

Abstract
Context
Cerebrotendinous xanthomatosis (CTX) is an autosomal recessively inherited lipid storage disease caused by mutation in the CYP27A1 gene. Spinal form CTX is a rare clinical subgroup of CTX and only 14 patients from 11 families have been reported to date. Here, we report the first Asian patient with spinal form CTX showing characteristic radiological findings.
Findings
The patient, a 46-year-old Japanese male, developed sensory disturbance of the lower legs at 39 and spastic gait at 46 years of age. Spinal cord magnetic resonance imaging (MRI) revealed a long hyperintense lesion involving lateral corticospinal tracts and gracile tracts in the cervical and thoracic cord on T2-weighted images. Gallium-67 (67Ga) scintigraphy revealed abnormal uptake in the Achilles tendons and the serum cholestanol level was elevated. CYP27A1 gene analysis identified homozygous missense mutation, c.1214G>A (p.R405Q). The patient was treated with atorvastatin monotherapy, which reduced serum cholestanol to less than 50% of the pretreatment level.
Conclusion
Spinal form CTX should be considered in the differential diagnosis of cryptogenic myelopathy, especially in patients with a long spinal cord lesion, as treatment with chenodeoxycholic acid and/or competitive inhibitors of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase reverse the metabolic derangement and prevent the neurologiccal dysfunction.
Keywords: Cerebrotendinous xanthomatosis, Cholestanol, CYP27A1gene, Long spinal cord lesion, Myelopathy
Introduction
Cerebrotendinous xanthomatosis (CTX, MIM# 213700) is an autosomal recessively inherited lipid storage disease caused by mutation in the CYP27A1 gene encoding sterol 27-hydroxylase (CYP27A1, EC 1.14.13.15), a key enzyme in the synthesis of chenodeoxycholic acid (CDCA), a primary bile acid.1,2 CYP27A1 catalyzes the initial oxidation in bile acids in the liver, therefore its deficiency results in an impaired bile acid synthesis and increased production of cholesterol metabolites, such as cholestanol, which subsequently accumulate in many tissues, especially the lenses of the eye, brain, tendons, vessels, and bones. Typical disease onset consists of bilateral cataracts and diarrhea in childhood, followed by progressive cerebellar and pyramidal signs, cognitive impairment, seizures, and the development of tendon xanthomas in late adolescence or early adulthood (classic form). In addition to the classic form, another phenotype of CTX, designated as spinal form, has been reported.3,4 Patients with spinal form CTX show a slowly progressive myelopathy, including pyramidal and dorsal column signs,3,4 however, detailed clinical findings of spinal form CTX have not been described yet. Here, we report detailed clinical, neuroradiological, and molecular biological findings of a Japanese spinal form CTX patient homozygous for p.R405Q mutation in the CYP27A1 gene.
Case report
The patient, a 46-year-old Japanese male, was the second child of non-consanguineous healthy parents and had a healthy sibling. He was born at term after a normal pregnancy and showed normal development. He graduated from a regular community high school and was employed as a furniture worker. At 39 years of age, he developed dysesthesia of the lower legs, which deteriorated gradually. At 46 years of age, he developed gait disturbance and was admitted to our hospital.
On admission, general examination of the patient was unremarkable with neither cataract nor Achilles tendon xanthomas. On neurological examination, he was alert and well-oriented. There were no abnormal findings in the cranial nerves. Mild weakness was observed in the distal portions of the lower limbs but was absent in the upper limbs. Decreased light touch, pain, and vibration sense were observed in the lower legs. Position sense was intact and Romberg's sign was negative. Increased tendon reflex of the extremities, ankle clonus, and positivity for Wartenberg, Babinski, and Chaddock signs were observed on both sides. His gait was spastic.
