

Abstract
The phosphatidylinositol‐3 kinase (PI3K)–AKT pathway is one of the most commonly dysregulated pathways in all of cancer, with somatic mutations, copy number alterations, aberrant epigenetic regulation and increased expression in a number of cancers. The carefully maintained homeostatic balance of cell division and growth on one hand, and programmed cell death on the other, is universally disturbed in tumorigenesis, and downstream effectors of the PI3K–AKT pathway play an important role in this disturbance. With a wide array of downstream effectors involved in cell survival and proliferation, the well‐characterized direct interactions of AKT make it a highly attractive yet elusive target for cancer therapy. Here, we review the salient features of this pathway, evidence of its role in promoting tumorigenesis and recent progress in the development of therapeutic agents that target AKT.
Keywords: clinical oncology, medical oncology, phosphatidylinositol 3‐kinases, protein kinase B, proto‐oncogene proteins c‐AKT, proto‐oncogene proteins c‐AKT/genetics, proto‐oncogene proteins c‐AKT/metabolism, proto‐oncogene proteins c‐AKT/physiology, signal transduction/physiology
Overview of the phosphatidylinositol‐3 kinase (PI3K)–AKT pathway
A number of intrinsic and extrinsic cell survival signals are transduced downstream through the PI3K–AKT pathway, ultimately resulting in increased proliferation, a loss of apoptosis signalling and cell growth, earning it the nickname of the ‘survival pathway’ 1, 2. The pathway can be activated upstream by a wide variety of receptor protein tyrosine kinases [RTKs; including the epidermal growth factor receptor (EGFR), vascular endothelial growth factor receptor (VEGFR2) and platelet‐derived growth factor receptor (PDGFR)], cytokine receptors, G‐protein coupled receptors (GPCRs), intracellular tyrosine kinases and intracellular small GTPases such as Ras 2 (the nomenclature used is as per Alexander et al. 3). In the case of RTKs, activation by ligand binding results in the noncovalent association of PI3Ks with phosphotyrosine consensus motifs on the intracellular domain of the RTK (Figure 1) 4. One or two N‐terminal src‐homology 2 (SH2) domains on the regulatory subunit of PI3K participate in this interaction, which results in allosteric changes to the catalytic subunit of PI3K and thus functional activation of its kinase domain 4, 5.
Figure 1.
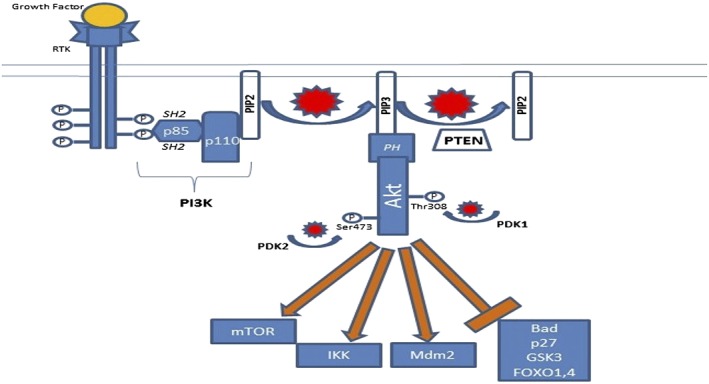
The phosphatidylinositol‐3 kinase (PI3K)–AKT PI3K‐AKT pathway is activated upstream by ligand binding to a growth factor receptor, in this case a receptor tyrosine kinase (RTK). Activated phosphotyrosine residues of the RTK interact with src‐homology 2 (SH2) domains on PI3K, as well as other SH2‐containing proteins. This leads to generation of the important lipid second messenger phosphatidylinositol 3,4,5‐trisphosphate (PIP3). AKT localizes to the cell membrane through interactions of its pleckstrin homology (PH) domain with PIP3, which ultimately leads to phosphorylation and activation of AKT by phosphoinositide‐dependent kinase (PDK) 1 and PDK2. AKT activates and inhibits a number of effector proteins via phosphorylation, including mammalian target of rapamycin (mTOR), IκB kinase (IKK), mouse double minute 2 homolog (Mdm2), Bad, p27, glycogen synthase kinase‐3 (GSK3) and forkhead family of transcription factors (FOXO) 1,4. The net result is increased cell survival and proliferation. P, phosphate; PIP2, phosphatidylinositol 4,5‐bisphosphate; PTEN, phosphatase and tensin homologue deleted on chromosome 10
PI3Ks are a family of lipid kinases which are involved in the phosphorylation of the 3′‐OH group on the inositol ring of inositol‐containing phospholipids. The PI3K family is divided into four classes, with the first three classes involved in lipid phosphorylation and class IV PI3Ks, such as ataxia telangiectasia mutated (ATM), are serine–threonine protein kinases 5. Class I PI3Ks are most strongly implicated in growth factor‐mediated signalling and cancer. They are heterodimers that consist of a catalytic alpha subunit (p110) and a regulatory beta subunit (p85, p65, p55 or p101), and specifically add a 3′‐OH group to phosphatidylinositol 4,5‐bisphosphate (PIP2) 1, 5. Class I PI3Ks are further subdivided into subclass Ia proteins that consist of one of three alternative forms of p110 (α, β or δ) encoded by the PIK3 catalytic subunit alpha (PIK3CA), ‐beta (PIK3CB) or ‐delta (PIK3CD) genes, combined with a regulatory subunit (p85, p65 or p55), and subclass Ib proteins consisting of a p110Υ catalytic subunit and the p101 regulatory subunit. Subclass Ia PI3Ks are most commonly involved in downstream signalling from tyrosine kinases, while subclass Ib PI3Ks are involved in downstream signalling from GPCRs 6. Dynamic expression of the various isoforms of PI3K appears to be tissue and cancer specific; for example, PI3Ks with the p110δ isoform appear to be predominantly expressed in leukocytes and neural tissue, and have become an important therapeutic target in B‐cell malignancies 7.
The important lipid second messenger phosphatidylinositol 3,4,5‐trisphosphate (PIP3) is generated as a result of phosphorylation by activated PI3K, and resides on the cytosolic side of the plasma membrane. PIP3 is then able to activate a number of proteins that contain pleckstrin homology (PH) or phenylalanine–tyrosine–valine–glutamate (FYVE) domains, including phosphoinositide‐dependent kinase (PDK) 1, AKT and other serine/threonine kinases 1, 6. The PH domain of AKT interacts with PIP3, resulting in transient localization of AKT to the cell membrane and subsequent phosphorylation of the Thr308 and Ser473 residues by PDK1 and ‐2, respectively. PDK1 itself is activated by PIP3, while PDK2 has been recently identified as a member of the mammalian target of rapamycin complex (mTORC) 2 8. Phosphorylated AKT (p‐AKT) represents the active form. The tumour suppressor phosphatase and tensin homologue deleted on chromosome 10 (PTEN) is an important negative regulator of the PI3K–AKT pathway as it functions to convert PIP3 back to PIP2 rapidly and thus limits the downstream activation of PDK1 and AKT 9. The breadth of pleiotropic downstream effects of AKT, as discussed in detail in the next section, makes it a natural target for drug development.
AKT as an oncogene
Aberrant overexpression of p‐AKT is a common feature in both early and advanced cancers 6. AKT was first discovered in the 1970s as an oncogene transduced by the transforming retrovirus (AKT‐8), which was isolated from an AKR mouse thymoma cell line and first molecularly cloned in 1991. AKT (also known as protein kinase B) is an evolutionarily conserved serine threonine kinase with three different isoforms in mammalian species encoded by three different genes: AKT1, AKT2 and AKT3 2, 6. AKT belongs to the cAMP‐dependent, cGMP‐dependent and protein kinase C (AGC) subfamily of protein kinases, which consist of over 60 members in humans 6. The AKT1, AKT2 and AKT3 isoforms share a conserved structure that includes an N‐terminus PH domain, a central kinase domain and a C‐terminus regulatory domain containing a hydrophobic motif (Figure 2) 10. Both the central kinase domain and the hydrophobic motif are highly conserved among AGC family proteins, and demonstrate 85–90% sequence identity between the three isoforms of AKT.
Figure 2.
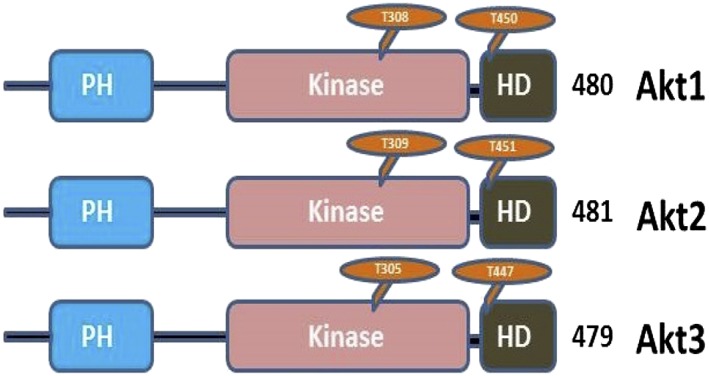
AKT1, AKT2 and AKT3 share common domain architecture with other members of the cAMP‐dependent, cGMP‐dependent and protein kinase C (AGC) subfamily of protein kinases, which consists of an N‐terminus pleckstrin homology (PH) domain, a large central kinase domain and a C‐terminus hydrophobic domain (HD). The position of threonine and serine residues involved in phosphorylation varies only slightly between the three different isoforms
AKT is an important signalling hub with well over 100 downstream target substrates, affecting cell metabolism, growth, survival and proliferation 2, 6. For example, AKT promotes glucose uptake and the storage of energy in the form of glycogen through its effects on glycogen synthase kinase‐3 (GSK‐3) 11 (Table 1). AKT‐mediated phosphorylation of GSK‐3A at Ser21 and GSK‐3B at Ser9 results in a loss of GSK‐3 kinase activity, an important early step in the activation of glycogen synthase. This inactivation of GSK3 is also thought to promote cell cycle progression in cancer cells. AKT promotes cell growth through the induction of insulin‐stimulated protein synthesis by phosphorylating tuberous sclerosis 2 (TSC2) at Ser939 and thus activating mTORC1 signalling 12. This activation of mTORC1 leads to the phosphorylation of 4E‐binding protein 1 (4E‐BP1) and P70‐S6 kinase 1, which are both crucial to ribosomal protein synthesis 6 (Figure 3). AKT contributes to impaired apoptosis and cell cycle progression in cancer cells via inhibition of Bad and caspase 9, as well as the phosphorylation of mouse double minute 2 homolog (Mdm2), leading to p53 ubiquitination 13, 14. AKT further promotes cell survival through the phosphorylation of the apoptosis signal‐related kinase [mitogen‐activated protein kinase kinase kinase 5 (MAP3K5)] at Ser83, which decreases the kinase activity of MAP3K5 that is normally induced by oxidative stress 15. Another important substrate of AKT is the GTPase‐activating protein deleted in liver cancer 1 (DLC1), whose phosphorylation also promotes cell growth and proliferation 16.
