

Abstract
Aims
The aim of the study was to compare the pharmacokinetics (PK), safety and tolerability of the proposed adalimumab biosimilar MSB11022 (Merck) with Humira® (AbbVie), sourced from both the US (US reference product [US‐RP]) and Europe (European reference medicinal product [EU‐RMP]).
Methods
In this phase 1 double‐blind, parallel group trial (EMR200588‐001), 213 healthy volunteers were randomized 1 : 1 : 1 to receive a single dose (40 mg) of MSB11022, US‐RP or EU‐RMP in order to achieve 80% power assuming a 5% difference among groups and a 10% dropout rate. Following a preplanned blinded sample size re‐assessment after more than 50% of the originally planned subjects had been observed, the sample size was increased to 237 (79 per arm) to ensure 213 completers. Primary PK endpoints analyzed by non‐compartmental methods, were area under the curve (AUC) from time 0 extrapolated to infinity (AUC(0,∞)), maximum observed concentration (C max), and AUC from time 0 to the last quantifiable concentration (AUC(0,t last)). PK equivalence was declared if the 90% CI for the test : reference ratio lay within the 80–125% equivalence margin. Bioequivalence was demonstrated if all three PK parameters met the PK equivalence criteria. Safety and tolerability were also evaluated.
Results
Mean serum concentration–time profiles for the three treatments were similar. MSB11022 demonstrated PK equivalence to US‐RP and EU‐RMP for all primary endpoints. The geometric means of AUC(0,∞), C max and AUC(0,t last) following a single dose of MSB11022 were 2276.05 μg ml–1 h, 3.44 μg ml–1 and 1983.90 μg ml–1 h, respectively. Adverse events (AEs) were similar across all groups, with treatment‐emergent AEs (TEAEs) reported by 62.8%, 56.3% and 62.0% of subjects within the MSB11022, US‐RP and EU‐RMP groups, respectively. Most of the TEAEs were considered mild and unrelated to study drug. No deaths or severe AEs related to the study drug were reported.
Conclusions
Bioequivalence between MSB11022, US‐RP and EU‐RMP was demonstrated. Safety, tolerability and immunogenicity profiles were similar between subjects receiving MSB11022 and US‐RP or EU‐RMP. These data support the further clinical evaluation of MSB11022 as a proposed biosimilar of adalimumab.
Keywords: adalimumab, biosimilar, pharmacokinetics, safety
What is Already Known about this Subject
Adalimumab is a fully human monoclonal antibody that targets tumour necrosis factor.
Adalimumab is approved for the treatment of rheumatoid arthritis, psoriasis, Crohn's disease, ulcerative colitis, psoriatic arthritis, juvenile idiopathic arthritis, ankylosing spondylitis and hidradenitis suppurativa.
What this Study Adds
This was the first clinical report of a new proposed adalimumab biosimilar, MSB11022.
This phase 1 randomized, controlled study compared PK and safety of MSB11022 vs. the US‐ and European‐approved adalimumab products.
The results demonstrate bioequivalence between MSB11022 and the US/European reference products, as well as comparable safety and tolerability profiles.
Introduction
Humira® (adalimumab) is a recombinant, fully human immunoglobulin (Ig) G1 monoclonal antibody (mAb) that has high specificity and affinity for tumour necrosis factor (TNF) and a terminal half‐life (t ½) comparable with that of human IgG1 (~2 weeks) 1, 2, 3. The inflammatory response associated with certain autoimmune diseases is partly due to the binding of TNF to its receptors and adalimumab exerts its therapeutic effect by neutralizing the activity of TNF, thus blocking the interaction of this cytokine with p55 and p75 cell surface receptors 2, 3.
Humira® (adalimumab) was first licensed for the treatment of rheumatoid arthritis (RA) by the US Food and Drug Administration in 2002 and the European Medicines Agency in 2003 and is now indicated across a multitude of immune‐mediated conditions (RA, juvenile idiopathic arthritis, psoriatic arthritis, ankylosing spondylitis, adult Crohn's disease, paediatric Crohn's disease, ulcerative colitis, adult plaque psoriasis, paediatric plaque psoriasis and hidradenitis suppurativa) 2, 3. Its safety/tolerability profile is consistent with the immunosuppressive effects of the anti‐TNF class generally, with the most frequently reported adverse events (AEs) seen in adalimumab recipients being infection, injection site reactions, headache and rash 2, 3. Analysis of a global clinical database including 23 458 patients showed infection to be the most common AE overall, as might be expected with an immunosuppressive agent, while overall malignancy rates were typical for the general population 4. The reported incidence of antidrug antibodies (ADAs) with adalimumab varies widely 2, 3, 5, largely owing to the absence of a standardized ADA assay. While increased ADA may lead to lower systemic drug levels and reduced clinical response to treatment 6, it remains unclear whether this is the case for adalimumab.
