

The high-resolution (1.57 Å) structure and kinetic data of the key fire blight phosphatase AmsI are reported.
Keywords: hydrolases, amylovoran, low-molecular-weight protein tyrosine phosphatase, kinetics, fire blight, Enterobacteriaceae, Erwinia amylovora
Abstract
AmsI is a low-molecular-weight protein tyrosine phosphatase that regulates the production of amylovoran in the Gram-negative bacterium Erwinia amylovora, a specific pathogen of rosaceous plants such as apple, pear and quince. Amylovoran is an exopolysaccharide that is necessary for successful infection. In order to shed light on AmsI, its structure was solved at 1.57 Å resolution at the same pH as its highest measured activity (pH 5.5). In the active site, a water molecule, bridging between the catalytic Arg15 and the reaction-product analogue sulfate, might be representative of the water molecule attacking the phospho-cysteine intermediate in the second step of the reaction mechanism.
1. Introduction
Fire blight is a devastating disease affecting rosaceous plants such as apple, pear and quince (Vanneste, 2000 ▸). The Gram-negative bacterium Erwinia amylovora is the aetiological agent of the disease. Currently, the main methods to control fire blight are the use of biological and chemical pesticides, antibiotics and resistant cultivars obtained through classical breeding or genetic engineering. Infected cultivars commonly require quarantine, pruning and/or eradication of the plants (Gusberti et al., 2015 ▸). Within the E. amylovora genome, the ams operon is necessary for pathogenicity and is responsible for the synthesis of amylovoran (Bugert & Geider, 1995 ▸), which is a complex branched heteropolysaccharide that is essential for virulence and pathogenicity, and is the major component of the exopolysaccharide (EPS) capsule of the bacterium together with levan (Caputi, Cianci et al., 2013 ▸; Caputi, Nepogodiev et al., 2013 ▸; Geier & Geider, 1993 ▸; Nimtz et al., 1996 ▸; Bernhard et al., 1993 ▸; Wuerges et al., 2015 ▸).
The AmsI protein, encoded by the amsI gene, is a cytoplasmatic key enzyme of amylovoran metabolism, and thus a potential drug target for the control of fire blight. A balanced amount of AmsI is essential for the synthesis of amylovoran. Overproduction of AmsI leads to considerable inhibition of EPS synthesis, while the amsI knockout is deficient in EPS (Bellemann & Geider, 1992 ▸; Bugert & Geider, 1995 ▸, 1997 ▸). AmsI is a cysteine-based protein tyrosine phosphatase (PTP). PTPs putatively catalyse the dephosphorylation of a cognate kinase on phospho-tyrosine residues. The kinase–phosphatase dual system plays a critical role in bacterial virulence and cell signalling (Bechet et al., 2009 ▸; Byrne et al., 2011 ▸; Cozzone et al., 2004 ▸; Grangeasse et al., 2007 ▸, 2010 ▸, 2012 ▸; Kolot et al., 2008 ▸; Lacour et al., 2008 ▸; Morona et al., 2006 ▸; O’Riordan & Lee, 2004 ▸). In particular, their involvement in the synthesis and export of polysaccharides responsible for biofilm and capsule formation, antibiotic resistance, lysogenization and DNA metabolism has been demonstrated. PTPs can be divided into three families: high-molecular-weight PTPs (HMW-PTPs; receptor-like and nonreceptor), dual-specificity PTPs (DS-PTPs; specificity for phospho-tyrosine and phospho-serine/threonine) and low-molecular-weight PTPs (LMW-PTPs), with a molecular mass of about 18 kDa (Walton & Dixon, 1993 ▸; Charbonneau & Tonks, 1992 ▸; Tabernero et al., 2008 ▸; Zhang & Dixon, 1994 ▸; Blenis, 1993 ▸; Yuvaniyama et al., 1996 ▸; Denu et al., 1995 ▸; Zhang et al., 1995 ▸; Raugei et al., 2002 ▸; Caselli et al., 2016 ▸). The three PTP families share no sequence homology apart from a signature motif C(X)5R(S/T) contributing to the phosphate-binding loop (P-loop) in the active site and a catalytic aspartate residue followed by a proline and often a tyrosine, which are part of the so-called D-loop.
AmsI is a member of the LMW-PTPs, which are found both in prokarya and eukarya (Su et al., 1994 ▸; Tabernero et al., 2008 ▸; Zhang, Van Etten et al., 1994 ▸; Zhang et al., 1997 ▸, 1998 ▸; Zabell et al., 2006 ▸; Stehle et al., 2012 ▸; Wang et al., 2000 ▸; Lescop et al., 2006 ▸; Madhurantakam et al., 2005 ▸; Vega et al., 2011 ▸; Gustafson et al., 2005 ▸; Hagelueken et al., 2009 ▸; Xu et al., 2006 ▸). The LMW-PTPs share a conserved P-loop motif consisting of a CXGNXCRSP consensus sequence, where X corresponds to a leucine, isoleucine, threonine or phenylalanine residue (see the alignment in Fig. 1 ▸).
Figure 1.

Alignment of LMW-PTPs. Sequence alignment of LMW-PTPs for which X-ray diffraction structures are available (the sequence names consist of the protein name followed by the source organism). The secondary-structural elements in AmsI are shown at the top. Conserved residues are highlighted in red. The characters in red represent the conservative residues. This figure was created using the ESPript server (Robert & Gouet, 2014 ▸).
From a structural point of view there are strong similarities among all of the LMW-PTPs. They share an overall fold, the P-loop in the N-terminal region and the same catalytic residues. Significant variability is observed among the residues forming the protein surface around the active site, and it is becoming clear that these residues have great importance in the interaction with the cognate kinase, such as the central positions of the W-loop (Grangeasse et al., 2003 ▸; Temel et al., 2013 ▸; Bechet et al., 2010 ▸). The structural determination of the interaction between Wzb (an analogue of AmsI) and Wzc (an analogue of AmsA, the cognate kinase of AmsI) has been described (Temel et al., 2013 ▸).