Table 1.
Downstream substrates of AKT
| Substrate | Description | Functional effect of AKT | Biological effect |
|---|---|---|---|
| GSK‐3β | Involved in phosphorylation and destruction of β‐catenin, cyclin D1 and Myc | Inhibition | Increased proliferation/cell cycle progression |
| FOXO4 | Transcription factor that induces expression of the CDK inhibitor p27, as well as proapoptosis genes | Inhibition (phosphorylated FOXO4 is bound by inhibitory 14‐3‐3 proteins) | |
| p21CIP1, p27KIP1 | CIP–KIP family of CDK inhibitors | Inhibition | |
| Bad | Bcl‐2 protein family member; promotes apoptosis via mitochondrial membrane pore formation | Inhibition | Antiapoptosis |
| Caspase‐9 | Part of the intrinsic apoptosis pathway | Inhibition | |
| CREB1 | Controls the transcription of antiapoptosis genes, including bcl‐2 and mcl‐1 | Activation | |
| IκB kinase | Phosphorylates and inactivates IκB, a protein that sequesters NFκB | Activation | |
| FOXO1, FOXO3 | Ubiquitous transcription factors involved in the expression of proapoptosis genes, differentiation and cell metabolism | Inhibition (phosphorylated FOXO1/3 are bound by 14‐3‐3 proteins) | |
| Mdm2 | Activated Mdm2 promotes ubiquitination and degradation of p53 | Activation | |
| TSC2 | TSC1/TSC2 complex functions to inhibit mTOR activity | Inhibition | Protein synthesis and cell growth |
| 4E‐BP1 | Broad regulator of translational activity | Inhibition |
3E‐BP1, 4E‐binding protein 1; CDK, cyclin‐dependent kinase; CIP, cyclin‐dependent kinase interacting protein 1; CREB, cAMP response element binding protein; FOXO, forkhead family of transcription factors; GSK, glycogen synthase kinase; KIP, kinase inhibitory protein 1; Mdm2, mouse double minute 2 homolog; mTOR, mammalian target of rapamycin; NFκB, nuclear factor κB; TSC2, tuberous sclerosis 2
Figure 3.
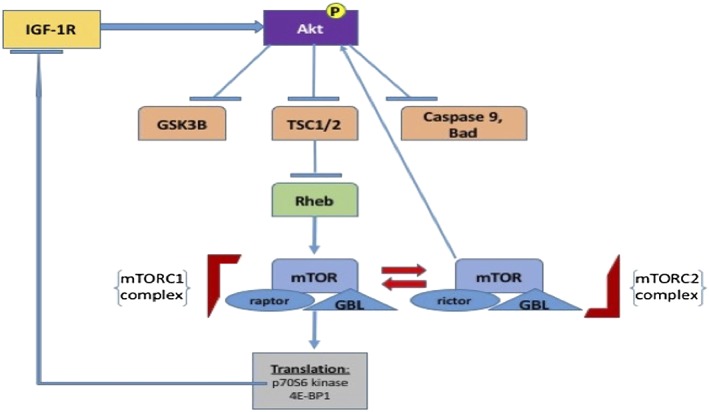
A selection of downstream targets of AKT is displayed. Following upstream activation, in this case by insulin‐like growth factor receptor 1 (IGF‐1R) signalling, AKT phosphorylates and inactivates three key downstream effectors: glycogen synthase kinase‐3 beta (GSK3B), tuberous sclerosis 1 and 2 (TSC1/2) and the proapotosis proteins caspase 9 and Bad. Inhibition of TSC1/2 leads to subsequent inhibition of Ras homologue enriched in brain (Rheb) and the downstream conversion of mammalian target of rapamycin (mTORC) 2 to mTORC1. This ultimately leads to the phosphorylation of p70S6 kinase and 4E‐BP1, which favours an increase in ribosomal protein translation and cell growth. Phosphorylated p70S6 kinase normally exerts negative feedback by inhibiting IGF‐1R signalling; loss of this negative feedback loop occurs in cancer cells following treatment with mTOR inhibitors. GBL, G protein beta protein subunit‐like
In addition to its effects on downstream signalling proteins, AKT activates and inactivates several transcription factors, resulting in more long‐lasting effects on cellular phenotype. AKT phosphorylates several members of the forkhead family of transcription factors (FOXO), leading to the binding of 14‐3‐3 proteins and preventing nuclear translocation of FOXO 17. FOXO3 and FOXO4 in particular are thought to play a significant role as tumour suppressors, and their expression is commonly downregulated in tumour samples from The Cancer Genome Atlas (TCGA) 18. AKT also has an important role in the regulation of nuclear factor (NF) κB‐dependent gene transcription and upregulates the activity of the cAMP response element binding protein (CREB) 1 19, 20. CREB1 phosphorylation induces the binding of accessory proteins that are necessary for the transcription of antiapoptosis genes, including bcl‐2 and mcl‐1.
While over 100 substrate candidates have been experimentally validated for AKT, little is known about the isoform specificity of the majority of these interactions. This issue has been a significant hurdle to the development of therapeutics that target AKT 6. For some of the biological functions carried out by the pathway, there is clear redundancy between the different isoforms of AKT as well as other AGC subfamily protein kinases 21. However, there is reason to believe that the different isoforms may also have selective, context‐specific, synergistic and possibly antagonistic biological effects. Thus, targeting specific isoforms may be of therapeutic importance. For example, AKT2 increases glucose uptake by mediating insulin‐induced translocation of the glucose transporter isoform 4 (GLUT4) glucose transporter to the cell surface in insulin‐sensitive cells. On the other hand, AKT1 potentiates this effect through the phosphorylation of protein tyrosine phosphatase nonreceptor type 1 (PTPN1) at Ser50, resulting in a decrease in its phosphatase activity and ultimately persistent phosphorylation of the active insulin receptor 21, 22. This difference in biological impact should be considered, as a number of agents targeting AKT are pan‐AKT inhibitors.
Notably, there are multiple mechanisms of cross‐talk and feedback regulation in the PI3K–AKT pathway. For example, a negative feedback loop exists in which p70S6 kinase‐1, a downstream effector of mTORC1, phosphorylates and inactivates insulin receptor substrate (IRS) proteins, which in turn reduces signalling through PI3K–AKT via the insulin‐like growth factor type 1 receptor (IGF‐1R) 23. When mTORC1 is inhibited, phosphorylated p70S6 levels decrease, and this negative feedback mechanism on IGFR–PI3K–AKT signalling is lost, possibly reducing the clinical efficacy of mTOR inhibitors in cancer. Moreover, this cross‐talk also explains some of the toxicities that result from inhibition of the pathway, including hyperglycaemia.
Mouse models of AKT
Knockout of AKT1, AKT2 and AKT3 has been achieved by targeted homologous recombination in murine models and has provided some insight into their respective functional roles 6, 21. Homozygous deletion of AKT1 leads to significant early mortality and growth retardation as a consequence of defects in placental development. AKT2 knockout mice display insulin resistance and develop a severe form of diabetes, along with mild growth retardation and reduced platelet aggregation. Simultaneous knockout of both AKT1 and AKT2 results in embryonic lethality or death immediately after birth 24. AKT3 knockout mice are characterized by a 25% reduction in brain mass but no other discernible abnormalities 24.
The mechanism underlying tumorigenesis secondary to constitutive activation of AKT appears to be complicated and depends on the AKT isoform that is expressed, AKT expression level, target tissue and molecular context. Despite the proposed canonical PI3K–AKT pathway that is directly opposed by PTEN activity, transgenic activated AKT mouse models do not consistently behave like PTEN knockout models or consistently have a pro‐oncogenic phenotype 25, 26. Constitutive activation of AKT in transgenic models is generally achieved by the addition of a myristoyl or palmitoyl‐group to the N‐terminus, resulting in its localization to the plasma membrane in a PIP3‐independent manner. Once phosphorylated at the plasma membrane, activated AKT can translocate to the cytosol or the nucleus.
While transgenic mice that specifically express constitutively active AKT in the mammary gland using an epithelial‐specific mouse mammary tumour virus (MMTV) promoter do not demonstrate an increase in breast tumour development, activation of the AKT pathway results in mammary gland involution defects, similar to those seen in PTEN knockout mice 25, 26, 27, 28, 29, 30, 31, 32. It has been proposed that the lack of tumour development is because the optimal level of AKT activation has not yet been generated in these models. An activation level that is too low will not activate the oncogenic phenotype, whereas an activation level that is too high triggers p53‐ and p27‐dependent cell senescence pathways. Consistent with this hypothesis, the coexpression of a nonfunctional p53 mutant with activated AKT significantly increases the size of mammary carcinomas, compared with the p53 mutant alone 25, 32. In addition, mice with mammary gland‐specific AKT1 expression under the control of the MMTV promoter that are systemically treated with the carcinogen 7,12‐dimethylbenz(a)anthracene (DMBA) develop oestrogen receptor‐positive (ER+) mammary cancers 33.
Compared with mammary tissue, other tissues are more susceptible to carcinogenesis by constitutively activated AKT. The introduction of myr‐AKT to keratinocytes using a skin‐specific viral vector results in the development of squamous cell cancers 34. In addition, the expression of either human wild‐type AKT or myr‐AKT in the basal layer of the epidermis results in significantly enhanced susceptibility to topically applied chemical carcinogens such as DMBA 35. In a germline model, mice with the highest levels of AKT activity develop spontaneous epithelial carcinomas in multiple organs after several months of follow‐up 25. Mice expressing constitutively active AKT in combination with loss of p53 in the epithelial layer of the oral cavity develop squamous cell oral cavity cancers 36. Expression of activated AKT in the prostate increases the proliferative capacity of cells, resulting in premalignant prostate intraepithelial neoplasia, although these lesions do not progress to cancer with follow‐up, in the absence of introducing additional oncogenes or knocking out tumour suppressors 37. Consistent with this, the coexpression of activated Ras and activated AKT in astrocytes causes glioblastoma in mice, whereas introduction of one or the other alone does not result in glioblastoma formation 38. These findings suggest that AKT alone is not sufficient for oncogenesis in most contexts, but that it demonstrates significant synergistic effects with other pro‐oncogenic abnormalities.
