation suitable for specific binding only by binding Cu2+ (8). Thus, in absence of externally supplied Cu2+, the fraction of Ace1p in activated conformation must be low, as confirmed by lack of visual Ace1p-3xGFP binding to the CUP1 array. In addition, Ace1p may bind specifically only to five sites in the whole genome (CUP1, CRS5, FET3, FTR1 and SOD1) (7). Thus, a great majority of Ace1p binding that we may observe in SMT experiments must be non-specific. Previously, we demonstrated by FRAP that Ace1p is capable of nonspecific binding to nucleoplasmic targets, as opposed to GFP (22). Therefore, by quantifying the behavior of Ace1p we may obtain the baseline for the nonspecific binding. By SMT we observe 20 ± 0.9% of bound molecules of Ace1p; 86 ± 0.7% of those have a residence time of 0.13 ± 0.003 s. A small fraction (14 ± 0.7%) of Ace1p is bound with an average residence time of 1.3 ± 0.07 s.
To compare the non-specific residence time of Ace1p to other proteins we estimated residence times of the similar inducible TF – heat-shock factor, encoded in yeast by a single HSF1 gene. We tracked Hsf1p-HaloTag with and without HS. Our data show that upon HS the estimated fraction of freely diffusing molecules decreases by a factor of 1.4 from 68% to 48%, and the average residence time of long binding events increases from 1.1 ± 0.07 s to 2.2 ± 0.06 s.
Our data lead to interesting conclusions: the estimated residence time of the short-residence molecules is essentially the same for Hht1p, Ace1p and Hsf1p (No HS), equalling 0.12–0.32 s. These three DNA-binding proteins are very different in their structure, function and intracellular concentration. This suggests that (i) short-residence molecules are bound to DNA non-specifically, and (ii) that non-specific binding shares common characteristics between vastly different DNA-bound proteins and thus may have a common underlying mechanism.
A fraction of long-residence H3 may represent dynamics of nucleosome assembly
We believe that the extracted population and residence time parameters from Ace1p represent characteristic values for proteins that only bind transiently to DNA, because very few long binding events, if any, are expected from Ace1p in the absence of Cu2+. As the time intervals adopted in this study were optimized for study of non-specific binding, we do not expect to extract the correct residence time for specific binding. It is known that the observed residence time strongly depends on the imaging conditions, particularly the imaging rate. This is likely due at least in part to the fact that fluorophores exhibit both reversible and irreversible photobleaching. Because a slower imaging rate allows the reversibly photobleached molecules to recover to their fluorescent state, longer intervals between images typically result in longer observed residence times (27–29). We believe that the residence time of H3 that we have extracted underestimates the true residence time. Because we expect histones to exhibit extremely stable binding lasting much longer than the extracted 1.7 s, this number represents the upper limit on the average observable residence time that can be obtained with the imaging conditions used due to photobleaching.
Regarding the fraction of bound molecules, we expected that as most of H3 is incorporated into the nucleosomes, the great majority of this protein would be stably associated with DNA. By SMT we observe that a sizeable fraction of H3 is bound, as expected (60 ± 0.9%). Unexpectedly, we observe not one but two fractions among those bound molecules. Our data agree with those reported for histone H2 in mammalian cells: fitting of the particle motion in SMT experiments required three components: freely (10%), moderately (19%) and slowly diffusing (71%) molecules (27). For H3 predictions may be derived from FRAP: by FRAP, 16% of H3 in mammalian cells diffuses freely, 16% exchanges with half-time of 130 min and 68% exchanges with half time of a few hours (30). Thus, 84% of the H3 population should be totally immobile for the time of observation of this study (9 s). It is unclear why we still observe two distinct populations of bound histone molecules while imaging cells at much shorter time intervals than (30). It is possible that (i) as the yeast cells have a very gene-dense genome, and (ii) the great majority of those genes undergoes dynamic nucleosomal repositioning (31), we are registering (i) and (ii) at the single molecule level as a fraction of molecules with higher exchange rate and lower residence time than those for H3 stably associated with nucleosomes. In addition, cells in logarithmically growing cultures typically assayed in yeast are actively dividing, thus chromatin replication may further contribute to nucleosomal dynamics (32).