The catalytic mechanism of the LMW-PTPs has been extensively investigated. According to the current model, the reaction starts with the formation of a covalent phospho-enzyme intermediate (Davis et al., 1994 ▸). The nucleophile is the Sγ atom of a cysteine present in the P-loop. This first step is assisted by the protonation of the leaving group (i.e. dephosphorylated protein) O atom by a general acid consisting of the conserved aspartate residue of the D-loop (Zhang & Van Etten, 1991 ▸). A P-loop arginine is involved both in substrate binding and in the stabilization of the reaction intermediate (Zhang, Wang et al., 1994 ▸). The catalytic cysteine and arginine, together with other residues of the P-loop, form a cradle providing critical hydrogen-bonding interactions with the phosphate group of the substrate, holding it in place for the subsequent nucleophilic attack (Evans et al., 1996 ▸). In fact, the amide groups in the P-loop point towards the interior of the cradle and form a network of hydrogen bonds with the phosphate O atoms. During the second step, which is the rate-limiting step, the phospho-cysteine intermediate is attacked by a water molecule. The same aspartate that acted as a general acid in the first step serves as a general base during hydrolysis of the phospho-enzyme by accepting a proton from the water molecule and by assisting the conversion of the phospho-cysteine enzyme to its resting state, regenerating the free enzyme (Zhang, Harms et al., 1994 ▸; Denu & Dixon, 1995 ▸; Denu et al., 1996 ▸). Finally, inorganic phosphate is released from the enzyme.
In order to investigate the key fire blight phosphatase AmsI, we solved its crystal structure at 1.57 Å resolution, we studied its oligomerization state using static light scattering and we studied its activity using steady-state kinetic analysis.
2. Materials and methods
2.1. Macromolecule production
Chemicals were purchased from Sigma–Aldrich unless stated otherwise. AmsI (UniProt D4I6U1_ERWAE; EC 3.1.3.48) from E. amylovora (strain ATCC 49946/CCPPB 0273/Ea273/27-3) was cloned, expressed and purified as described previously (Benini et al., 2014 ▸). Briefly, the amsI gene was amplified by PCR from the genomic DNA and inserted into a pETM-11 (EMBL) vector that fuses an N-terminal His6 tag and a TEV protease cleavage site to the N-terminus of the protein (Dümmler et al., 2005 ▸). AmsI was expressed in Escherichia coli strain BL21 (DE3). The purification consisted of two strategies, depending on whether protein with or without the tag was needed. To obtain protein with the tag, immobilized metal-affinity chromatography (IMAC) was followed by size-exclusion chromatography (SEC). To obtain protein without the tag, an initial IMAC step, TEV protease cleavage, buffer exchange with a HiPrep 26/10 column (when only buffer exchange is required, this enables the process to be sped up and the loss of protein to be decreased with respect to size exclusion), a second IMAC step and a final SEC step. The obtained final fractions were in 20 mM Tris–HCl pH 7.5, 150 mM NaCl, 0.5 mM TCEP. For crystallization purposes, the fractions were pooled and concentrated to 15 mg ml−1 by ultrafiltration using a Vivaspin 20 Centricon with 5 kDa cutoff (Sartorius). In order to compare the elution profiles of AmsI with and without the histidine tag, we ran analytical size-exclusion chromatography using a Superdex 75 10/300 GL column (GE Healthcare, Sweden) in 20 mM HEPES pH 7.5, 150 mM NaCl, 2 mM TCEP at room temperature.
2.2. Crystallization
AmsI was crystallized from a 15 mg ml−1 protein solution using a microbatch-under-oil setup at 293 K in 96-well MRC plates (Molecular Dimensions) and volatile oil, as described previously (Benini et al., 2014 ▸).
The crystallization wells were protected from drying using adhesive ClearView sheets (Molecular Dimensions). Drops of 1 µl precipitant solution were added to 15 µl volatile oil, immediately followed by 1 µl protein solution. Crystals grew within two weeks to maximum dimensions of about 0.2 × 0.2 × 0.2 mm in 1.7 M ammonium sulfate, 100 mM sodium acetate pH 5.5. Crystals were obtained using the His6-tagged AmsI.
2.3. Data collection and processing
Diffraction data were collected at 100 K on a Dectris PILATUS 6M detector on EMBL beamline P13 (PETRA III, DESY, Hamburg, Germany). The wavelength was set to 1.033 Å. The data were processed using XDS (Kabsch, 2010 ▸). Space-group determination was carried out using POINTLESS (Evans, 2011 ▸) and the data were scaled with AIMLESS (Evans, 2006 ▸). The crystal belonged to space group P3121 and diffracted to a maximum resolution of 1.57 Å (Benini et al., 2014 ▸).
2.4. Structure solution and refinement
The structure was solved by molecular replacement using BALBES (Long et al., 2008 ▸). BALBES automatically searched the PDB and found the E. coli LMW-PTP Wzb (PDB entry 2wmy; 51% sequence identity; Hagelueken et al., 2009 ▸) to be the best model for molecular replacement. Model building was carried out and water molecules were added using Coot (Emsley et al., 2010 ▸). The structure was refined with REFMAC5 to a final R and R free of 0.169 and 0.179, respectively. The asymmetric unit contains one molecule of AmsI with a solvent content of 55.36% and a Matthews coefficient of 2.75 Å3 Da−1. Table 1 ▸ reports a summary of refinement and model quality (PDB entry 4d74).
Table 1. Structure solution and refinement.
Values in parentheses are for the outer shell.
| Resolution range (Å) | 57.18–1.57 (1.611–1.570) |
| Completeness (%) | 99.9 |
| No. of reflections, working set | 24575 (1785) |
| No. of reflections, test set | 1318 (97) |
| Final R cryst | 0.169 (0.208) |
| Final R free † | 0.179 (0.232) |
| Cruickshank DPI‡ | 0.069 |
| No. of non-H atoms | |
| Protein | 1157 |
| Ion | 15 |
| Ligand | 0 |
| Water | 100 |
| Total | 1272 |
| R.m.s. deviations | |
| Bonds (Å) | 0.012 |
| Angles (°) | 1.591 |
| Average B factors (Å2) | |
| Protein | 23.1 |
| Ion | 37.7 |
| Ligand | 0.0 |
| Water | 31.2 |
| Ramachandran plot | |
| Most favoured (%) | 96 |
| Allowed (%) | 4 |
To calculate R free, a subset of reflections (5.0%) was randomly chosen as a test set.