Mutations in the PI3K–AKT pathway in cancer
Mutations in genes encoding the subunits of PI3K represent some of the most common mutations in oncology, with kinase‐domain‐activating point somatic mutations in PIK3CA (which encodes the catalytic subunit p110α) second only to TP53 as the most commonly recurring mutation across all cancer types in TCGA 39. PTEN loss‐of‐function mutations (nonsense mutations, gene deletions and large‐scale chromosomal deletions) and PTEN silencing by methylation and other epigenetic modulation are also among the most common aberrations seen across many different cancer types (Table 2) 39, 40. Germline mutations in PTEN are responsible for the Cowden familial cancer syndrome 41. Mutations in PTEN and PIK3CA do not appear to be completely mutually exclusive, underscoring the complex functioning of the various components of the PI3K–AKT pathway 39, 42.
Table 2.
Phosphatidylinositol‐3 kinase (PI3K)–AKT pathway genes with significantly increased somatic mutation rates (% of tumour samples) compared with background mutation rate in tumour specimens, by tumour type (The Cancer Genome Atlas data)
| Cancer type | PIK3CA | PTEN | PIK3R1 | PIK3CG | AKT1 |
|---|---|---|---|---|---|
| Breast | 33.6 | 3.8 | 2.5 | 0.4 | 2.5 |
| Lung adenocarcinoma | 4.4 | 2.2 | 1.3 | 0.7 | 0.5 |
| Lung‐squamous | 14.9 | 8.1 | 0.6 | 7.5 | 0.6 |
| Colorectal | 17.6 | 1.0 | 2.1 | 0.5 | 0.0 |
| Endometrial | 52.2 | 63.5 | 39 | 1.3 | 1.3 |
| Ovarian | 0.6 | 0.6 | 0.3 | 1.0 | 0.0 |
| Head and neck | 20.6 | 1.3 | 1.7 | 2.7 | 0.7 |
| Renal | 2.9 | 4.3 | 0.5 | 0.7 | 0.5 |
| Glioblastoma | 11.0 | 30.7 | 11.4 | 2.4 | 0.3 |
| AML | 0 | 0 | 0.0 | 0.0 | 0.0 |
| Bladder | 17.4 | 3.1 | 1.0 | 2.0 | 0.0 |
| Combined | 17.8 | 9.7 | 4.4 | 1.7 | 0.9 |
Adapted from Kandoth et al. 39, based on N = 3281 total specimens. The results shown here are in whole based upon data generated by The Cancer Genome Atlas Research Network: (http://cancergenome.nih.gov/). AML, acute myeloid leukaemia; PIK3CA, PIK3 catalytic subunit alpha; PIK3CG, phosphatidylinositol‐4, 5‐bisphosphate 3‐kinase catalytic subunit gamma isoform; PIK3R1, phosphoinositide‐3‐kinase, regulatory subunit 1; PTEN, phosphatase and tensin homologue deleted on chromosome 10
By contrast, mutations in AKT genes are found in human cancers at a lower rate 6, 43. Activating mutations have been described in a small percentage of breast cancers, head and neck squamous cell carcinomas, endometrial cancer, non‐small cell lung cancer and renal cancers. An AKT1 point mutation in the PH domain that replaces a glutamic acid with lysine (E17K) at residue 17 is the most commonly reported mutation and confers increased activity by promoting constitutive localization of AKT1 to the plasma membrane 44. Other reported activating mutations include the E49K (AKT1) and G171R (AKT3) substitutions, which occur in the PH domain and the kinase domain, respectively, although their mechanism of conferring activation is not as well established 45, 46. Measurement of p‐AKT levels in both tumours and cancer cell lines confirms a quantitative increase in activated AKT as a consequence of these point mutations, and overall levels of p‐AKT appear to correlate with sensitivity to AKT inhibition 45, 46, 47, 48.
In spite of the biological rationale that demonstrates the involvement of AKT in promoting tumorigenesis, it has been difficult to establish the significance of these mutations as drivers owing to the relative infrequency of AKT mutations. In one study of 547 human breast cancer specimens and 41 breast cancer cell lines, AKT1 mutations were found in only 1.4% of tumour specimens, with all mutations restricted to the hormone receptor‐positive subtype 43, 48. None of the 41 breast cancer cell lines demonstrated an AKT1 mutation, which is one factor that has further hampered attempts at studying AKT mutations in vitro. In this same study, PIK3CA mutations were common, but were less consistently linked to increased p‐AKT expression and activation of downstream substrates of the pathway compared with PTEN and AKT mutations. After the initial discovery of the AKT1 E17K mutation in breast, ovarian and colorectal cancers, a study was conducted on 731 cancer specimens to determine the frequency of this mutation across different cancer types using a single‐strand conformation polymorphism assay 49. In this study, 4.3% of the 93 breast cancer specimens had the E17K mutation in AKT1, but none of the lung, colorectal, gastric, acute leukaemia and hepatocellular carcinoma specimens harboured this mutation. A targeted search for comparable PH domain mutations in AKT2 and AKT3 was unrevealing. Further large‐scale mutational analysis in the TCGA (N = 3281 specimens) revealed that only breast, endometrial, head and neck, and lung cancers have nonsynonymous AKT1 mutation rates greater than 0.5% 39. Rates of mutations in AKT2 and AKT3 did not reach statistical significance compared with the background mutation rate. Given the infrequency of AKT mutations in human cancers, it is not clear if mutational status has an impact on clinical prognosis. The frequency of PI3K–AKT pathway gene mutations from a subset of samples is reported in Table 2.
In addition to the E17K mutation, large‐scale, high‐resolution sequencing studies in breast cancer have recently identified additional somatic variants in the PH domain of AKT 50, 51, 52. One study evaluated the functional significance of the L52R, D32Y, K39N, P42T, C77F and Q79K substitutions in AKT1 using an MCF‐7 cell line which normally expresses an activating PIK3CA mutation (E545K). Somatic cell gene targeting was used to replace the mutant PIK3CA alleles with wild‐type PIK3CA 53. Replacement of mutant PIK3CA with wild‐type PIK3CA leads to a drastic reduction in p‐AKT and downstream targets such as FOXO3 compared with parental MCF‐7 cells. When E17K mutant AKT1 is knocked in to this cell construct, there is an increase in p‐AKT back to levels seen in the parent PIK3CA mutant cells. Introduction of mutant L52R, C77F and Q79K also substantially increased p‐AKT, similar to the E17K mutant, while the D32Y, K39N and P42T variants did not activate AKT. The cells with E17K, L52R, C77F and Q79K mutant AKT1 also demonstrated increased phosphorylation of downstream AKT targets, including FOXO1/3 and proline‐rich AKT substrate of 40kDa (PRAS40). Similar to the E17K mutation that confers increased activity by causing AKT to localize to the plasma membrane through increased interactions between its PH domain and PIP3, the L52R, C77F and Q79K mutant AKT1 also demonstrated increased localization to the plasma membrane. The D32Y, K39N and P42T variants did not. The clinical significance of these additional AKT mutations remains unclear.
Dysregulation of AKT in human cancers
While AKT mutations are infrequent, AKT gene amplification occurs at a higher rate and has been described in gastric, breast, ovarian, pancreatic, colon, oesophageal and thyroid cancers, with amplifications most commonly involving the AKT2 isoform 6, 54. Post‐translational modification of AKT, including tyrosine phosphorylation, O‐GlcNAcylation, lysine modifications, sumoylation and acetylation, are also thought to play a critical role in maintaining AKT hyperactivation in cancers, even in the presence of normal PI3K and PTEN activity 6, 55, 56, 57. Moreover, aberrant activity of AKT can occur via numerous mechanisms that effect elements upstream of AKT. Activating mutations or amplifications in PI3K, loss of PTEN, and activating mutations of growth factor or cytokine receptors and intracellular oncogenes like Ras are common findings in cancer and result in increased expression and activity of one, two or all three isoforms of AKT 6.
Although the subset of downstream effectors of AKT that are most crucial to tumour development has not been entirely elucidated, hyperactivation and overexpression of the different isoforms of AKT occur in a number of cancer types, and the evidence is summarized below. In addition to promoting initial tumour development, there is growing evidence that AKT is involved in acquired resistance to traditional chemotherapy as well as targeted therapies including trastuzumab, gefitinib, tamoxifen and all‐trans‐retinoic acid 58, 59.
Lung cancer
Phosphorylated AKT is noted to be increased in smokers with lung cancer as well as in bronchial metaplasias and dysplasias, suggesting a role in early carcinogenesis 60. In addition, significant increases in p‐AKT expression are noted in malignant and premalignant bronchial epithelial cell lines. AKT1 amplification has been established as a mechanism for in vitro acquired resistance to cisplatin in the A549 cell line 61.
Breast cancer
AKT is activated in a subset of premalignant breast lesions. In one study, p‐AKT was overexpressed in 38 of 114 (33%) ductal carcinoma in situ (DCIS) lesions and in 52 of 136 (38%) invasive breast cancers. The majority of tumours in this study expressed the oestrogen receptor (79%) 62. AKT activation did not appear to increase with tumour progression, whereas loss of PTEN expression correlated with disease progression. PTEN expression was lost in 12% of DCIS and in 25% of invasive carcinomas, with a correlation between loss of PTEN and higher tumour grade.
Prostate cancer
Studies have demonstrated that AKT is activated in prostatic intraepithelial neoplasias (PINs) 63. In particular, p‐AKT overexpression by immunohistochemistry was observed in the majority of human specimens of high‐grade PIN as well as invasive carcinomas 64. PTEN mutations occur in about 15% of prostate adenocarcinomas, and homozygous loss of PTEN is frequently found in metastatic lesions. In prostate epithelial cell lines, simultaneous expression of AKT and the androgen receptor is sufficient for transformation, and results in cells that are resistant to the effects of androgen ablation 65. Additionally, AKT3 is overexpressed in androgen‐insensitive prostate cancer cell lines such as DU145 66.