Development of the test for the physiological effects of organic dye incorporation
Physiological tests are necessary to ensure that single molecule labeling is benign to the transcription and thus the produced estimates of residence time are accurate. Such tests are difficult to develop even in such a simple model organism as yeast, because they require monitoring of the transcription initiation. We developed a biological test of transcription initiation based on microscopic visualization of the binding of a transcriptional activator Ace1p to CUP1 promoters. Due to high concentration of Ace1p binding sites in the naturally occurring tandem array of CUP1 genes, induced binding of Ace1p-3xGFP to its target sequences may be monitored in real time. Dynamics of this binding have been previously well characterized (2), and therefore one may assay whether labeling perturbs this process either by preventing or by over-stimulating Ace1p binding. Application of this test to staining procedures permitted us to establish that the pdr5Δ-facilitated labeling procedure developed in this study is benign and does not affect the dynamics of TA binding to DNA. However, electroporation, applied to labeling, leads to adverse effects on TA binding. These results validate the test. We recommend this test for evaluation of any manipulation of yeast cells for the transcription studies.
Development of a new and robust labeling procedure based on mutation of a single yeast transporter gene
SMT has not been yet successfully applied to yeast TFs due to technical challenges, such as a small size of the nucleus and imperfect labeling. We adapted SMT to yeast by developing a benign labeling procedure, and by improving the tracking and quantification procedures.
Development of benign organic dye incorporation procedure for HaloTag fusions in yeast. SMT requires bright photostable labels such as HaloTag ligands. However, labeling of HaloTag fusion proteins in wild-type yeast cells is problematic because they pump out xenobiotic compounds very efficiently and hence the stable-state intracellular concentration of HaloTag ligands is very low. We explored the possibility of increasing intracellular ligand concentration by impairing the process of ligand export. Deletion of ABC-MDR transporter (PDR5) proved to be sufficient for successful labeling of HaloTag proteins. We demonstrated that pdr5Δ-enhanced staining procedure does not interfere with Ace1p binding and thus it is applicable to TF studies.
Alternatively, HaloTag ligands may be electroporated into yeast cells (12). However, we demonstrated that electroporation interferes with normal physiology of Ace1p binding and thus is unsuitable for studies of transcription. Also, intracellular SNAP-tag fusions were successfully labeled in strains with triple deletions of the ABC-MDR transporters PDR5, SNQ2 and YOR1, but this approach cannot be universally recommended as triple deletions strongly reduce the cell's viability (13,14). Instead, we recommend pdr5Δ-enhanced staining procedure, which is benign and easy to apply.
Addressing blinking of the dyes in SMT analysis. The ligands we tested exhibit different rates of photobleaching and blinking. By using computational simulations, we established that the TrackRecord software performs a very good automatic correction for photobleaching (19). However, blinking is not corrected adequately if the setting for the maximal number of frames for the gap in the track generated by blinking is arbitrarily set to one frame. Although the residence time is not that much affected, in simulations we observed that the long binding events were completely ignored, evidenced by the fact that the data analyzed with the 1-frame gap setting was adequately fit with a single-component exponential function. Thus, the correct parameters for blinking gap correction have to be determined from real samples. By testing naturally immobile molecules of histone H3 we observed that although the bleaching rate changes for fixed molecules of the ligand-bound protein, blinking rate remains the same. Therefore, one may extract the blinking rate from fixed samples of the cells. It is worth noting, that we observed slightly different blinking rates for different HaloTag fusion proteins stained with the same ligand JF646. For the gap setting we used 4 frames for Hht1p, Ace1p and Hsf1p, as this accounted for ∼75% of the gaps in fixed cells. Thus, for each set of fusions, ligands and imaging rates this parameter has to be established in preliminary experiments.
New techniques developed in this study allowed us to introduce SMT to studies of yeast transcription factor dynamics and to obtain important information for the characteristics of non-specific binding.
Supplementary Material
Acknowledgments
The authors would like to thank Dr Luke D. Lavis and Dr Jonatan B. Grimm (Janelia Research Campus, Howard Hughes Medical Institute, Ashburn, VA 20147, USA) for JF HaloTag ligands. The authors acknowledge Euroscarf for providing pUG6 plasmid.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Intramural Research Program of National Institutes of Health (NIH), National Cancer Institute (NCI) and Center for Cancer Research (CCR). Funding for open access charge: Intramural Research Program of National Institutes of Health (NIH).
Conflict of interest statement. None declared.