Diffraction-component precision indicator of the atom position.
2.5. Static light-scattering experiments (SEC-QELS)
SLS measurements were carried out using a Wyatt QELS apparatus (Dawn EOS, Wyatt Technology) coupled to an HPLC pump (Knauer Smartline Pump 1000). The SLS device was equipped with a laser beam operating at λ = 690 nm. Light-scattering intensities recorded at 18 angles between 14.2 and 163.5° were obtained using the ASTRA software with a gyration radius of between 0.5 and 10 nm. This procedure consists of plotting Kc/R θ versus sin2(θ/2) + kc, where K is an optical constant which contains the specific refractive-index increment dn/dc (0.18 ml g−1 in this case), c is the concentration of protein (g ml−1), R θ is the Rayleigh ratio, k is a constant for the graphical representation of the Zimm diagram and θ is the angle of observation. Extrapolations to zero angle and concentration provided the values of molecular mass and R g,z.
To study the oligomerization state of the protein, AmsI deprived of the His6 tag was assessed at concentrations ranging from 2 to 20 mg ml−1 in the presence and absence of 5 mM sulfate ion. The sulfate ion was used to evaluate its contribution to the dimerization observed in the crystal structure. In order to separate and analyse different-sized species present in solution, a Superdex 75 10 300 GL column connected to the HPLC pump was used. The running-buffer formulations were 20 mM Tris–HCl, 150 mM NaCl, 2 mM TCEP and, alternatively, the same buffer with the addition of 5 mM sodium sulfate. The buffers were extensively filtered and degassed. The pure protein was centrifuged at 16 000g for 15 min at 4°C before experiments and 100 µl was injected into the column.
2.6. Steady-state kinetics
The protein used in all of the experiments derived from the same batch as utilized for crystallization and structure determination. AmsI showed a purity of greater than 99% as indicated by SDS–PAGE analysis. The activity of AmsI was studied by stopped-assay steady-state kinetic experiments (Gardossi et al., 2010 ▸; Bisswanger, 2011 ▸, 2014 ▸; Tipton et al., 2014 ▸) following the formation of p-nitrophenyl (pNP) from the hydrolysis of p-nitrophenyl phosphate (pNPP) at 405 nm using an ∊c of 18 000 M −1 cm−1 (Davis et al., 1994 ▸). The quantification of pNP was calculated using a path length of 0.65 cm, corresponding to 250 µl of solution in a well of 3.48 mm radius, as stated by the plate-manufacturing company (VWR International). The errors on V max and K m were calculated as standard deviations by GraphPad Prism v.5.03 (GraphPad Software). The relative statistical error propagation was taken into account and errors were rounded in excess.
Experiments were carried out at 25°C using 100 mM buffers: citrate pH 4.8, citrate pH 5.5, citrate pH 6.5 and bicine pH 8.5. The reactions were set up in transparent 96-well plates (Greiner Bio-One, VWR International, USA). The total reaction volume per well was 200 µl. For each buffer, an experiment consisting of 96 conditions was carried out at least in triplicate. For each experiment, 12 substrate concentrations were used ranging from 0.5 to 30 mM and positioned from plate columns 1 to 12. Row A was used for blank solutions where no enzyme was added. Rows B–H were identical and each was used for a different quenching time, giving a total of seven timing points. The reactions were started by the addition of 20 µl enzyme prepared by the dilution of a 20 mg ml−1 solution in the same buffer as the experiment. The enzyme concentration was 50 ng ml−1 (2.65 nM) and therefore 10 ng was used per well. The protein concentration was estimated in 6 M urea by spectrophotometry using the parameters M r = 19 kDa and ∊c = 17 mM −1 cm−1 as estimated by ProtParam (Gasteiger et al., 2005 ▸). The reactions were quenched by adding 50 µl 1 M NaOH to a final concentration of 0.2 M. The pH increase owing to NaOH also enhances the pNP colour development, as accurate measurement requires that the 4-nitrophenol product is fully deprotonated.
The 96-well plates were read using a Tecan Infinite 200 PRO (Tecan Group AG, Männedorf, Switzerland) immediately after the end of the experiment and also the day after to check that no changes owing to incomplete colour development or incomplete quenching occurred (no difference was measured). For each protein sample at least nine measurements were taken and the resulting values were averaged. Every well was read in four different positions using 25 flashes per reading point and the values were then averaged out. The exclusion of one reading point was allowed when it was not consistent with the other three.
3. Results and discussion
3.1. Tertiary structure
The X-ray diffraction structure of AmsI from E. amylovora was solved at 1.57 Å resolution (final R = 0.16; R free = 0.17; see Table 1 ▸ for refinement statistics) in complex with the product analogue sulfate (PDB entry 4d74).
The residue numbering in the PDB file starts at −7. Residues −7 to −1 correspond to the TEV cleavage site that is part of the N-terminal His6 fusion tag. No electron density was visible for the first 12 residues owing to disorder or multiple conformations. The N-terminus is opposite the active site and has been reported not to influence catalysis (Ostanin et al., 1995 ▸; Wang et al., 2000 ▸). Residue 1 corresponds to the first amino acid of the wild-type sequence and residue 2 is a conservative mutation Ile to Val as a consequence of the cloning strategy.
The enzyme has the typical βαβ fold of the LMW-PTPs characterized by β1α1β2 and β3α4β4 motifs that create a central four-stranded β-sheet (Fig. 2 ▸).
Figure 2.

AmsI structure. Cartoon representation of the E. amylovora low-molecular-weight protein tyrosine phosphatase AmsI. The secondary-structure elements are named and coloured according to their N- to C-terminal position within the protein chain (e.g. from α1 to α5). This image was prepared with the PyMOL molecular-graphics system (v.1.7; Schrödinger).