Melanoma
AKT activation increases during melanoma formation and progression. In one study of 293 patient specimens, strong p‐AKT expression was observed in only 17% of benign nevi, in 49% of localized melanomas and in 77% of metastatic melanoma lesions 67. A statistically significant difference was identified between each progressive stage. In low‐risk thin lesions (<1.5 mm thickness), strong p‐AKT expression was an independent poor prognostic factor for 5‐year outcomes. AKT3 expression and activity specifically, and not AKT1 and AKT2, have been associated with the formation of sporadic melanoma, particularly when combined with loss of PTEN 68. Interestingly, in vitro work demonstrates that constitutively active AKT transforms melanocytes in a mildly hypoxic environment, a state which simulates normal skin conditions 69. Activation of mTORC1 and expression of hypoxia‐inducible factor‐1 alpha are required to maintain transformation in these cells. There is also cross‐talk between the AKT and BRAF signalling pathways; BRAF is mutated in about half of all melanomas, and the presence of an AKT1 Q79K mutation has been reported in cases of acquired resistance to BRAF inhibitors 70.
Endometrial and cervical cancers
Somatic mutations and deletions of PTEN are the most common genetic abnormalities in endometrial cancer, and both PIK3CA and PTEN mutations are common in cervical cancer 39, 71, 72. Studies have also demonstrated a link between inactivation of retinoblastoma by the E7 transforming protein of human papilloma virus (HPV) and increases in AKT activity seen in cervical intraepithelial neoplasia 73. A similar increase in AKT activity is seen in HPV‐positive head and neck squamous cell cancer cell lines 74.
Targeting AKT
In spite of the importance of the growth factor–PI3K–AKT–mTOR axis in human cancers, the optimal approach to targeting it has yet to be discovered. The major mechanism of resistance to upstream targeted agents that function at the level of the growth factor receptor, such as cetuximab, an inhibitor of EGFR, and trastuzumab, an inhibitor of human epidermal growth factor receptor 2 (HER2)/neu, is compensatory activation of several intracellular cross‐talk pathways that ultimately lead to downstream activation of AKT 6, 75. The mTOR inhibitors temsirolimus and everolimus are the first drugs approved by the US Food and Drug Administration for an oncological indication that directly target the PI3K–AKT pathway. Both drugs have indications for the treatment of metastatic renal cell carcinoma, while everolimus is also approved for the treatment of advanced well‐ or moderately differentiated pancreatic neuroendocrine tumours, advanced ER+ breast cancer and subependymal giant cell tumours, which are associated with tuberous sclerosis. Unfortunately, downstream inhibition of mTORC1 leads to a loss of the negative feedback loop mediated by p70S6, and thus can lead to increased upstream signalling in the PI3K–AKT pathway and ultimately an increase in p‐AKT 23. More recently, idelalisib, a selective inhibitor of the delta isoform of PI3K that is involved in downstream signalling from the B‐cell receptor, was approved for use in chronic lymphocytic leukaemia and advanced follicular lymphoma 76. Buparlisib, a pan‐isoform PI3K inhibitor, has entered late‐stage clinical trials in ER+ breast cancer 77. The results of the phase III Buparlisib brEast cancer cLinicaL Evaluation (BELLE) 2 trial were presented at the 2015 San Antonio Breast Cancer Symposium. The addition of buparlsib 100 mg daily to fulvestrant in 1147 patients with advanced endocrine‐resistant ER+/HER2‐negative breast cancer demonstrated a modest 1.9‐month increase in progression‐free survival compared with fulvestrant alone (P < 0.001) 78. A more substantial progression‐free survival benefit of 3.8 months was seen in the stratum of 587 patients with PIK3CA mutations identified in circulating tumour DNA prior to treatment, with 18.4% of those patients administered the combination achieving an objective response compared with only 3.5% treated with fulvestrant alone. Unfortunately, neither selective nor nonselective PI3K inhibitors have demonstrated robust responses in most solid tumour malignancies and can exhibit dose‐limiting toxicities (DLTs) that lessen their efficacy 79. AKT represents an attractive target to shut down signalling through this pathway. As many of the cross‐talk pathways signal through AKT, a direct AKT inhibitor could potentially be less susceptible to resistance mechanisms.
Targeting AKT has been a pharmacological challenge, and to date there has not been a single positive randomized phase III trial with an AKT inhibitor. Nonetheless, several promising AKT inhibitors have been developed that either selectively or nonselectively inhibit the three isoforms of AKT by binding to the kinase or PH domains, and are currently in clinical trials. In addition, an antisense oligonucleotide directed at AKT, as well as inhibitors of the lipid second messenger PIP3 have been developed. It remains an open question whether it is best to use pan‐AKT inhibitors or isoform‐specific inhibitors in specific tumour types and whether there is a synergistic effect when AKT inhibitors are combined with cytotoxic chemotherapy or hormone therapy.
AKT inhibitors in clinical trials
AKT inhibitors fall under four general categories (Table 3) 80. The first category consists of competitive inhibitors of the ATP binding site on the kinase domain. Examples include drugs that specifically target isoforms of AKT, including CCT128930, which targets AKT2, and BAY‐1125976, which targets AKT1 and AKT2. Pan‐AKT kinase inhibitors include afuresertib, AZD5363, GDC‐0068, GSK690693 and AT7867. The second category is allosteric inhibitors of the AKT kinase domain, the best known example of which is MK‐2206. Competitive inhibitors appear to have more efficacy in cell lines with AKT mutations, whereas the allosteric inhibitors may have a more broad antitumour effect, with increased efficacy in cell lines harbouring PIK3CA mutations or loss of PTEN function, although this is not a consistent finding for all drugs 81. The third category is lipid‐based inhibitors that prevent the generation of PIP3 from PIP2 by PI3K and thus indirectly inhibit the activation of all isoforms of AKT. Examples from this category include PX‐866 and perifosine. These drugs also inhibit the NFkB pathway and c‐Jun. The fourth category comprises drugs that interact with the PH domain of AKT and thus prevent localization of AKT to the cell membrane, where it is normally activated. The best known examples in this category include triciribine and PX‐316. As there is less sequence identity in the PH domains of the three isoforms of AKT compared with the kinase domains, drugs designed to target the PH domains perhaps represent the best opportunity to develop highly selective isoform‐specific inhibitors in the future 6. In addition to small molecule inhibitors, antisense oligonucleotides directed at AKT mRNA (RX‐0201) have also entered clinical trials. Representative drugs from each category that have been evaluated in clinical trials are discussed in detail below.
Table 3.
Ongoing clinical trials involving AKT inhibitors as registered on clinicaltrials.gov as of October 2015
| Drug | Company | Alternative names | Targets | Trial phase | Cancer‐specific trials |
|---|---|---|---|---|---|
| GSK2141795 | GlaxoSmithKline | Uprosertib | Pan‐AKT isoforms (competitive) | I/II | Advanced solid tumours with trametinib, dabrafenib |
| II | AML with trametinib | ||||
| II | Multiple myeloma with trametinib | ||||
| II | Triple negative BC with trametinib | ||||
| AZD5363 | AstraZeneca | Pan‐AKT isoforms (competitive) | I | Solid tumours with olaparib | |
| I/II | Castrate resistant prostate Ca with docetaxel | ||||
| I/II | Endometrial and ovarian Ca with mTORC1/2 inhibitor AZD2014 | ||||
| II | Castrate Resistant Prostate Ca with enzalutamide | ||||
| II | ER+ breast cancer | ||||
| II | Triple negative BC with paclitaxel | ||||
| GSK2110183 | GlaxoSmithKline | Afuresertib | Pan‐AKT isoforms (competitive) | Ib | Gastric Ca with paclitaxel |
| I/II | Multiple myeloma with carfilzomib | ||||
| II | Continuation study for responders in solid/haematological malignancies | ||||
| II | CLL with ofatumumab | ||||
| GDC‐0068 | Genentech | Ipatasertib | Pan‐AKT isoforms (competitive) | I/II | Glioblastoma |
| II | Neoadjuvant treatment of triple negative BC with paclitaxel | ||||
| II | Metastatic triple negative BC with paclitaxel (first‐line) | ||||
| MSC‐2363318A | Merck Sorono | Pan‐AKT isoforms and p70S6K inhibitor (competitive) | I | Solid tumours | |
| LY2780301 | Lilly | Pan‐AKT isoforms and p70S6 kinase inhibitor (competitive) | Ib | Solid tumours and NHL with gemcitabine | |
| Ib/II | HER2+ metastatic BC with paclitaxel | ||||
| MK‐2206 | Merck | Pan‐AKT isoforms (allosteric) | I | Pancreatic Ca‐ with dinaciclib | |
| II | HR+ breast cancer with anastrozole/goserelin | ||||
| II | Lung or thymic Ca; only patients with AKT, PTEN, or PIK3CA mutations | ||||
| BAY‐1125976 | Bayer | AKT1/2 isoform inhibitor (allosteric) | I | Solid tumours | |
| ARQ 092 | ArQule | Pan‐AKT isoforms (allosteric) | I | Solid tumours and lymphoma. | |
| Ib | Triple negative BC and gynaecological cancers with carboplatin and paclitaxel | ||||
| KRX‐0401 | Aeterna Zentaris | Perifosine | Pan‐AKT (lipid‐based/PIP3 inhibitor) | II | Gliomas combined with temsirolimus |
| PTX‐200 | Prescient Therapeutics | Triciribine | PH domain (AKT) inhibitor | I/II | Advanced or metastatic HER2‐negative BC with paclitaxel, doxorubicin and cyclophosphamide |
| AG1343 | Roche | Nelfinavir; Viracept | HIV protease inhibitor, AKT | I/II | Multiple myeloma with lenalidomide + dexamethasone |
| ‐‐‐ | Multiple trials in multiple Ca types as radiosensitizing agent | ||||
| ONC‐201 | Oncoceutics | TRAIL pathway inducing compound/indirect AKT inhibitor | I | Solid tumours | |
| I/II | Relapsed/refractory NHL | ||||
| I/II | Relapsed/refractory AML and high‐risk MDS | ||||
| Archexin | Rexahn | AKT1 antisense oligonucleotide | I/II | Renal cell Ca with everolimus | |
AML, acute myeloid leukaemia; BC, breast cancer; Ca, cancer; CLL, chronic lymphocytic leukaemia; DLBCL, diffuse large B‐cell lymphoma; ER, oestrogen receptor; HER2, human epidermal growth factor receptor 2; HIV, human immunodeficiency virus; HR, hormone receptor; MDS, myelodysplastic syndrome; mTOR, mammalian target of rapamycin; NHL, non‐Hodgkin's lymphoma; PH, pleckstrin homology; PIP3, phosphatidylinositol 3,4,5‐trisphosphate; PIK3CA, PIK3 catalytic subunit alpha; PTEN, phosphatase and tensin homologue deleted on chromosome 10; TRAIL, tumour necrosis factor‐related apoptosis‐inducing ligand.