Findings of routine blood examination, cerebrospinal fluid, chest roentgenography, and electrocardiography were all normal, but serum cholestanol level was markedly elevated (24.1 μg/mL; normal, 1.91–3.51 μg/mL). Serum vitamin B12, folic acid, copper, and ceruloplasmin, and plasma very long chain fatty acid were normal. Bone mineral density at the lumbar spine assessed by dual-energy X-ray absorptiometry was markedly decreased (0.696 g/cm2, T-score –4.1). Gallium-67 (67Ga) scintigraphy revealed abnormal uptake in the Achilles tendons, although xanthoma was not detected on magnetic resonance imaging (MRI) (Figs. 1A–C). Brain MRI revealed slightly increased signal intensity in the bilateral cerebral white matter on T2-weighted and fluid-attenuated inversion recovery (FLAIR) images. Spinal cord MRI showed long hyperintense lesions involving lateral corticospinal tracts and gracile tracts in the cervical and thoracic cord extending from C1 to Th4 level on T2-weighted images (Figs. 2A–D).
Figure 1.
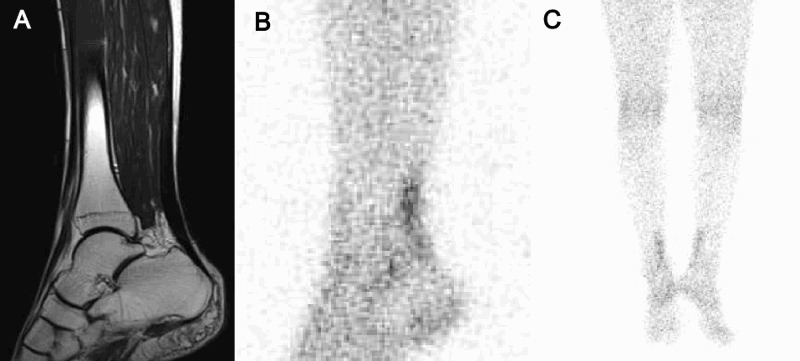
Achilles tendon of the patient. (A) Magnetic resonance imaging (MRI), T2-weighted image. (B, C) Gallium-67 (67Ga) scintigraphy. Achilles tendon xanthomas were not observed on physical examination or MRI; however, abnormal uptake of 67Ga was observed in the Achilles tendons.
Figure 2.
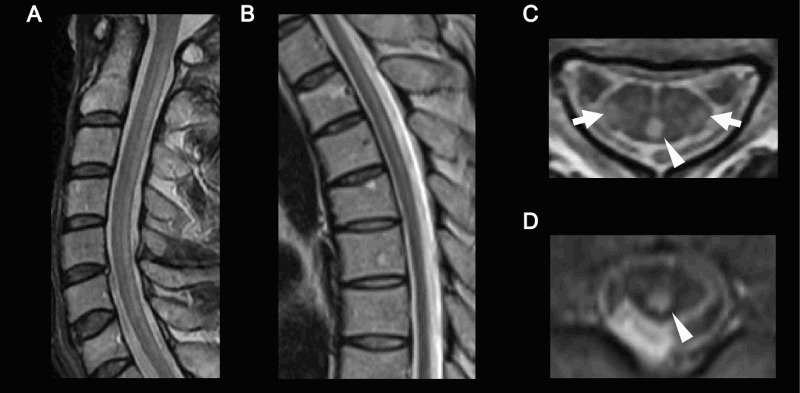
Spinal cord magnetic resonance imaging (MRI) of the patient (T2-weighted image). (A) Cervical cord, sagittal view. (B) Thoracic cord, sagittal view. (C) Cervical cord, axial view. (D) Thoracic cord, axial view. Long hyperintense lesions involving lateral corticospinal tracts (arrows) and gracile tracts (arrow heads) were observed.
As clinical findings of the patient were suggestive of spinal form CTX, genetic analysis for this disorder was performed with informed consent. DNA was extracted from peripheral leukocytes of the patient and his parents according to the standard protocol. All nine exons of the CYP27A1 gene were amplified by polymerase chain reaction (PCR) as described previously.5 Direct sequence analysis of the PCR-amplified DNA from the patient identified homozygous missense mutation, c.1214G > A, which resulted in amino acid alteration of p.R405Q. The parents were heterozygous for the c.1214G > A (p.R405Q) mutation.