REFERENCES
- 1.McNally J.G., Muller W.G., Walker D., Wolford R., Hager G.L. The glucocorticoid receptor: Rapid exchange with regulatory sites in living cells. Science. 2000;287:1262–1265. doi: 10.1126/science.287.5456.1262. [DOI] [PubMed] [Google Scholar]
- 2.Karpova T.S., Kim M.J., Spriet C., Nalley K., Stasevich T.J., Kherrouche Z., Heliot L., McNally J.G. Concurrent fast and slow cycling of a transcriptional activator at an endogenous promoter. Science. 2008;319:466–469. doi: 10.1126/science.1150559. [DOI] [PubMed] [Google Scholar]
- 3.Voss Ty.C., Hager G.L. Dynamic regulation of transcriptional states by chromatin and transcription factors. Nat. Rev. 2014;15:69–81. doi: 10.1038/nrg3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morisaki T., Muller W.G., Golob N., Mazza D., McNally J.G. Single-molecule analysis of transcription factor binding at transcription sites in live cells. Nat. Commun. 2014;5:4456. doi: 10.1038/ncomms5456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mazza D., Abernathy A., Golob N., Morisaki T., McNally J.G. A benchmark for chromatin binding measurements in live cells. Nucleic Acid Res. 2012;40:e119. doi: 10.1093/nar/gks701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Izeddin I., Recamier V., Bosanac L., Cisse I.I., Boudarene L., Dugast-Darzacq C., Proux F., Benichou O., Voituriez R., Bensaude O., et al. Single-molecule tracking in live cells reveals distinct target-search strategies of transcription factors in the nucleus. Elife. 2014;3:e02230. doi: 10.7554/eLife.02230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gross C., Kelleher M., Iyer V.R., Brown P.O., Winge D.R. Identification of the copper regulon in Saccharomyces cerevisiae by DNA microarrays. J. Biol. Chem. 2000;275:32310–32316. doi: 10.1074/jbc.M005946200. [DOI] [PubMed] [Google Scholar]
- 8.Furst P., Hu S., Hackett R., Hamer D. Copper activates metallothionein gene transcription by altering the conformation of a specific DNA binding protein. Cell. 1988;55:705–717. doi: 10.1016/0092-8674(88)90229-2. [DOI] [PubMed] [Google Scholar]
- 9.Kolberg K., Puettmann C., Pardo A., Fitting J., Barth S. SNAP-tag technology: A general introduction. Curr. Pharm. Des. 2013;19:5406–5413. doi: 10.2174/13816128113199990514. [DOI] [PubMed] [Google Scholar]
- 10.England C.G., Luo H., Cai W. HaloTag technology: A versatile platform for biomedical applications. Bioconjug. Chem. 2015;26:975–986. doi: 10.1021/acs.bioconjchem.5b00191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grimm J.B., English B.P., Chen J., Slaughter J.P., Zhang Z., Revyakin A., Patel R., Macklin J.J., Normanno D., Singer R.H., et al. A general method to improve fluorophores for live-cell and single-molecule microscopy. Nat. Methods. 2015;12:244–250. doi: 10.1038/nmeth.3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stagge F., Mitronova G.Y., Belov V.N., Wurm C.A., Jakobs S. SNAP-, CLIP- and Halo-tag labelling of budding yeast cells. PLoS One. 2013;8:e78745. doi: 10.1371/journal.pone.0078745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McMurray M.A., Thorner J. Septin stability and recycling during dynamic structural transitions in cell division and development. Curr. Biol. 2008;18:1203–1208. doi: 10.1016/j.cub.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chidley C., Haruki H., Pedersen M.G., Muller E., Johnsson K. A yeast-based screen reveals that sulfasalazine inhibits tetrahydrobiopterin biosynthesis. Nat. Chem Biol. 2011;7:375–383. doi: 10.1038/nchembio.557. [DOI] [PubMed] [Google Scholar]
- 15.Kolaczkowski M., Kolaczowska A., Luczynski J., Witek S., Goffeau A. In vivo characterization of the drug resistance profile of the major ABC transporters and other components of the yeast pleiotropic drug resistance network. Microb. Drug Resist. 1998;4:143–158. doi: 10.1089/mdr.1998.4.143. [DOI] [PubMed] [Google Scholar]