Perifosine
Perifosine, an oral lipid‐based inhibitor of AKT, was the furthest along in clinical trials prior to two recently published negative phase III trials. Perifosine in combination with antimetabolites such as capecitabine and 5‐fluorouracil (5‐FU) demonstrated substantial synergistic activity in colorectal cancer cell lines. The combination of perifosine with capecitabine was first investigated in a phase I study of multiple tumour types that were heavily pretreated, and demonstrated some activity, with no dose‐limiting toxicities at the doses tested 82. In the subsequent randomized phase II trial of 38 patients with metastatic colorectal cancer who had been treated with at least one prior line of therapy, the combination of capecitabine 825 mg twice daily with perifosine 50 mg once daily improved the primary endpoint of median time to progression from 11 weeks to 28 weeks, compared with capecitabine alone (P = 0.0012) 83. Similar to the phase I trial, no significant dose‐limiting toxicities were reported. There was also an unexpected statistically significant increase in median overall survival from 10.9 months to 17.7 months with the combination (P = 0.016). This increase in survival was even seen in the subset of patients who previously progressed on 5‐FU therapy (6.6 months vs. 15.1 months). There was one complete response and three partial responses by Response Evaluation Criteria in Solid Tumours (RECIST) criteria in the arm that received perifosine. Results from this randomized phase II trial led to the larger phase III X‐PECT trial, which, again, compared the combination of perifosine and capecitabine with capecitabine alone in a similar population of 468 patients with refractory metastatic colorectal cancer 84. The results of this larger trial were negative. The median overall survival in the two arms was 6.4 months and 6.8 months, respectively (P = 0.315), and survival was similar for both Kirsten Ras gene (KRAS) mutant and wild‐type tumours.
Perifosine was also studied in multiple myeloma. In a phase I study that assessed the safety of perifosine in combination with lenalidomide and dexamethasone in 32 patients with relapsed and heavily pretreated multiple myeloma, the combination was well tolerated, with the exception of some grade III neutropenia in 26% and thrombocytopenia in 18% of patients, as well as grade II fatigue and diarrhoea occurring in about half of patients. There was a minimal response rate (or better) of 73% and a median overall survival of 30.6 months in the study population 85. A phase I/II study assessed the combination of bortezomib and perifosine in 73 patients with relapsed, heavily pretreated, bortezomib‐refractory multiple myeloma 86. In this study, the overall response rate was 41% and an additional 41% of patients had stable disease. The median overall survival was 23 months. These results led to a phase III trial in multiple myeloma comparing bortezomib plus dexamethasone with perifosine or placebo. This trial was discontinued at interim analysis as it was felt unlikely that the treatment arm would achieve a benefit in progression‐free survival, although no new safety concerns were raised. There is currently only one registered and actively recruiting trial of perifosine in the USA, a trial investigating the combination of perifosine and temsirolimus in recurrent or progressive malignant glioma (clinicaltrials.gov identifier: NCT02238496).
MK‐2206
MK‐2206 is an oral allosteric inhibitor of the AKT kinase domain which has been well studied in the clinical setting, particularly in breast cancer. In preclinical studies, the drug demonstrated synergy with erlotinib in nonsmall cell lung cancer cell lines and with lapatinib in breast cancer cell lines 87. In a presurgical study of women with stage I‐III breast cancer, the effect of two doses of MK‐2206 given 9 days and 2 days prior to surgery on tumour p‐AKT expression and peripheral blood biomarkers was evaluated 88. A total of 12 patients were enrolled prior to study termination owing to drug toxicity. Four patients were treated at a dose of 200 mg, three patients received a dose of 135 mg and five patients received a dose of 90 mg, with 3/4, 2/3 and 1/5 patients developing grade III rash or pruritus in the respective dose cohorts. Initial biomarker assessment suggested effective target modulation with a decrease in p‐AKT and p70S6 kinase (and an increase in the upstream tyrosine kinase IGF‐1R) in peripheral blood mononuclear cells. In a phase I study in patients with advanced solid tumour malignancies, MK‐2206 resulted in central tumour necrosis on imaging in several patients 89. In a phase I study of 34 patients with heavily pretreated HER2 amplified breast or gastric cancer, doses of MK‐2206 60 mg daily or 135 mg weekly were well tolerated in combination with trastuzumab, and trastuzumab did not appear to alter the pharmacokinetic profile of MK‐2206 90. One patient with breast cancer had a complete response, while one patient had a partial response and five patients demonstrated stable disease.
Preclinical studies have also demonstrated synergy between antioestrogens and MK‐2206 in oestrogen‐sensitive breast cancer cell lines, thus leading to a phase I dose‐escalation trial combining MK‐2206 with the aromatase inhibitors anastrozole or letrozole, or with both anastrozole and the selective oestrogen receptor downregulator fulvestrant 91. Four of the 30 patients enrolled discontinued treatment early owing to a grade III rash, with skin biopsies suggestive of a hypersensitivity drug rash. The choice of anastrozole, letrozole or the combination of anastrozole and fulvestrant did not appear to influence the tolerability of MK‐2206, although the authors note that a dose‐limiting rash occurred at lower doses than in some prior MK‐2206 monotherapy trials. The recommended phase II dose was MK‐2206 150 mg once weekly in combination with antiestrogen therapy at standard doses, with prednisone 20 mg prescribed for three daily doses beginning on the day before treatment for rash prophylaxis. Of the 26 patients evaluable for response, two patients demonstrated a partial response while another nine patients derived clinical benefit defined as stable disease at the 6‐month assessment, for a clinical benefit rate of 42%. The clinical benefit rate was higher in those patients who were receiving their first treatment in the metastatic setting, with 67% deriving benefit, with a median time to progression of 14 months. While this modest clinical benefit rate compares with that seen with other drugs that are approved in ER+ breast cancer, such as everolimus, the authors hypothesized that the relatively low dose of MK‐2206 is inadequate for a complete pharmacodynamic effect. A phase II neoadjuvant trial for patients with stage II or III ER+/HER2‐ breast cancer combining anastrozole and MK‐2206 150 mg weekly with prednisone has been opened by the same study group to explore pharmacodynamic biomarkers and pathological response rates further. A different phase II trial investigated MK‐2206 in the treatment of patients with breast cancer harbouring either mutations in PIK3CA or AKT, or with loss of PTEN function; the results of this study have not yet been published.
A phase II study of MK‐2206 in 59 patients with relapsed or refractory lymphoma of any subtype other than Burkitt lymphoma assessed the safety and response rate of MK‐2206 200 mg given orally once weekly during 28‐day cycles, for a maximum of 12 cycles 92. Dose escalation to 300 mg was allowed. In the study, eight patients (14%) had an objective response, while a total of 29 patients (49%) demonstrated at least some reduction in tumour measurements on imaging. Another phase II study evaluating MK‐2206 as second‐line therapy in advanced gastric and gastro‐oesophageal junction cancers enrolled 70 patients, with a primary endpoint of overall survival 93. Patients were administered a dose of 60 mg once every other day. Grade III and IV toxicities, including hyperglycaemia, anaemia and infection, occurred in less than 5% of patients. There were two deaths potentially related to the study drug from cardiac arrest and respiratory failure. Common grade I or II adverse events included fatigue (50%), maculopapular rash (30%), acneiform rash (13%), hyperglycaemia (30%) and nausea (40%). The objective response rate was only 1%, with a progression‐free survival of only 1.8 months, and the median overall survival was 5.1 months, which did not meet the prespecified cutoff of 6.5 months to pursue further investigation of the drug in gastric cancers. A phase II trial in advanced non‐small cell lung cancer investigated the combination of erlotinib 150 mg daily and MK‐2206 45 mg every other day in patients who previously had had clinical benefit from erlotinib but subsequently developed progression of disease on the drug 94. A total of 80 patients were enrolled: 45 with tumours harboring activating mutations in EGFR and 35 with wild‐type EGFR. The objective response rate and 12‐week stable disease rate were 9% and 40% in the EGFR‐mutated cohort and 3% and 47% in the EGFR‐wild‐type cohort, respectively. The trial did not meet the prespecified cutoff of a 20% objective response rate for patients with an activating EGFR mutation, but did meet the prespecified cutoff of a 40% clinical benefit rate in the EGFR wild‐type group.
Finally, the results from a phase II biomarker‐driven study of the combination of MK‐2206 and the oral mitogen‐activated protein kinase kinase (MEK) inhibitor selumetinib in patients with colorectal cancer has been reported and provides some further insight on the biological activity of MK‐2206 95. Pharmacodynamic inhibition of phosphorylated extracellular signal‐regulated kinase (p‐ERK) and p‐AKT in paired pre‐ and post‐treatment tumour biopsies was the primary endpoint. Initially, 12 patients were enrolled and received MK‐2206 90 mg weekly along with selumetinib 75 mg daily in continuous 28‐day cycles. Following interim safety analysis, the doses were increased to MK‐2206 135 mg weekly and selumetinib 100 mg daily. Common grade I–III toxicities included gastrointestinal disturbances, liver function test abnormalities, skin toxicity and myelosuppression. There were no objective responses among the 21 patients enrolled. Target modulation did not meet the prespecified criteria of dual 70% inhibition of p‐ERK and p‐AKT in any of the 21 patients. The p‐AKT reduction in post‐treatment biopsies ranged from 2% to 77%, with a median decrease of only 28%. Overlapping toxicities limited the ability to dose escalate further.