The patient was treated with 750 mg/day of chenodeoxycholic acid (CDCA) and 10 mg/day of atorvastatin calcium hydrate. However, CDCA was discontinued due to drug-induced liver injury and therefore, the patient was treated with atorvastatin monotherapy. After 2 months of treatment with atorvastatin, the serum concentration of cholestanol decreased to 9.1 μg/mL.
Discussion
Spinal form CTX, also called spinal xanthomatosis, is a rare clinical subgroup of CTX and only 14 patients from 11 families have been reported to date.3,4 Patients with spinal form CTX show slowly progressive myelopathy, including pyramidal and dorsal column signs, and have a relatively mild clinical course compared with the classic form of CTX, in which most patients show cerebellar ataxia, dementia, and tendon xanthomas in the disease process. Verrips et al.3 reported that extensive white matter lesions in the lateral corticospinal tracts and in the gracile tracts are characteristic MRI findings in spinal form CTX, which were also seen in the present patient (Fig. 2). Pathologically, extensive symmetric loss of myelin and axons accompanied by gliosis and perivascular accumulation of macrophages are present in the white matter of the spinal cord, especially in the lateral corticospinal tracts and gracile tracts.3 In the present patient, Achilles tendon xanthomas were not observed on physical examination or MRI; however, abnormal uptake of 67Ga was observed in the Achilles tendons (Fig. 1). The uptake of 67Ga by xanthoma is considered to result from proliferation of histiocytes that phagocytose transferrin combined with 67Ga.6,7 Our observations indicate that 67Ga scintigraphy may be more sensitive for detecting tiny xanthomas than MRI and may be useful for monitoring the effects of treatment.
Molecular genetic analysis of the CYP27A1 gene revealed that the patient was homozygous for c.1214G>A (p.R405Q) mutation, confirming the diagnosis of CTX. The R405 residue is located near the adrenodoxin-binding site of the sterol 27-hydroxylase enzyme and would be expected to affect the activity of the enzyme. In addition, transient expression study of CYP27A1 cDNA revealed that p.R405Q protein did not show enzymatic activity.8 The p.R405Q mutation was reported previously in five CTX families.8–11 Chen et al. reported a Japanese family homozygous for p.R405Q mutation, who showed typical CTX manifestations, including tendon xanthomas, cataracts, neurological dysfunctions, elevated serum cholestanol level, and undetectable sterol 27-hydroxylase activities.8 The other four families were compound heterozygous and all patients showed classic form CTX phenotype.8–11 Similar to the p.R405Q mutation, all mutations observed in spinal form CTX were also found in classic form CTX, indicating that none of these mutations is preferentially associated with either the classic or spinal form of the disease.3,4
Although CTX can mimic neurodegenerative diseases, such as spastic paraplegia and spinocerebellar degeneration, early diagnosis of CTX is crucial because treatment with CDCA reverses metabolic derangement and can prevent or even improve neurological dysfunction.12–14 CDCA is one of the major biliary bile acids in humans and has been used as medical therapy to dissolve gallstones. CDCA is a relatively safe drug, but gastrointestinal manifestations (e.g. diarrhea, abdominal pain or discomfort, heartburn, and bloating) and liver injury are frequent side effects. Competitive inhibitors of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase were also administered to several CTX patients.15–19 The patient discussed here was treated with atorvastatin monotherapy, as CDCA induced liver injury. There are limited data on the clinical benefits of treatment with HMG-CoA reductase in CTX patients, and the results reported to date remain controversial.15–19 Lewis et al.19 reported that mevinolin (13 mg/day) normalized serum cholestanol level within four days after commencement of treatment and reduced the size of the xanthoma. On the other hand, Batta et al.17 reported that lovastatin (40 mg/day) did not affect abnormal bile acid synthesis or reduce plasma cholestanol level. Three studies15,16,18 indicated that HMG-CoA reductase add-on treatment to CDCA is effective to further reduce serum cholestanol level15,16 and normalize bile alcohol excretion in urine18 in CTX patients. In addition, Nakamura et al.15 and Luyckx et al.18 reported that CTX patients who previously responded to CDCA showed further improvement in clinical symptoms and neurophysiological findings with administration of HMG-CoA reductase. However, clinical symptoms and laboratory data were re-exacerbated after switching from combination therapy to HMG-CoA reductase monotherapy.15,18
The present patient was treated with atorvastatin monotherapy, which reduced serum cholesterol to less than 50% of the pretreatment level, although whether this monotherapy will be effective for long-term prevention of clinical deterioration remains to be established. Spinal form CTX should be considered in any individual presenting cryptogenic spastic paraplegia, especially with a long spinal cord lesion involving lateral corticospinal tracts and gracile tracts on MRI.