This β-sheet together with helices α1, α2 and α5 sets up the hydrophobic core of the protein. The large hydrophobic moment of helix 5 (calculated with DS Visualizer 4.0; Accelrys Software Inc.) indicates its incisive role in AmsI folding (Supplementary Fig. S1). Three long loops characterize the structure: loop β2–α2 (the W-loop), loop α2–α3 and loop β5–α4 (the D-loop). In particular, the D-loop, connecting β4 and α5 and bearing the conserved catalytic Asp115 and Pro116 residues, is defined by an N-terminal type I β-turn and a C-terminal type II β-turn and is kept stretched by its interactions with helix α5. The overall fold of AmsI is very well conserved among the LMW-PTPs (a three-dimensional representation of their structural conservation is available as Supplementary Fig. S2). E. coli Wzb (PDB entry 2wmy; 51% sequence identity) is the structure closest to AmsI, with an r.m.s.d. of 0.8 Å.
No residues were found in disallowed regions of the Ramachandran plot (Supplementary Fig S3). Asn12 adopts a left-handed conformation, which is a conserved feature of the P-loop. Asp62 and Lys118 are also in a left-handed conformation and are part of the long loop connecting helices α2 and α3 and of the turn connecting α5 and the D-loop, respectively. Pro95, Ser96 and Val97 form a 310-helix turn and are part of the portion of the protein connecting helix α4 and strand β4. The dihedral angles of the central amino acids in the D-loop (residues 112–117) are in a β-strand conformation, apart from Pro116, which is located in the α-helical region of the Ramachandran plot.
The thermal displacements (B factors) of the structure were refined isotropically. The protein mean isotropic displacement (ID) is 23.1 Å2. As expected, the sulfate and the water in the active site show higher displacements of about 26.7 and 33.5 Å2, respectively. Not surprisingly, the highest IDs were found for an arginine and for a lysine residue: Arg92 (ID = 46.87 Å2) is part of helix α4 and Lys118 (ID = 43.45 Å2) is part of the turn that ends at helix α5.
3.2. Quaternary structure
In the crystallographic unit cell, the AmsI monomer is packed in a dimeric form by symmetry. Considering only the native residues, upon dimerization each monomer has a buried surface of about 424 Å2 from a total monomer surface of about 7465 Å2, as calculated by the PBDePISA server (Krissinel & Henrick, 2007 ▸; CSS score = 0.03; the complexation significance score, CSS, scores how significant the interface is for assembly formation). The buried area accounts for about 6% of the surface of each AmsI monomer. Considering that the interaction surfaces in protein complexes range from ∼600 to ∼4800 Å2 (representing 6–24% of the accessible surface area of the individual monomers, with an average of about 12%), the stability of the AmsI dimer appears to be weak (Janin et al., 1988 ▸; Kleanthous, 2000 ▸). It is worth noting that a sulfate anion, the purification tag (residues Gln−2, Tyr−4, Phe−3) and residues Lys84 and Asp109 contribute to the monomer–monomer interaction. Hence, in order to evaluate the oligomerization state of AmsI in solution, SEC-QELS experiments at different concentrations of AmsI without a His6 tag in the absence and presence of sulfate were carried out. The AmsI samples were highly monodisperse. The molecular mass was calculated to be about 16 kDa and the R g,z about 1.8 nm for every measured sample. The results reveal that in each case AmsI elutes as a monomer in solution (Supplementary Fig. S4, Supplementary Table S1). Moreover, comparing the analytical size-exclusion chromatography elution profiles of AmsI with and without the tag it is clear that His6-tagged AmsI is also a monomer in solution (Supplementary Fig. S5).
3.3. Active site
The active site is located in a crevice directly exposed to the solvent. The P-loop forms half of the active site and contains part of the consensus sequence C(X)5R(S/T), here 9CIGNICRS16, which is putatively responsible for interacting with the phosphoryl moiety of the substrate (Fig. 3 ▸). In fact, residues Cys9–Ser16 bind a sulfate molecule that keeps AmsI in a closed conformation (Tabernero et al., 2008 ▸), in contrast to the open conformation observed in a homologous structure when no ligand is present (Stehle et al., 2012 ▸).
Figure 3.
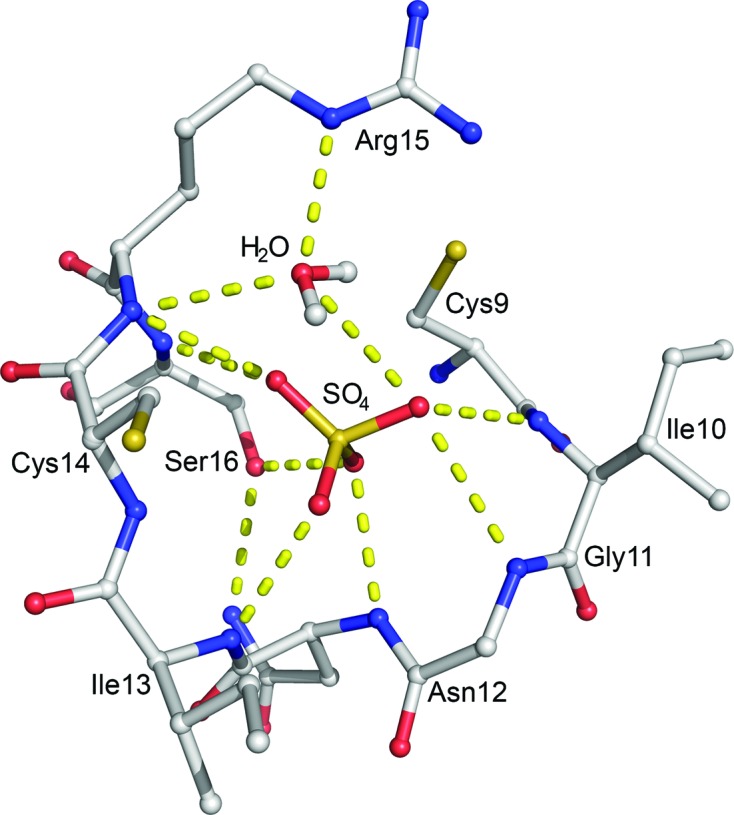
AmsI active site. Residues 10–16 of the P-loop, the sulfate (SO4) and the water molecule (H2O; HOH2017) in the active site are shown in ball-and-stick representation. Atoms are coloured as follows: carbon, grey; oxygen, red; nitrogen, blue; sulfur, yellow; hydrogen, white. The water H atoms are included in the model. Hydrogen bonds are represented as dotted yellow lines. This image was prepared with the PyMOL molecular-graphics system (v.1.7; Schrödinger).