Afuresertib
Afuresertib is an oral competitive pan‐AKT kinase domain inhibitor. In a phase I dose finding study of the combination of afuresertib with the MEK inhibitor trametinib in 20 patients with advanced solid tumours and multiple myeloma, safety and best response were evaluated 96. Exclusion criteria included diabetes, active gastrointestinal disease, leptomeningeal disease and a history of retinopathy or retinal vein occlusion, due to the concern of MEK inhibitor‐related retinal toxicities. At the starting dose of afuresertib 50 mg daily with trametinib 1.5 mg daily continuously, several patients developed DLTs, including grade II oesophagitis, grade III liver function test abnormalities, mucosal inflammation and hypokalaemia. Subsequent lower‐dose combinations with 25 mg/1.5 mg and 50 mg/1.0 mg continuously or 50 mg/1.5 mg on days 1–10 of 28‐day cycles were determined to be the maximum tolerated doses (MTD). Common adverse events included diarrhoea (60%), acneiform rash (55%), maculopapular rash (45%), fatigue (30%), dry skin (25%), nausea (25%), dyspnoea (20%) and vomiting (20%). There was only one objective response (i.e. a partial response that occurred in a patient with BRAF wild‐type metastatic melanoma), and there were four additional patients with stable disease.
Afuresertib was also evaluated as monotherapy in an open‐label phase I study in 73 patients with advanced haematological malignancies, including 34 patients with multiple myeloma 97. Patients were treated with doses of afuresertib ranging from 25 mg to 150 mg daily, with 125 mg daily being established as the MTD. Liver function test abnormalities were the main DLT. The most common adverse events were nausea (35%), diarrhoea (33%) and dyspepsia (24%). Three of the 34 patients with multiple myeloma demonstrated a partial response, while another three patients had a minimal response. There were few objective responses in patients with non‐Hodgkin's lymphoma, Hodgkin's lymphoma and Langerhans cell histiocytosis.
Ongoing clinical trials
In addition to some of the agents previously mentioned, several new AKT inhibitors have recently entered clinical trials (Table 3). In preclinical studies of enzalutamide‐resistant prostate cancer cell lines and LNCaP xenograft models, AZD5363, an oral competitive pan‐AKT kinase domain inhibitor, has demonstrated the ability to restore sensitivity to enzalutamide with impressive tumour regression in the xenograft model 98. AZD5363 also delays the onset of castrate‐resistant disease when combined with androgen deprivation as part of the initial therapy. There has been activity in trastuzumab‐resistant HER2‐positive breast cancer models, and a significant relationship between the presence of PIK3CA or PTEN mutations and sensitivity to AZD5363 99. The results of a phase I pharmacodynamically driven study of AZD5363 were reported at the American Society of Clinical Oncology 2015 annual meeting 100. The dose‐finding component of the study recruited 90 patients with advanced solid tumours and established AZD5363 480 mg twice daily given four out of seven days each week as the dosing schedule associated with the optimal pharmacokinetic, pharmacodynamic and toxicity profiles. The most common grade III toxicities were hyperglycaemia (20%), rash (10%) and diarrhoea (10%).The expansion phase component of the study enrolled 27 patients with PIK3CA‐mutant ER‐positive breast cancer and 18 patients with gynaecological cancers, with AZD5363 monotherapy resulting in a partial response rate of 20% and 7%, respectively. Several disease‐specific phase I/II trials are also currently enrolling patients (Table 3). In addition, there are plans to include an AKT inhibitor arm in the National Cancer Institute Molecular Analysis for Therapy Choice (MATCH) trial. Thus, patients with known AKT‐mutated tumours will be eligible to participate in a phase II subprotocol, through which they will receive an AKT inhibitor.
GSK2141795, another oral competitive pan‐AKT inhibitor, has demonstrated in vitro activity in head and neck, prostate, colon and gynaecological malignancies 80. A phase I dose‐escalation study of GSK2141795 in combination with BRAF inhibition is ongoing in solid tumours, and phase II studies in acute myeloid leukaemia, multiple myeloma and triple negative breast cancer. Toxicities thus far include grade II–III skin rash and diarrhoea 101.
Future directions
The PI3K–AKT pathway has important physiological functions, and pathway activity is commonly upregulated in cancer. While mTOR inhibitors and PI3K inhibitors have demonstrated modest clinical success in renal cell carcinoma, breast cancer and B‐cell malignancies, resulting in the approval of temsirolimus, everolimus and idelalisib, the full potential of modulating the pathway has not yet been harnessed. AKT is the ultimate signalling hub within the pathway, with over 100 downstream substrates. Successful inhibition of AKT is likely to have a significant antineoplastic effect. Unfortunately, dose‐limiting toxicities related to the normal physiological function of AKT and an incomplete understanding of the unique effects of the various isoforms of AKT have made it an elusive pharmacological target. Studies investigating pharmacodynamic markers at the MTD of several of the AKT inhibitors have demonstrated inadequate target modulation. There are currently no validated biomarkers that predict the response to AKT inhibitors before or after initiation of the drug, and it is not currently possible to predict which patients are likely to have significant adverse effects from AKT inhibition. Given the dose‐limiting toxicities at higher doses, it is likely that AKT inhibitors will need to be combined with additional targeted or cytotoxic agents that are synergistic in order to provide clinical benefit at a tolerable dose. Targeting AKT remains an important area of active clinical oncological research.
Competing Interests
All authors have completed the Unified Competing Interest form at www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare: no support from any organization for the submitted work; no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years; no other relationships or activities that could appear to have influenced the submitted work.
Mundi, P. S. , Sachdev, J. , McCourt, C. , and Kalinsky, K. (2016) AKT in cancer: new molecular insights and advances in drug development. Br J Clin Pharmacol, 82: 943–956. doi: 10.1111/bcp.13021.
References
- 1. Vivanco I, Sawyers C. The phosphatidylinositol 3‐kinase–AKT pathway in human cancer. Nat Rev Cancer 2002; 7: 489–501. [DOI] [PubMed] [Google Scholar]
- 2. Vara J, Casado E, de Castro J, Cejas P, Belda‐Iniesta C, Gonzalez‐Baron M. PI3K/AKT signaling pathway and cancer. Cancer Treat Rev 2004; 2: 193–204. [DOI] [PubMed] [Google Scholar]
- 3. Alexander SP, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 5th edition. Br J Pharmacol 2011; 164 Suppl 1: S1–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Crowell JA, Steele VE, Fay JR. Targeting the AKT protein kinase for cancer chemoprevention. Mol Cancer Ther 2007; 8: 2139–48. [DOI] [PubMed] [Google Scholar]
- 5. Camero A, Paramio J.The PTEN/PI3K/AKT pathway in vivo, cancer mouse models. Front Oncol 2014; 4: 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell 2007; 7: 1261–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Blachly JS, Baiocchi RA. Targeting the PI3‐kinase (PI3K), AKT, and mTOR axis in lymphoma. Br J Hematol 2014; 167: 19–32. [DOI] [PubMed] [Google Scholar]
- 8. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of AKT/PKB by the rictor‐mTOR complex. Science 2005; 307: 1098–1101. [DOI] [PubMed] [Google Scholar]
- 9. Blanco‐Aparicio C, Renner O, Leal JF, Carnero A. PTEN, more than the AKT pathway. Carcinogenesis 2007; 7: 1379–86. [DOI] [PubMed] [Google Scholar]
- 10. Feng J, Park J, Cron P, Hess D, Hemmings BA. Identification of a PKB/AKT hydrophobic motif ser‐473 kinase as DNA‐dependent protein kinase. J Biol Chem 2004; 39: 41189–96. [DOI] [PubMed] [Google Scholar]
- 11. Roberts MS, Woods AJ, Dale TC, Van Der Sluijs P, Norman JC. Protein kinase B/AKT acts via glycogen synthase kinase 3 to regulate recycling of alpha v beta 3 and alpha 5 beta 1 integrins. Mol Cell Bio 2004; 24: 1505–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Huang J, Manning BD. The TSC1–TSC2 complex: a molecular switchboard controlling cell growth. Biochem J 2008; 412: 179–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gottlieb TM, Leal JF, Seger R, Taya Y, Oren M. Cross‐talk between AKT, p53 and Mdm2: possible implications for the regulation of apoptosis. Oncogene 2002; 21: 1299–303. [DOI] [PubMed] [Google Scholar]
- 14. Mayo LD, Donner DB. A phosphatidylinositol 3‐kinase/AKT pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc Natl Acad Sci U S A 2001; 98: 11598–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kim AH, Khursigara G, Sun X, Franke TF, Chao MV. AKT phosphorylates and negatively regulates apoptosis signal‐regulating kinase 1. Mol Cell Bio 2001; 3: 893–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ko FC, Chan LK, Tung EK, Lowe SW, Ng IO, Yam JW. AKT phosphorylation of deleted in liver cancer 1 abrogates its suppression of liver cancer tumorigenesis and metastasis. Gastroenterology 2010; 139: 1397–1407. [DOI] [PubMed] [Google Scholar]
- 17. Tzivion G, Dobson M, Ramakrishnan G. FoxO transcription factors: regulation by AKT and 14‐3‐3 proteins. Mol Cell Res 2011; 1813: 1938–45. [DOI] [PubMed] [Google Scholar]
- 18. Ni D, Ma X, Li HZ, Gao Y, Li XT, Zhang Y, et al. Downregulation of FOXO3a promotes tumor metastasis and is associated with metastasis‐free survival of patients with clear cell renal cell carcinoma. Clin Can Res 2014; 20: 1779–90. [DOI] [PubMed] [Google Scholar]
- 19. Du K, Montminy M. CREB is a regulatory target for the protein kinase AKT/PKB. J Biol Chem 1998; 49: 32377–9. [DOI] [PubMed] [Google Scholar]