Disclaimer statements
Contributors The authors thank Ms. E. Nomura for her technical support.
Funding None.
Conflicts of interest None.
Ethics approval No ethical approval was required.
References
- 1.Cali JJ, Hsieh CL, Francke U, Russell DW. Mutations in the bile acid biosynthetic enzyme sterol 27-hydroxylase underlie cerebrotendinous xanthomatosis. J Biol Chem 1991;266(12):7779–83. [PMC free article] [PubMed] [Google Scholar]
- 2.Cali JJ, Russell DW. Characterization of human sterol 27-hydroxylase. A mitochondrial cytochrome P-450 that catalyzes multiple oxidation reaction in bile acid biosynthesis. J Biol Chem 1991;266(12):7774–8. [PubMed] [Google Scholar]
- 3.Verrips A, Nijeholt GJ, Barkhof F, Van Engelen BG, Wesseling P, Luyten JA, et al. Spinal xanthomatosis: a variant of cerebrotendinous xanthomatosis. Brain 1999;122(8):1589–95. doi: 10.1093/brain/122.8.1589 [DOI] [PubMed] [Google Scholar]
- 4.Pilo-de-la-Fuente B, Jimenez-Escrig A, Lorenzo JR, Pardo J, Arias M, Ares-Luque A, et al. Cerebrotendinous xanthomatosis in Spain: clinical, prognostic, and genetic survey. Eur J Neurol 2011;18(10):1203–11. doi: 10.1111/j.1468-1331.2011.03439.x [DOI] [PubMed] [Google Scholar]
- 5.Nozue T. Genetic analysis of cerebrotendinous xanthomatosis patients and the effect of treatment with chenodeoxycholic acid. J Juzen Med Soc 2002;111(1):20–34. [Google Scholar]
- 6.Larson SM, Rasey JS, Allen DR, Grunbaum Z. A transferrin-mediated uptake of gallium-67 by EMT-6 sarcoma. II. Studies in vivo (BALB/c mice): concise communication. J Nucl Med 1979;20(8):843–6. [PubMed] [Google Scholar]
- 7.Okada J, Oonishi H, Tamada H, Kizaki T, Yasumi K, Matuo T. Gallium uptake in cerebrotendinous xanthomatosis. Eur J Nucl Med 1995;22(9):1069–72. doi: 10.1007/BF00808420 [DOI] [PubMed] [Google Scholar]
- 8.Chen W, Kubota S, Kim KS, Cheng J, Kuriyama M, Eggertsen G, et al. Novel homozygous and compound heterozygous mutations of sterol 27-hydroxylase gene (CYP27) cause cerebrotendinous xanthomatosis in three Japanese patients from two unrelated families. J Lipid Res 1997;38(5):870–9. [PubMed] [Google Scholar]
- 9.Suh S, Kim HK, Park HD, Ki CS, Kim MY, Jin SM, et al. Three siblings with cerebrotendinous xanthomatosis: a novel mutation in the CYP27A1 gene. Eur J Med Genet 2012;55(1):71–4. doi: 10.1016/j.ejmg.2011.08.003 [DOI] [PubMed] [Google Scholar]
- 10.Kuriyama M, Fujiyama J, Yoshidome H, Takenaga S, Matsumuro K, Kasama T, et al. Cerebrotendinous xanthomatosis: clinical and biochemical evaluation of eight patients and review of the literature. J Neurol Sci 1991;102(2):225–32. doi: 10.1016/0022-510X(91)90073-G [DOI] [PubMed] [Google Scholar]