A biosimilar medicinal product must demonstrate comparable physicochemical, non‐clinical and clinical efficacy and safety to the authorized biological drug (known as the reference medicinal product, RMP [EU] or reference product, RP [US]) through a series of strictly regulated preclinical, clinical, immunogenicity and safety studies 7, 8. MSB11022 (Merck), a proposed biosimilar to adalimumab, is a recombinant fully human IgG1 mAb that is directed against human TNF. MSB11022 shares an identical amino acid sequence to the US‐licensed adalimumab reference product Humira® (US‐RP) and the EU‐approved adalimumab reference product Humira® (EU‐RMP) with all products produced in Chinese hamster ovary cells. Extensive biochemical/biophysical analytical methods were used to confirm the primary structure, post‐translational modifications and high order structure of MSB11022 in comparison with US‐RP/EU‐RMP. The similarity assessment also included relevant mAb production attributes, such as purity and impurities and product variants. In order to confirm the similarity in functional characteristics, extensive in vitro testing was performed. MSB11022 showed comparable physicochemical and in vitro primary pharmacodynamic properties to US‐RP/EU‐RMP. By testing several commercial scale drug product lots of MSB11022 and US‐RP/EU‐RMP, the Fab activities for binding to human soluble and membrane bound forms of TNF and for the inhibition of TNF‐induced cytotoxicity, as well as the Fc binding to neonatal Fc receptor, Fc‐γ receptors and C1q complement protein, were shown to result in affinities and potencies for MSB11022 that fall within the range established by the US‐RP/EU‐RMP 9. In addition to the physicochemical and in vitro functional testing, a comparative, repeat‐dose, toxicity study in Cynomolgus monkeys was conducted. The results indicate similar exposure and immunogenicity profiles for MSB11022 and US‐RP and show no adverse findings for either drug (unpublished data on file, Merck Biosimilars, Aubonne, Switzerland). The aim of the present study was to compare the pharmacokinetic (PK) profile, safety, tolerability and immunogenicity of MSB11022, US‐RP, and EU‐RMP in healthy subjects.
Methods
Study population and design
This trial was conducted in accordance with the principles of the International Conference on Harmonization requirements for Good Clinical Practice, the Declaration of Helsinki and with the approval of a National Health Service Ethics Review Committee. All subjects gave written informed consent. The trial is reported according to CONSORT guidelines.
The study was conducted at two sites in the United Kingdom. To be included in the trial, subjects were required to weigh between 60.0 and 94.9 kg with body mass index (BMI) 20.0–29.9 kg m– 2. Vital signs, physical examination, clinical laboratory tests and 12‐lead electrocardiogram (ECG) had to be within the normal range or at least considered clinically non‐significant by the investigator. Female volunteers had to have been of non‐childbearing potential (confirmed at screening as either post‐menopausal or irreversibly sterilized). Subjects with a history of cancer including lymphoma, leukaemia and skin cancer were excluded from the trial. Subjects with a positive hepatitis C antibody test or hepatitis B surface antigen test and/or core antibody test for IgG and/or IgM as well as subjects with a positive test for human immunodeficiency virus at screening were excluded from the trial. Smoking more than 10 cigarettes per day or an inability to refrain from smoking or nicotine‐containing products during the residential stay at the trial site were also exclusion criteria. Other key exclusion criteria were history and/or current presence of clinically significant atopic allergy; known or suspected clinically relevant drug hypersensitivity, active or latent tuberculosis; history of invasive systemic fungal infections; recurrent or chronic local fungal infections, serious infection (defined as an infection that required hospitalization and/or which required anti‐infectives or antibiotics) within 6 months prior to trial drug administration, infection within 2 weeks of screening or during the screening period (unless the infection resolved completely within 2 weeks of admission) and previous treatment with adalimumab or another recombinant human mAb.
Subjects were admitted to the trial site and remained resident there for 8 days following dosing. After 10 subjects per arm had been observed for a minimum of 8 days, an interim safety analysis was conducted with data accumulated to that point. If the study drugs were well tolerated as per the investigator's judgement and reflected the expected safety profile, the confinement period was shortened to 4 or 5 days for the remainder of the subjects (and outpatient visits increased accordingly).
An initial sample size of 213 randomized subjects was estimated to achieve 80% power to show bioequivalence among MSB11022, US‐RP and EU‐RMP assuming a coefficient of variation (CV) for maximum observed concentration (C max) of 33% for US‐RP/EU‐RMP, a difference of 5% between groups and a 10% drop‐out rate. The planned number of subjects was increased following a slightly higher than expected inter‐subject CV of 34.3% for area under the curve (AUC) from time 0 to the last quantifiable concentration (AUC(0,t last)), during a pre‐specified blinded sample size re‐assessment (when >50% of subjects had been observed). To ensure 213 evaluable subjects, the sample size of the trial was increased to 237 randomized subjects (79 total subjects per arm).
Subjects were randomized 1 : 1 : 1 to receive a single 40 mg dose of MSB11022, US‐RP or EU‐RMP via a subcutaneous injection in the lower abdomen (Figure S1). Allocation of randomization numbers occurred immediately prior to study drug administration according to the randomization list prepared by the randomization statistician.
Study objectives
The primary objective of this trial was to demonstrate bioequivalence of MSB11022, US‐RP and EU‐RMP in healthy subjects. Secondary objectives included comparing the safety, tolerability and immunogenicity of the three products.