- 20. Hussain AR, Ahmed SO, Ahmed M, Khan OS, Al Abdulmohsen S, Platanias LC, et al. Cross‐talk between NFkB and the PI3‐kinase/AKT pathway can be targeted in primary effusion lymphoma (PEL) cell lines for efficient apoptosis. PLoS One 2012; 7: e39945. doi: 10.1371/journal.pone.0039945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gonzalez E, McGraw TE. The AKT kinases: isoform specificity in metabolism and cancer. Cell Cycle 2009; 8: 2502–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ji W, Chen X, Lv J, Wang M, Ren S, Yuan B, et al Liraglutide exerts antidiabetic effect via PTP1B and PI3K/AKT2 signaling pathway in skeletal muscle of KKAy mice. Int J Endocrinol 2014; 2014: 312452. doi: 10.1155/2014/312452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Paz‐Ares L, Blanco‐Aparicio C, Garcia‐Carbonero R, Camero A. Inhibiting PI3K as a therapeutic strategy against cancer. Clin Transl Oncol 2009; 11: 572–9. [DOI] [PubMed] [Google Scholar]
- 24. Dummler B, Tschopp O, Hynx D, Yang ZZ, Dimhofer S, Hemmings BA. Life with a single isoform of AKT: mice lacking AKT2 and AKT3 are viable but display impaired glucose homeostasis and growth deficiencies. Mol Cell Biol 2006; 26: 8042–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Inoue K, Fry EA, Taneja P. Recent progress in mouse models for tumor suppressor genes and its implications in human cancer. Clin Med Insights Oncol 2013; 7: 103–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Suzuki A, de la Pompa JL, Stambolic V, Elia AJ, Sasaki T, del Carco Barrantes, I , et al High cancer susceptibility and embryonic lethality associated with mutation of the PTEN tumor suppressor gene in mice. Curr Biol 1998; 8: 1169–78. [DOI] [PubMed] [Google Scholar]
- 27. Di Cristofano A, Pesce B, Cordon‐Cardo C, Pandolfi PP. Pten is essential for embryonic development and tumour suppression. Nat Genet 1998; 19: 348–55. [DOI] [PubMed] [Google Scholar]
- 28. Hutchinson J, Jin J, Cardiff RD, Woodgett JR, Muller WJ. Activation of AKT (protein kinase B) in mammary epithelium provides a critical cell survival signal required for tumor progression. Mol Cell Biol 2001; 21: 2203–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ackler S, Ahmad S, Tobias C, Johnson MD, Glazer RI. Delayed mammary gland involution in MMTV‐AKT1 transgenic mice. Oncogene 2002; 21: 198–206. [DOI] [PubMed] [Google Scholar]
- 30. Schwertfeger KL, Richert MM, Anderson SM. Mammary gland involution is delayed by activated AKT in transgenic mice. Mol Endocrinol 2001; 15: 867–81. [DOI] [PubMed] [Google Scholar]
- 31. Blanco‐Aparicio C, Canamero M, Cecilia Y, Pequeno B, Renner O, Ferrer I, et al. Exploring the gain of function contribution of AKT to mammary tumorigenesis in mouse models. PLoS One 2010; 5: e9305. doi: 10.1371/journal.pone.0009305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Deng CX, Brodie SG. Knockout mouse models and mammary tumorigenesis. Semin Cancer Biol 2001; 11: 387–94. [DOI] [PubMed] [Google Scholar]
- 33. Dunbar ME, Wysolmerski JJ. Mammary ductal and alveolar development: lesson learned from genetically manipulated mice. Microsc Res Tech 2001; 52: 163–70. [DOI] [PubMed] [Google Scholar]
- 34. Segrelles C, Moral M, Lara MF, Ruiz S, Santo M, Leis H, et al Molecular determinants of AKT‐induced keratinocyte transformation. Oncogene 2006; 25: 1174–85. [DOI] [PubMed] [Google Scholar]
- 35. Segrelles C, Lu J, Hammann B, Santos M, Moral M, Cascallana JL, et al Deregulated activity of AKT in epithelial basal cells induces spontaneous tumors and heightened sensitivity to skin carcinogenesis. Cancer Res 2007; 67: 10879–88. [DOI] [PubMed] [Google Scholar]
- 36. Moral M, Segrelles C, Lara MF, Martinez‐Cruz AB, Lorz C, Santos M, et al AKT activation synergizes with Trp53 loss in oral epithelium to produce a novel mouse model for head and neck squamous cell carcinoma. Cancer Res 2009; 69: 1099–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Majumder PK, Yeh JJ, George DJ, Febbo PG, Jum J, Zue Q, et al. Prostate intraepithelial neoplasia induced by prostate restricted AKT activation: the MPAKT model. Proc Natl Acad Sci U S A 2003; 100: 7841–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Holland EC, Celestino J, Dai C, Schaefer L, Sawaya RE, Fuller GN. Combined activation of Ras and AKT in neural progenitors induces glioblastoma formation in mice. Nat Genet 2000; 25: 55–7. [DOI] [PubMed] [Google Scholar]
- 39. Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, et al. Mutational landscape and significance across 12 major cancer types. Nature 2013; 502: 333–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kandel ES, Hay N. The regulation and activities of the multifunctional serine/threonine kinase AKT/PKB. Exp Cell Res 1999; 253: 210–29. [DOI] [PubMed] [Google Scholar]
- 41. Liaw D, Marsh DJ, Li, J , Dahia, PL , Wang, S , Zheng, Z , et al Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet 1997; 16: 64–7. [DOI] [PubMed] [Google Scholar]
- 42. Boyault S, Drouet Y, Navarro C, Bachelot T, Lasset C, Treilleux I, et al. Mutational characterization of individual breast tumors: TP53 and PI3K pathway genes are frequently and distinctively mutated in different subtypes. Breast Cancer Res Treat 2012; 132: 29–39. [DOI] [PubMed] [Google Scholar]
- 43. Stemke‐Hale K, Gonzalez‐Angulo AM, Lluch A, Neve RM, Kuo WL, Davies M, et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res 2008; 68: 6084–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kumar A, Purohit R. Cancer associated E17K mutation causes rapid conformational drift in AKT1 pleckstrin homology (PH) domain. PLoS One 2013; 8: e64364. doi: 10.1371/journal.pone.0064364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Askham JM, Platt F, Chambers PA, Snowden H, Taylor CF, Knowles MA. AKT1 mutations in bladder cancer: identification of a novel oncogenic mutation that can co‐operate with E17K. Oncogene 2010; 29: 150–5. [DOI] [PubMed] [Google Scholar]
- 46. Parikh C, Janakiraman V, Wu W, Foo CK, Kljavin NM, Chaudhuri S, et al. Disruption of PH–kinase domain interactions leads to oncogenic activation of AKT in human cancers. Proc Natl Acad Sci 2012; 109: 19368–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Davies MA, Stemke‐Hale K, Lin E, Tellez C, Deng W, Gopal YN, et al Integrated molecular and clinical analysis of AKT activation in metastatic melanoma. Clin Cancer Res 2009; 15: 7538–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Park ES, Rabinovsky R, Carey M, Hennessy BT, Agarwal R, Liu W, et al Integrative analysis of proteomic signatures, mutations, and drug responsiveness in the NCI 60 cancer cell line set. Mol Cancer Ther 2010; 9: 257–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kim MS, Jeong EG, Yoo NJ, Lee SH. Mutational analysis of oncogenic AKT E17K mutation in common solid cancers and acute leukaemias. Br J Cancer 2008; 98: 1533–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Perou C; Cancer Genome Atlas Network . Comprehensive portraits of human breast tumors. Nature 2012; 490: 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Landgraf KE, Pilling C, Falke JJ. Molecular mechanism of an oncogenic mutation that alters membrane targeting: Glu17Lys modifies the PIP lipid specificity of the AKT1 PH domain. Biochemistry 2008; 47: 12260–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, et al The landscape of cancer genes and mutational processes in breast cancer. Nature 2012; 486: 400–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yi KH, Axtmayer J, Gustin JP, Rajpurohit A, Lauring J. Functional analysis of non‐hotspot AKT1 mutants found in human breast cancers identifies novel driver mutations: implications for personalized medicine. Oncotarget 2013; 4: 29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Altomare DA, Testa JR. Perturbations of the AKT signaling pathway in human cancer. Oncogene 2005; 24: 7455–64. [DOI] [PubMed] [Google Scholar]
- 55. Chan CH, Jo U, Kohrman A, Rezaeian AH, Chou PC, Logothetis C, et al Posttranslational regulation of AKT in human cancer. Cell and Bioscience 2014; 4: 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ahmed NN, Franke TF, Bellacosa A, Datta K, Gonzalez‐Portal ME, Taguchi T, et al The proteins encoded by c‐AKT and v‐AKT differ in post‐translational modification, subcellular localization and oncogenic potential. Oncogene 1993; 8: 1957–63. [PubMed] [Google Scholar]
- 57. Yang WK, Wu CY, Wu J, Lin HK. Regulation of AKT signaling activation by ubiquitination. Cell Cycle 2010; 9: 487–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sale EM, Sale GJ. Protein kinase B: signaling roles and therapeutic targets. Cell Mol Life Sci 2008; 65: 113–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Shao Y, Aplin AE. AKT3‐mediated resistance to apoptosis in B‐RAF‐targeted melanoma cells. Cancer Res 2010; 70: 6670–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. West KA, Linnoila IR, Belinsky SA, Harris CC, Dennis PA. Tobacco carcinogen‐induced cellular transformation increases activation of the phosphatidylinositol 3′‐kinase/AKT pathway in vitro and in vivo . Cancer Res 2004; 64: 446–51. [DOI] [PubMed] [Google Scholar]
- 61. Chun KH, Kosmeder JW, Sun S, Pezzuto JM, Lotan R, Hong WK, et al Effects of deguelin on the phosphatidylinositol 3‐kinase/AKT pathway and apoptosis in premalignant human bronchial epithelial cells. J Natl Cancer Inst 2003; 95: 291–302. [DOI] [PubMed] [Google Scholar]
- 62. Bose S, Chandran S, Mirocha JM, Bose N. The AKT pathway in human breast cancer: a tissue‐array‐based analysis. Mod Pathol 2006; 19: 238–45. [DOI] [PubMed] [Google Scholar]
- 63. Majumder PK, Febbo PG, Bikoff R, Berger R, Xue Q, McMahon LM, et al mTOR inhibition reverses Akt‐dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF‐1‐dependent pathways. Nat Med 2004; 10: 594–601. [DOI] [PubMed] [Google Scholar]