The P-loop binds the sulfate O atoms through its peptidic amino groups. The P-loop is further stabilized by a hydrogen-bond network with residues 18–20, 34–40, Asp46 and His63. The conserved Asn12 shows the typical left-handed conformation stabilized by either the P-loop or by hydrogen bonds to His63 N∊, Ser34 Oγ, Gln66 N and Ser16 Oγ. The Arg15 side chain binds the P-loop backbone O atom of Ile113 and Glu83 O∊. The conserved Cys14 features two distinct conformations (modelled with 50% occupancy each), both pointing away from the phosphate pocket. The amino acids involved in catalysis are highly conserved and consist of Cys9, Arg15 and Asp115. Arg15 is a strictly conserved catalytic residue and has been shown to be involved in the binding of sulfate/phosphate O atoms (Wang et al., 2000 ▸; Zhang et al., 1997 ▸, 1998 ▸; Hagelueken et al., 2009 ▸; Lescop et al., 2006 ▸; Stehle et al., 2012 ▸). Interestingly, we observed an electron density that was undoubtedly modelled as a water molecule (HOH2017 in the PDB entry; a figure showing the active site with electron density is available as Supplementary Fig. S6) bridging Arg15 and the sulfate, which are shifted 1.2–1.6 Å apart compared with the corresponding arginine and sulfates or phosphates of the other reported X-ray diffraction structures (see Supplementary Fig. S7). This is also reflected in the main difference observed in the P-loop between AmsI and E. coli Wzb. Wzb Cys13 (the residue affording the phospho-cysteine intermediate during the reaction) points towards the sulfate, while in AmsI the corresponding Cys9 points away from the sulfate as it is otherwise too close. The HOH2017 molecule makes hydrogen bonds to both the sulfate O atoms and the Arg15 N∊ and backbone N atoms, and it is 3.5 Å from the Oδ atom of Asp115, which is the acid–base catalyst of the reaction.
Asp115 forms a hydrogen bond through its Oδ1 atom to the backbone N atom of Tyr117; it is placed parallel to the guanidinium group of Arg15 and establishes electrostatic interactions with it. In eukarya, the residue corresponding to Tyr117 is followed by another tyrosine that is involved in the regulation by phosphorylation (Tabernero et al., 2008 ▸). In AmsI Tyr117 is uniquely followed by two lysine residues that could be involved in the interaction with the cognate kinases. In AmsI, Met40 plays a corresponding role to the Tyr/Trp residue of the studied eukaryotic LMW-PTPs (e.g. Tyr49 in human Acp1). The entrance to the active site is dominated by the residues Ile10, Gly11, Ile13, Arg15, Met40, Asp115 and Tyr117. These amino acids form a doughnut-like structure around the entrance. Half of this structure is formed by polar residues, while the other half is formed by hydrophobic residues (Supplementary Fig. S8a). A similar pattern characterizes the entire protein surface around the active site (Supplementary Fig. S8b). Specifically, the polar part is characterized by Glu83, Lys85, Asp115, Tyr117, Lys118 and Lys119, while the hydrophobic moiety by the residues Ile10, Gly11, Leu37, Met40 and Val41. Interestingly, the hydrophobic area around the active site is interrupted by Lys38 protruding from the middle. According to our docking model, the residues in bold above are important for the AmsI–AmsA interaction.
3.4. AmsI kinetics
Stopped-assay steady-state kinetic experiments were carried out at 25°C using para-nitrophenyl phosphate (pNPP) as a substrate (Table 2 ▸).
Table 2. Kinetic parameters of AmsI.
Measurements were taken at 25°C, in 200 µl reaction volumes, using 2.65 nM protein and pNPP as substrate.
| Buffer (100 mM) | pH | Calculated ionic strength (mM) | K m (mM) | k cat (s−1) | k cat/K m (×10−3 M −1 s−1) | Fit R 2 |
|---|---|---|---|---|---|---|
| Citrate | 5.5 | 235 | 1.3 ± 0.2 | 34.4 ± 0.8 | 28 ± 5 | 0.97 |
| Citrate | 6.5 | 248 | 1.9 ± 0.4 | 10.0 ± 0.4 | 5 ± 2 | 0.93 |
| Bicine | 8.5 | 44 | 10 ± 4 | 0.8 ± 0.1 | 0.08 ± 0.03 | 0.92 |
Three acidic pH values and one basic condition were tested. Below pH 5 the proteins tended to aggregate and precipitate, no matter which buffer was used (data not shown). In citrate at pH 5.5 AmsI showed a k cat of 34.4 s−1 and a k cat/K m of 28 × 10−3 M −1 s−1. Citrate at pH 6.5 gave a k cat of 10.0 s−1 and a k cat/K m of 5 × 10−3 M −1 s−1. As expected from an acidic phosphatase, AmsI showed a lower activity rate in bicine at pH 8.5, with a k cat of 0.8 s−1.
4. Discussion and conclusions
Fire blight is a major global threat to commercial apple and pear production and is caused by the bacterium E. amylovora, which has been included in the top ten plant pathogenic bacteria (Mansfield et al., 2012 ▸). AmsI is an LMW-PTP that is necessary for E. amylovora pathogenicity by regulating the biosynthesis of the pathogenicity factor amylovoran. AmsI folds into a structure akin to all other known LMW-PTPs, emphasizing the high structural conservation in this type of phosphatase (Su et al., 1994 ▸; Tabernero et al., 2008 ▸; Zhang, Van Etten et al., 1994 ▸; Zhang et al., 1997 ▸, 1998 ▸; Zabell et al., 2006 ▸; Stehle et al., 2012 ▸; Wang et al., 2000 ▸; Lescop et al., 2006 ▸; Madhurantakam et al., 2005 ▸; Vega et al., 2011 ▸; Gustafson et al., 2005 ▸; Hagelueken et al., 2009 ▸; Xu et al., 2006 ▸). The oligomerization state of LMW-PTPs has been debated for a long time, and it is thought to regulate the enzymatic activity by competing with the substrate (Blobel et al., 2009 ▸). The dominant oligomerization process is the formation of dimers. In mammalian LMW-PTPs, the active monomers are in equilibrium with inactive dimers (Tabernero et al., 1999 ▸; Åkerud et al., 2002 ▸; Blobel et al., 2009 ▸). In prokaryotic LMW-PTPs, weak dimerization has been reported for E. coli Wzb, Bacillus subtilis YwlE and Staphylococcus aureus PtpB (Nath et al., 2014 ▸). In general, dimerization inactivates the enzyme and involves residues in the active site and tyrosines in the D-loop. Recently, Nath and coworkers revealed that VcLMWPTP from Vibrio cholerae dimerizes in a novel fashion, involving new interactions and leaving the enzyme active (Nath et al., 2014 ▸). AmsI is present as a dimer in the crystal unit cell, with a previously unreported dimerization interface which involves part of the His6 tag and a sulfate ion. However, we demonstrate that AmsI is a permanent fully active monomer in solution and that the dimer is a crystal-packing artefact that is not functionally relevant.