- 11.Yoshinaga T, Sekijima Y, Koyama S, Maruyama K, Yoshida T, Kato T, et al. Clinical and radiological findings of a cerebrotendinous xanthomatosis patient with a novel p.A335 V mutation in the CYP27A1 gene. Intern Med 2014;53(23):2725–9. doi: 10.2169/internalmedicine.53.2996 [DOI] [PubMed] [Google Scholar]
- 12.Berginer VM, Salen G, Shefer S. Long-term treatment of cerebrotendinous xanthomatosis with chenodeoxycholic acid. N Engl J Med 1984;311(26):1649–52. doi: 10.1056/NEJM198412273112601 [DOI] [PubMed] [Google Scholar]
- 13.van Heijst AF, Verrips A, Wevers RA, Cruysberg JR, Renier WO, Tolboom JJ. Treatment and follow-up of children with cerebrotendinous xanthomatosis. Eur J Pediatr 1998;157(4):313–6. doi: 10.1007/s004310050818 [DOI] [PubMed] [Google Scholar]
- 14.Mondelli M, Sicurelli F, Scarpini C, Dotti MT, Federico A. Cerebrotendinous xanthomatosis: 11-year treatment with chenodeoxycholic acid in five patients. An electrophysiological study. J Neurol Sci 2001;190(1–2):29–33. doi: 10.1016/S0022-510X(01)00563-9 [DOI] [PubMed] [Google Scholar]
- 15.Nakamura T, Matsuzawa Y, Takemura K, Kubo M, Miki H, Tarui S. Combined treatment with chenodeoxycholic acid and pravastatin improves plasma cholestanol levels associated with marked regression of tendon xanthomas in cerebrotendinous xanthomatosis. Metabolism 1991;40(7):741–6. doi: 10.1016/0026-0495(91)90094-D [DOI] [PubMed] [Google Scholar]
- 16.Verrips A, Wevers RA, Van Engelen BG, Keyser A, Wolthers BG, Barkhof F, et al. Effect of simvastatin in addition to chenodeoxycholic acid in patients with cerebrotendinous xanthomatosis. Metabolism 1999;48(2):233–8. doi: 10.1016/S0026-0495(99)90040-9 [DOI] [PubMed] [Google Scholar]
- 17.Batta AK, Salen G, Tint GS. Hydrophilic 7 beta-hydroxy bile acids, lovastatin, and cholestyramine are ineffective in the treatment of cerebrotendinous xanthomatosis. Metabolism 2004;53(5):556–62. doi: 10.1016/j.metabol.2003.12.003 [DOI] [PubMed] [Google Scholar]
- 18.Luyckx E, Eyskens F, Simons A, Beckx K, Van West D, Dhar M. Long-term follow-up on the effect of combined therapy of bile acids and statins in the treatment of cerebrotendinous xanthomatosis: a case report. Clin Neurol Neurosurg 2014;118(3):9–11. doi: 10.1016/j.clineuro.2013.12.008 [DOI] [PubMed] [Google Scholar]
- 19.Lewis B, Mitchell WD, Marenah CB, Cortese C, Reynolds EH, Shakir R. Cerebrotendinous xanthomatosis: biochemical response to inhibition of cholesterol synthesis. Br Med J (Clin Res Ed) 1983;287(6384):21–2. doi: 10.1136/bmj.287.6384.21-a [DOI] [PMC free article] [PubMed] [Google Scholar]