- 64. Malik SN, Brattain M, Ghosh PM, Troyer DA, Prihoda T, Bedolla R, et al Immunohistochemical demonstration of phospho‐AKT in high Gleason grade prostate cancer. Clin Cancer Res 2002; 8: 1168–71. [PubMed] [Google Scholar]
- 65. Xin L, Teitell MA, Lawson DA, Kwon A, Mellinghoff IK, Witte ON. Progression of prostate cancer by synergy of AKT with genotropic and nongenotropic actions of the androgen receptor. Proc Natl Acad Sci U S A 2006; 103: 7789–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Majumder PK, Sellers WR. AKT‐regulated pathways in prostate cancer. Oncogene 2005; 24: 7465–74. [DOI] [PubMed] [Google Scholar]
- 67. Dai DL, Martinka M, Li G. Prognostic significance of activated AKT expression in melanoma: a clinicopathologic study of 292 cases. J Clin Oncol 2005; 23: 1473–82. [DOI] [PubMed] [Google Scholar]
- 68. Stahl JM, Sharma A, Cheung M, Zimmerman M, Cheng JQ, Bosenberg MW, et al Deregulated AKT3 activity promotes development of malignant melanoma. Cancer Res 2004; 64: 7002–10. [DOI] [PubMed] [Google Scholar]
- 69. Bedogni B, Welford SM, Cassarino DS, Nickoloff BJ, Giaccia AJ, Powell MB. The hypoxic microenvironment of the skin contributes to AKT‐mediated melanocyte transformation. Cancer Cell 2005; 8: 443–54. [DOI] [PubMed] [Google Scholar]
- 70. McCubrey JA, Steelman LS, Abrams SL, Lee JT, Chang F, Bertrand FE, et al Roles of the RAF/MEK/ERK and PI3K/PTEN/AKT pathways in malignant transformation and drug resistance. Adv Enzyme Regul 2006; 46: 249–79. [DOI] [PubMed] [Google Scholar]
- 71. Ojesina AI, Lichtenstein L, Freeman SS, Pedamallu CS, Imaz‐Rosshandler I, Pugh TJ, et al Landscape of genomic alterations in cervical carcinomas. Nature 2014; 506: 371–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Cancer Genome Atlas Research Network , Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H, et al Integrated genomic characterization of endometrial carcinoma. Nature 2013; 497: 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Du CX, Wang Y. Expression of P‐AKT, NFkappaB and their correlation with human papillomavirus infection in cervical carcinoma. Eur J Gynaecol Oncol 2012; 33: 274–7. [PubMed] [Google Scholar]
- 74. Gupta AK, Lee JH, Wilke WW, Quon H, Smith G, Maity A, et al Radiation response in two HPV‐infected head‐and‐neck cancer cell lines in comparison to a non‐HPV‐infected cell line and relationship to signaling through AKT. Int J Radiat Oncol Biol Phys 2009; 74: 928–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Grabinski N, Mollmann K, Milde‐Langosch K, Müller V, Schumacher U, Brandt B, et al AKT3 regulates ErbB2, ErbB3 and estrogen receptor α expression and contributes to endocrine therapy resistance of ErbB2(+) breast tumor cells from Balb‐neuT mice. Cell Signal 2014; 26: 1021–9. [DOI] [PubMed] [Google Scholar]
- 76. Furman RR, Sharman JP, Coutre SE, Cheson BD, Pagel JM, Hillmen P, et al Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med 2014; 370: 997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Geuna E, Milani A, Martinello R, Aversa C, Valabrega G, Scaltriti M, et al. Buparlisib, an oral pan‐PI3K inhibitor for the treatment of breast cancer. Expert Opin Investig Drugs 2015; 24: 421–31. [DOI] [PubMed] [Google Scholar]
- 78. Baselga J. PI3K inhibitor improves PFS in BELLE‐2 trial. Cancer Discov 2016; 6: 115–6. (San Antonio Breast Cancer Symposium 2015 abstract and oral session). [DOI] [PubMed] [Google Scholar]
- 79. Dienstmann R, Rodon J, Serra V, Tabernero J. Picking the point of inhibition: a comparative review of PI3K/AKT/mTOR pathway inhibitors. Mol Cancer Ther 2014; 13: 1021–31. [DOI] [PubMed] [Google Scholar]
- 80. Pal SK, Reckamp K, Yu H, Figlin RA. AKT inhibitors in clinical development for the treatment of cancer. Expert Opin Investig Drugs 2010; 19: 1355–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Lindsley CW, Barnett SF, Yaroschak M, Bilodeau MT, Layton ME. Recent progress in the development of ATP‐competitive and allosteric AKT kinase inhibitors. Curr Top Med Chem 2007; 7: 1349–63. [DOI] [PubMed] [Google Scholar]
- 82. Crul M, Rosing H, de Klerk GJ, Dubbelman R, Traiser M, Reichert S, et al. Phase I and pharmacological study of daily oral administration of perifosine (D‐21266) in patients with advanced solid tumours. Eur J Cancer 2002; 38: 1615–21. [DOI] [PubMed] [Google Scholar]
- 83. Bendell JC, Nemunaitis J, Vukelja SJ, Hagenstad C, Campos LT, Hermann RC, et al Randomized placebo‐controlled phase II trial of perifosine plus capecitabine as second‐ or third‐line therapy in patients with metastatic colorectal cancer. J Clin Oncol 2011; 29: 4394–400. [DOI] [PubMed] [Google Scholar]
- 84. Bendell JC, Ervin TJ, Senzer NN, Richards DA, Firdaus I, Lockhart AC, et al. Results of the X‐PECT study: a phase III randomized double‐blind, placebo‐controlled study of perifosine plus capecitabine (P‐CAP) versus placebo plus capecitabine (CAP) in patients (pts) with refractory metastatic colorectal cancer (mCRC). J Clin Oncol 2012; American Society of Clinical Oncology Meeting Abstract. [Google Scholar]
- 85. Jakubowiak AJ, Richardson PG, Zimmerman T, Alsina M, Kaufman JL, Kandarpa M, et al. Perifosine plus lenalidomide and dexamethasone in relapsed and relapsed/refractory multiple myeloma: a Phase I Multiple Myeloma Research Consortium study. Br J Haematol 2012; 158: 472–80. [DOI] [PubMed] [Google Scholar]
- 86. Richardson PG, Wolf J, Jakubowiak A, Zonder J, Lonial S, Irwin D, et al. Perifosine plus bortezomib and dexamethasone in patients with relapsed/refractory multiple myeloma previously treated with bortezomib: results of a multicenter phase I/II trial. J Clin Oncol 2011; 29: 4243–9. [DOI] [PubMed] [Google Scholar]
- 87. Hirai H, Sootome H, Nakatsuru Y, Miyama K, Taguchi S, Tsujioka K, et al. MK‐2206, an allosteric AKT inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo . Mol Cancer Ther 2010; 9: 1956–67. [DOI] [PubMed] [Google Scholar]
- 88. Kalinsky KM, Sparano JA, Andreopoulou E, Taback B, Wiechmann LS, Feldman SM, et al. Presurgical evaluation of the AKT inhibitor MK‐2206 in patients with operable invasive breast cancer. J Clin Oncol 2014, American Society of Clinical Oncology Meeting Abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Tolcher AW, Yap TA, Fearen I, Taylor A, Carpenter C, Brunetto AT, et al. A phase I study of MK‐2206, an oral potent allosteric AKT inhibitor (AKTi), in patients (pts) with advanced solid tumor (ST). J Clin Oncol 2009, American Society of Clinical Oncology Meeting Abstract. [Google Scholar]
- 90. Hudis C, Swanton C, Janjigian YY, Lee R, Sutherland S, Lehman R, et al. A phase 1 study evaluating the combination of an allosteric AKT inhibitor (MK‐2206) and trastuzumab in patients with HER2‐positive solid tumors. Breast Cancer Res 2013; 15: R110. doi: 10.1186/bcr3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Oki Y, Fanale M, Romaguera J, Fayad L, Fowler N, Copeland A, et al. Phase II study of an AKT inhibitor MK2206 in patients with relapsed or refractory lymphoma. Br J Haematol 2015; 171: 463–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Ma CX1, Sanchez C2, Gao F3, Crowder R4, Naughton M5, Pluard T, et al. A phase I study of the AKT inhibitor MK‐2206 in combination with hormonal therapy in postmenopausal women with estrogen receptor positive metastatic breast cancer. Clin Cancer Res 2016; 22: 2650–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Ramanathan RK, McDonough SL, Kennecke HF, Iqbal S, Baranda JC, Seery TE, et al. Phase 2 study of MK‐2206, an allosteric inhibitor of AKT, as second‐line therapy for advanced gastric and gastroesophageal junction cancer: a SWOG cooperative group trial (S1005). Cancer 2015; 121: 2193–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Lara PN, Longmate J, Mack PC, Kelly K, Socinski MA, Salgia R, et al. Phase II study of the AKT inhibitor MK‐2206 plus erlotinib in patients with advanced non‐small cell lung cancer who previously progressed on erlotinib. Clin Cancer Res 2015; 21: 4321–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Do K, Speranza G, Bishop R, Khin S, Rubinstein L, Kinders RJ, et al. Biomarker‐driven phase 2 study of MK‐2206 and selumetinib (AZD6244, ARRY‐142886) in patients with colorectal cancer. Invest New Drugs 2015; 33: 720–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Tolcher AW, Patnaik A, Papadopoulos KP, Rasco DW, Becerra CR, Allred AJ, et al Phase I study of the MEK inhibitor trametinib in combination with the AKT inhibitor afuresertib in patients with solid tumors and multiple myeloma. Cancer Chemother Pharmacol 2015; 75: 183–9. [DOI] [PubMed] [Google Scholar]
- 97. Spencer A, Yoon SS, Harrison SJ, Morris SR, Smith, DA , Brigandi, RA , et al The novel AKT inhibitor afuresertib shows favorable safety, pharmacokinetics, and clinical activity in multiple myeloma. Blood 2014; 124: 2190–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Toren P, Kim S, Cordonnier T, Crafter C, Davies BR, Fazli L, et al. Combination AZD5363 with enzalutamide significantly delays enzalutamide‐resistant prostate cancer in preclinical models. Eur Urol 2015; 67: 986–90. [DOI] [PubMed] [Google Scholar]
- 99. Davies BR, Greenwood H, Dudley P, Crafter C, Yu DH, Zhang J, et al. Preclinical pharmacology of AZD5363, an inhibitor of AKT: pharmacodynamics, antitumor activity, and correlation of monotherapy activity with genetic background. Mol Cancer Ther 2012; 11: 873–87. [DOI] [PubMed] [Google Scholar]
- 100. Banerji U. A pharmacokinetically (PK) and pharmacodynamically (PD) driven phase I trial of the pan‐AKT inhibitor AZD5363 with expansion cohorts in PIK3CA mutant breast and gynecological cancers. J Clin Oncol 2015, American Society of Clinical Oncology Meeting Abstract. [Google Scholar]
- 101. Algazi AP, Muthukumar AH, Obrien K, Lencioni A, Tsai KK, Kadafour M, et al. Phase II trial of trametinib in combination with the AKT inhibitor GSK 2141795 in BRAF wild‐type melanoma. J Clin Oncol 2015, American Society of Clinical Oncology Meeting Abstract. [Google Scholar]