In the AmsI active-site pocket a water molecule bridges the catalytic arginine and the sulfate. Such a scenario has only been proposed for the NMR structure of B. subtilis YwlE and is not observed in the structures of other homologues (Xu et al., 2006 ▸). In contrast to the other LMW-PTP structural models, the sulfate in AmsI is more deeply nestled within the P-loop and is further from the Arg15 side chain. In our scenario, in which it is in close proximity to both the sulfate and the catalytic cysteine, could the water molecule be representative of that attacking the phospho-Cys9 during the second step of the reaction? In fact, during this step Arg15 may act as the base, abstracting a proton from the water and favouring the hydrolysis of the phospho-Cys9 intermediate phospho-thionate bond. Afterwards, Arg15 could donate the proton to the nearby Asp115. Further studies will be needed to address this hypothesis.
In AmsI the sulfate binds to the P-loop residues CX5RS (bold), to the abovementioned water molecule and to the side-chain O atom and the backbone N atom of the P-loop Ser16. This situation is not observed in the homologous LMW-PTPs, where the ligand is bound to Arg N∊, Arg Nη2 and sometimes to the catalytic cysteine. Another exception is found in Entamoeba histocolytica EhPTP, where there is a sulfate ligand bound both to the P-loop serine and the catalytic cysteine.
The kinetic study of AmsI shows a decay in activity at basic pH. In the conditions used in this study citrate pH 5.5 is the optimal buffer. This observation is consistent with the results obtained for other LMW-PTPs, such as those from B. subtilis, S. aureus and Yersinia enterocolitica, with the best conditions at pH 5.5–6.0, 6.2 and 5.5, respectively (Zhang, Wang et al., 1994 ▸; Musumeci et al., 2005 ▸; Soulat et al., 2002 ▸).
Structural and mutational studies on the AmsI–AmsA complex are planned to understand the protein–protein interaction between the two partners. To provide a hint towards a potential model of the interaction, we performed homology modelling of the AmsI cognate kinase AmsA, and based on the data published by Temel et al. (2013 ▸), and new partial data from the same group, we identified potential putative interaction patches on the surface of the two proteins. This information can be found in the Supporting Information (§S2, Supplementary Tables S2 and S3, Supplementary Figs. S9, S10, S11 and S12).
Supplementary Material
PDB reference: AmsI, 4d74
Supporting Information containing supplementary figures and tables: structural aspects, SEC-QELS chromatogram, molecular modeling and docking etc.. DOI: 10.1107/S2053230X16018781/dz5400sup1.pdf
Acknowledgments
We thank Dr M. Malnoy (Fondazione Edmund Mach, S. Michele all’Adige, Trento, Italy) for providing the genomic DNA from E. amylovora strain Ea273. We thank Professor R. Ghose of the Department of Chemistry and Biochemistry of the City College of New York for supplying the experimental NMR data used in the docking simulation. We thank the CERM (Centro di Risonanze Magnetiche), Sesto Fiorentino, Italy for making available the QELS facility, with special regard to Dr Leonardo Gonnelli for his precious help with QELS measurements and data analysis. Plasmid pETM-11 was obtained from the European Molecular Biology Laboratory under a signed Material Transfer Agreement. Data were collected under the European Molecular Biology Laboratory beam time award No. MX-152. The work was supported by the Fondazione Libera Università di Bolzano and by the Autonomous Province of Bolzano project: ‘A structural genomics approach for the study of the virulence and pathogenesis of Erwinia amylovora’. FM was supported by the University of Bologna through the FARB Program (Finanziamenti dell’Alma Mater Studiorum alla Ricerca di Base).
References
- Åkerud, T., Thulin, E., Van Etten, R. L. & Akke, M. (2002). J. Mol. Biol. 322, 137–152. [DOI] [PubMed]
- Bechet, E., Gruszczyk, J., Terreux, R., Gueguen-Chaignon, V., Vigouroux, A., Obadia, B., Cozzone, A. J., Nessler, S. & Grangeasse, C. (2010). Mol. Microbiol. 77, 1315–1325. [DOI] [PubMed]
- Bechet, E., Guiral, S., Torres, S., Mijakovic, I., Cozzone, A. J. & Grangeasse, C. (2009). Amino Acids, 37, 499–507. [DOI] [PubMed]
- Bellemann, P. & Geider, K. (1992). J. Gen. Microbiol. 138, 931–940. [DOI] [PubMed]
- Benini, S., Caputi, L. & Cianci, M. (2014). Acta Cryst. F70, 1693–1696. [DOI] [PMC free article] [PubMed]
- Bernhard, F., Coplin, D. L. & Geider, K. (1993). Mol. Gen. Genet. 239, 158–168. [DOI] [PubMed]
- Bisswanger, H. (2011). Practical Enzymology, 2nd ed. Weinheim: Wiley-VCH.
- Bisswanger, H. (2014). Perspect. Sci. 1, 41–55.