- 16.Zhao Y., Strope P.K., Kozmin S.G., McCusker J.H., Dietrich F.S., Kokoska R.J., Petes T.D. Structures of naturally evolved CUP1 tandem arrays in yeast indicate that these arrays are generated by unequal nonhomologous recombination. G3 (Bethesda) 2014;4:2259–2269. doi: 10.1534/g3.114.012922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burke D., Dawson D., Stearns T. Methods in Yeast Genetics. NY: Cold Spring Harbor Laboratory Press; 2000. [Google Scholar]
- 18.Tokunaga M., Imamoto N., Sakata-Sogawa K. Highly inclined thin illumination enables clear single-molecule imaging in cells. Nat. Methods. 2008;5:159–161. doi: 10.1038/nmeth1171. [DOI] [PubMed] [Google Scholar]
- 19.Mazza D., Ganguly S., McNally J.G. Monitoring dynamic binding of chromatin proteins in vivo by single-molecule tracking. Methods Mol. Biol. 2013;1042:117–137. doi: 10.1007/978-1-62703-526-2_9. [DOI] [PubMed] [Google Scholar]
- 20.Hoogenboom J.P., Hernando J., van Dijk E.M., van Hulst N.F., Garsia-Parajo M.F. Power-law blinking in the fluorescence of single organic molecules. Chem. Phys. Chem. 2007;8:823–833. doi: 10.1002/cphc.200600783. [DOI] [PubMed] [Google Scholar]
- 21.Leppert G., McDevitt R., Falco S.C., Van Dyk T.K., Ficke M.B., Golin J. Cloning by gene amplification of two loci conferring multiple drug resistance in Saccharomyces. Genetics. 1990;125:13–20. doi: 10.1093/genetics/125.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karpova T.S., Chen T.Y., Sprague B.L., McNally J.G. Dynamic interactions of a transcription factor with DNA are accelerated by a chromatin remodeller. EMBO Rep. 2004;5:1064–1070. doi: 10.1038/sj.embor.7400281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Y., Ye S., Erkine A.M. Analysis of Saccharomyces cerevisiae genome for the distributions of stress-response elements potentially affecting gene expression by transcriptional interference. In Silico Biol. 2009;9:379–389. doi: 10.3233/ISB-2009-0412. [DOI] [PubMed] [Google Scholar]
- 24.Yamamoto A., Mizukami Y., Sakurai H. Identification of a novel class of target genes and a novel type of binding sequence of heat shock transcription factor in Saccharomyces cerevisiae. J. Biol. Chem. 2005;280:11911–11919. doi: 10.1074/jbc.M411256200. [DOI] [PubMed] [Google Scholar]
- 25.Erkine A.M., Magrogan S.F., Sekinger E.A., Gross D.S. Cooperative binding of heat shock factor to the yeast HSP82 promoter in vivo and in vitro. Mol. Cell. Biol. 1999;19:1627–1639. doi: 10.1128/mcb.19.3.1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hahn J.S., Hu Z., Thiele D.J., Iyer V.R. Genome-wide analysis of the biology of stress responses through heat shock transcription factor. Mol. Cell Biol. 2004;24:5249–5256. doi: 10.1128/MCB.24.12.5249-5256.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gebhardt J.C., Suter D.M., Roy R., Zhao Z.W., Chapman A.R., Basu S., Maniatis T., Xie X.S. Single-molecule imaging of transcription factor binding to DNA in live mammalian cells. Nat. Methods. 2013;10:421–426. doi: 10.1038/nmeth.2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Normanno D., Boudarene L., Dugast-Darzacq C., Chen J., Richter C., Proux F., Benichou O., Voituriez R., Darzacq X., Dahan M. Probing the target search of DNA-binding proteins using TetR as model searcher. Nat. Commun. 2015;6:7357. doi: 10.1038/ncomms8357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Knight S.C., Xie L., Deng W., Guglielmi B., Witkowsky L.B., Bosanac L., Zhang E.T., El Beheiry M., Masson J.B., Dahan M., et al. Dynamics of CRISPR-Cas9 genome interrogation in living cells. Science. 2015;350:823–826. doi: 10.1126/science.aac6572. [DOI] [PubMed] [Google Scholar]
- 30.Kimura H., Cook P.R. Kinetics of core histones in living human cells: little exchange of H3 and H4 and some rapid exchange of H2B. J. Cell Biol. 2001;153:1341–1353. doi: 10.1083/jcb.153.7.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nocetti N., Whitehouse I. Nucleosome repositioning underlies dynamic gene expression. Genes Dev. 2016;30:660–672. doi: 10.1101/gad.274910.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deniz O., Flores O., Aldea M., Soler-Lopez M., Orozco M. Nucleosome architecture throughout the cell cycle. Sci. Rep. 2016;6:19729. doi: 10.1038/srep19729. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.