- Blenis, J. (1993). Proc. Natl Acad. Sci. USA, 90, 5889–5892. [DOI] [PMC free article] [PubMed]
- Blobel, J., Bernadó, P., Xu, H., Jin, C. & Pons, M. (2009). FEBS J. 276, 4346–4357. [DOI] [PubMed]
- Bugert, P. & Geider, K. (1995). Mol. Microbiol. 15, 917–933. [DOI] [PubMed]
- Bugert, P. & Geider, K. (1997). FEBS Lett. 400, 252–256. [DOI] [PubMed]
- Byrne, J. P., Morona, J. K., Paton, J. C. & Morona, R. (2011). J. Bacteriol. 193, 2341–2346. [DOI] [PMC free article] [PubMed]
- Caputi, L., Cianci, M. & Benini, S. (2013). Acta Cryst. F69, 570–573. [DOI] [PMC free article] [PubMed]
- Caputi, L., Nepogodiev, S. A., Malnoy, M., Rejzek, M., Field, R. A. & Benini, S. (2013). J. Agric. Food Chem. 61, 12265–12273. [DOI] [PubMed]
- Caselli, A., Paoli, P., Santi, A., Mugnaioni, C., Toti, A., Camici, G. & Cirri, P. (2016). Biochim. Biophys. Acta, 1864, 1339–1355. [DOI] [PubMed]
- Charbonneau, H. & Tonks, N. K. (1992). Annu. Rev. Cell Biol. 8, 463–493. [DOI] [PubMed]
- Cozzone, A. J., Grangeasse, C., Doublet, P. & Duclos, B. (2004). Arch. Microbiol. 181, 171–181. [DOI] [PubMed]
- Davis, J. P., Zhou, M.-M. & Van Etten, R. L. (1994). J. Biol. Chem. 269, 8734–8740. [PubMed]
- Denu, J. M. & Dixon, J. E. (1995). Proc. Natl Acad. Sci. USA, 92, 5910–5914. [DOI] [PMC free article] [PubMed]
- Denu, J. M., Lohse, D. L., Vijayalakshmi, J., Saper, M. A. & Dixon, J. E. (1996). Proc. Natl Acad. Sci. USA, 93, 2493–2498.
- Denu, J. M., Zhou, G., Guo, Y. & Dixon, J. E. (1995). Biochemistry, 34, 3396–3403. [DOI] [PubMed]
- Dümmler, A., Lawrence, A. M. & de Marco, A. (2005). Microb. Cell Fact. 4, 34. [DOI] [PMC free article] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Evans, P. R. (2011). Acta Cryst. D67, 282–292. [DOI] [PMC free article] [PubMed]
- Evans, B., Tishmack, P. A., Pokalsky, C., Zhang, M. & Van Etten, R. L. (1996). Biochemistry, 35, 13609–13617. [DOI] [PubMed]
- Gardossi, L., Poulsen, P. B., Ballesteros, A., Hult, K., Švedas, V. K., Vasić-Rački, Đ., Carrea, G., Magnusson, A., Schmid, A., Wohlgemuth, R. & Halling, P. J. (2010). Trends Biotechnol. 28, 171–180. [DOI] [PubMed]
- Gasteiger, E., Hoogland, C., Gattiker, A., Duvaud, S., Wilkins, M., Appel, R. & Bairoch, A. (2005). The Proteomics Protocols Handbook, edited by J. M. Walker, pp. 571–607. Totowa: Humana Press.
- Geier, G. & Geider, K. (1993). Physiol. Mol. Plant Pathol. 42, 387–404.
- Grangeasse, C., Cozzone, A. J., Deutscher, J. & Mijakovic, I. (2007). Trends Biochem. Sci. 32, 86–94. [DOI] [PubMed]
- Grangeasse, C., Nessler, S. & Mijakovic, I. (2012). Philos. Trans. R. Soc. B Biol. Sci. 367, 2640–2655. [DOI] [PMC free article] [PubMed]
- Grangeasse, C., Obadia, B., Mijakovic, I., Deutscher, J., Cozzone, A. J. & Doublet, P. (2003). J. Biol. Chem. 278, 39323–39329. [DOI] [PubMed]
- Grangeasse, C., Terreux, R. & Nessler, S. (2010). Biochim. Biophys. Acta, 1804, 628–634. [DOI] [PubMed]
- Gusberti, M., Klemm, U., Meier, M. S., Maurhofer, M. & Hunger-Glaser, I. (2015). Int. J. Environ. Res. Publ. Health, 12, 11422–11447. [DOI] [PMC free article] [PubMed]
- Gustafson, C. L., Stauffacher, C. V., Hallenga, K. & Van Etten, R. L. (2005). Protein Sci. 14, 2515–2525. [DOI] [PMC free article] [PubMed]
- Hagelueken, G., Huang, H., Mainprize, I. L., Whitfield, C. & Naismith, J. H. (2009). J. Mol. Biol. 392, 678–688. [DOI] [PMC free article] [PubMed]
- Janin, J., Miller, S. & Chothia, C. (1988). J. Mol. Biol. 204, 155–164. [DOI] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Kleanthous, C. (2000). Editor. Protein–Protein Recognition. Oxford University Press.
- Kolot, M., Gorovits, R., Silberstein, N., Fichtman, B. & Yagil, E. (2008). Virology, 375, 383–390. [DOI] [PubMed]
- Krissinel, E. & Henrick, K. (2007). J. Mol. Biol. 372, 774–797. [DOI] [PubMed]
- Lacour, S., Bechet, E., Cozzone, A. J., Mijakovic, I. & Grangeasse, C. (2008). PLoS One, 3, e3053. [DOI] [PMC free article] [PubMed]
- Lescop, E., Hu, Y., Xu, H., Hu, W., Xia, B. & Jin, C. (2006). J. Biol. Chem. 281, 19570–19577. [DOI] [PubMed]
- Long, F., Vagin, A. A., Young, P. & Murshudov, G. N. (2008). Acta Cryst. D64, 125–132. [DOI] [PMC free article] [PubMed]
- Madhurantakam, C., Rajakumara, E., Mazumdar, P. A., Saha, B., Mitra, D., Wiker, H. G., Sankaranarayanan, R. & Das, A. K. (2005). J. Bacteriol. 187, 2175–2181. [DOI] [PMC free article] [PubMed]
- Mansfield, J., Genin, S., Magori, S., Citovsky, V., Sriariyanum, M., Ronald, P., Dow, M., Verdier, V., Beer, S. V., Machado, M. A., Toth, I., Salmond, G. & Foster, G. D. (2012). Mol. Plant Pathol. 13, 614–629. [DOI] [PMC free article] [PubMed]
- Morona, J. K., Morona, R. & Paton, J. C. (2006). Proc. Natl Acad. Sci. USA, 103, 8505–8510. [DOI] [PMC free article] [PubMed]
- Musumeci, L., Bongiorni, C., Tautz, L., Edwards, R. A., Osterman, A., Perego, M., Mustelin, T. & Bottini, N. (2005). J. Bacteriol. 187, 4945–4956. [DOI] [PMC free article] [PubMed]
- Nath, S., Banerjee, R. & Sen, U. (2014). Biochem. Biophys. Res. Commun. 450, 390–395. [DOI] [PubMed]
- Nimtz, M., Mort, A., Domke, T., Wray, V., Zhang, Y., Qiu, F., Coplin, D. & Geider, K. (1996). Carbohydr. Res. 287, 59–76. [DOI] [PubMed]
- O’Riordan, K. & Lee, J. C. (2004). Clin. Microbiol. Rev. 17, 218–234. [DOI] [PMC free article] [PubMed]
- Ostanin, K., Pokalsky, C., Wang, S. & Van Etten, R. L. (1995). J. Biol. Chem. 270, 18491–18499. [DOI] [PubMed]
- Raugei, G., Ramponi, G. & Chiarugi, P. (2002). Cell. Mol. Life Sci., 59, 941–949. [DOI] [PMC free article] [PubMed]
- Robert, X. & Gouet, P. (2014). Nucleic Acids Res. 42, W320–W324. [DOI] [PMC free article] [PubMed]
- Soulat, D., Vaganay, E., Duclos, B., Genestier, A. L., Etienne, J. & Cozzone, A. J. (2002). J. Bacteriol. 184, 5194–5199. [DOI] [PMC free article] [PubMed]
- Stehle, T., Sreeramulu, S., Löhr, F., Richter, C., Saxena, K., Jonker, H. R. & Schwalbe, H. (2012). J. Biol. Chem. 287, 34569–34582. [DOI] [PMC free article] [PubMed]
- Su, X.-D., Taddei, N., Stefani, M., Ramponi, G. & Nordlund, P. (1994). Nature (London), 370, 575–578. [DOI] [PubMed]
- Tabernero, L., Aricescu, A. R., Jones, E. Y. & Szedlacsek, S. E. (2008). FEBS J. 275, 867–882. [DOI] [PubMed]
- Tabernero, L., Evans, B. N., Tishmack, P. A., Van Etten, R. L. & Stauffacher, C. V. (1999). Biochemistry, 38, 11651–11658. [DOI] [PubMed]
- Temel, D. B., Dutta, K., Alphonse, S., Nourikyan, J., Grangeasse, C. & Ghose, R. (2013). J. Biol. Chem. 288, 15212–15228. [DOI] [PMC free article] [PubMed]
- Tipton, K. F., Armstrong, R. N., Bakker, B. M., Bairoch, A., Cornish-Bowden, A., Halling, P. J., Hofmeyr, J., Leyh, T. S., Kettner, C., Raushel, F. M., Rohwer, J., Schomburg, D. & Steinbeck, C. (2014). Perspect. Sci. 1, 131–137.
- Vanneste, J. L. (2000). Fire Blight. New York: CABI Publishing.
- Vega, C., Chou, S., Engel, K., Harrell, M. E., Rajagopal, L. & Grundner, C. (2011). J. Mol. Biol. 413, 24–31. [DOI] [PMC free article] [PubMed]
- Walton, K. M. & Dixon, J. E. (1993). Annu. Rev. Biochem. 62, 101–120. [DOI] [PubMed]
- Wang, S., Tabernero, L., Zhang, M., Harms, E., Van Etten, R. L. & Stauffacher, C. V. (2000). Biochemistry, 39, 1903–1914. [DOI] [PubMed]
- Wuerges, J., Caputi, L., Cianci, M., Boivin, S., Meijers, R. & Benini, S. (2015). J. Struct. Biol. 191, 290–298. [DOI] [PubMed]
- Xu, H., Xia, B. & Jin, C. (2006). J. Bacteriol. 188, 1509–1517. [DOI] [PMC free article] [PubMed]
- Yuvaniyama, J., Denu, J. M., Dixon, J. E. & Saper, M. A. (1996). Science, 272, 1328–1331. [DOI] [PubMed]
- Zabell, A. P., Schroff, A. D. Jr, Bain, B. E., Van Etten, R. L., Wiest, O. & Stauffacher, C. V. (2006). J. Biol. Chem. 281, 6520–6527. [DOI] [PubMed]
- Zhang, M., Stauffacher, C. V., Lin, D. & Van Etten, R. L. (1998). J. Biol. Chem. 273, 21714–21720. [DOI] [PubMed]
- Zhang, M., Stauffacher, D. L. & Van Etten, R. L. (1995). Advances In Protein Phosphatases, Vol. 9, edited by W. Merlevede, pp. 1–23. Leuven University Press.
- Zhang, M., Van Etten, R. L. & Stauffacher, C. V. (1994). Biochemistry, 33, 11097–11105. [DOI] [PubMed]
- Zhang, M., Zhou, M., Van Etten, R. L. & Stauffacher, C. V. (1997). Biochemistry, 36, 15–23. [DOI] [PubMed]
- Zhang, Z., Harms, E. & Van Etten, R. L. (1994). J. Biol. Chem. 269, 25947–25950. [PubMed]
- Zhang, Z.-Y. & Dixon, J. E. (1994). Adv. Enzymol. Relat. Areas Mol. Biol. 68, 1–36. [DOI] [PubMed]
- Zhang, Z.-Y. & Van Etten, R. L. (1991). Biochemistry, 30, 8954–8959. [DOI] [PubMed]
- Zhang, Z.-Y., Wang, Y. & Dixon, J. E. (1994). Proc. Natl Acad. Sci. USA, 91, 1624–1627. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: AmsI, 4d74
Supporting Information containing supplementary figures and tables: structural aspects, SEC-QELS chromatogram, molecular modeling and docking etc.. DOI: 10.1107/S2053230X16018781/dz5400sup1.pdf