PK assessments and endpoints
Blood samples for PK evaluation were obtained at 0 (pre‐dose), 4, 8, 12 and 24 h post‐dose. Further samples were obtained every 24 h thereafter until day 9, then at all subsequent outpatient visits (outlined in Figure S1). Serum MSB11022, US‐RP and EU‐RMP concentrations were determined using a validated enzyme‐linked immunosorbent assay that employed a TNF coated plate, horseradish peroxidase‐conjugated anti‐human IgG antibody to detect bound analyte and 3,3′,5,5′‐tetramethylbenzidine for colorimetric readout. Colorimetric intensity was determined using a plate reader at 450 nm (detection) and 630 nm (reference) wavelengths. The lower limit of quantification (LLOQ) was 300 ng ml−1.
Inter‐assay precision and accuracy were calculated from quality controls (QCs) in 18 validation runs (low, mid and high QCs) and six validation runs (LLOQ, upper limit of quantitation [ULOQ] and back‐up LLOQ and ULOQ QCs) for MSB11022, and in six validation runs (each) for US‐RP and EU‐RMP. Results are summarized in Table S1. The post‐validation performance of the bioanalytical method during sample analysis of study EMR200588‐001 is presented in Table S2. Inter‐assay precision and accuracy confirmed the performance observed during method validation. Incurred sample re‐analysis demonstrated reproducible quantitation of the drug in study samples. Overall, 96.6% of the re‐analyzed samples met the incurred sample re‐analysis acceptance criteria.
Samples below the LLOQ before the last quantifiable data point were set to 0. Concentrations below the LLOQ after the last quantifiable data point were considered as missing. Primary PK endpoints were: C max, AUC from time 0 extrapolated to infinity (AUC(0,∞)) and AUC from time 0 to the last quantifiable concentration (AUC(0,t last)).
Other PK endpoints were time to reach C max (t max), apparent volume of distribution during the terminal phase (V z/F), terminal rate constant (λz), terminal half‐life (t ½ ) and apparent total clearance (CL/F).
PK parameters were derived using non‐compartmental methods with the validated computer programme Phoenix® WinNonlin® Version 6.3 (Certara LP, Princeton, NJ, USA). PK concentration–time curves were constructed using actual elapsed time from study drug administration.
Immunogenicity
Blood samples for immunogenicity analysis were obtained pre‐dose and on days 15, 29, 43 and 71 post‐dose. The immunogenicity of the administered drug was determined by measuring the incidence of ADAs. ADAs were measured using a single assay approach and acid‐dissociation with an electrochemiluminescent immunoassay validated to detect anti‐adalimumab in human serum. Chemiluminescence was measured in relative light units using the Meso Scale Discovery PRTM 6000 Plate Reader (Meso Scale Diagnostics, Rockville, MD, USA). The assay sensitivity was 86.4 ng ml−1 with a drug tolerance of 250 μg ml−1 at the LLOQ of 129.6 ng ml−1. Inter‐run assay precision for the QC samples and negative control was 10.3–19.7%. As anti‐drug antibodies to adalimumab have been shown to be largely neutralizing 10, 11, assays to detect the neutralizing capacity of antidrug antibodies to MSB11022, US‐RP and EU‐RMP were performed. These data will be published at a later date.
Safety and tolerability
All AEs were coded according to the Medical Dictionary for Regulatory Activities version 17.0 or higher and assigned to a system organ class and preferred term. Severity of AEs was graded in accordance with the National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.03. An AE was considered as a treatment‐emergent adverse event (TEAE) if it occurred after study drug administration on day 1 or if it was present prior to, but exacerbated after, study drug administration.
Additional safety assessments included: vital signs, clinical laboratory values (haematology, biochemistry, and urinalysis), 12‐lead ECGs, AEs, TEAEs and serious AEs (SAEs). Prespecified AEs of special interest were injection site reactions, serious infections and hypersensitivity reactions.
Statistical analysis
AUC(0,∞), AUC(0,t last) and C max were analyzed using a one way analysis of variance model with treatment as fixed effect. The PK parameters were log transformed for analysis and the 90% confidence interval (CI) for the difference in mean parameters among the three groups was calculated, then re‐expressed on the original ratio scale to assess equivalence. PK equivalence was declared if the 90% CI for the test : reference ratio lay entirely within the 80–125% equivalence margin for each comparison. Bioequivalence was demonstrated if all three PK parameters met the PK equivalence criteria. Other PK parameters, serum drug concentrations and safety/tolerability data were summarized using descriptive statistics.
Results
Subject disposition
A total of 237 healthy volunteers aged 18–55 years were enrolled in this phase 1 double‐blind, parallel‐group trial (trial number: EMR200588‐001). Subject disposition is outlined in Figure 1. Altogether, 237 subjects were randomized to receive MSB11022 (n = 78), US‐RP (n = 80) and EU‐RMP (n = 79) between 30 May 2014 and 23 December 2014. All randomized subjects received one dose of their allocated treatment and were included in the safety analysis set (n = 237). One subject in the US‐RP group was excluded from the PK analysis due to withdrawal of consent (PK analysis set, n = 236). Two additional subjects (one in each of the US‐RP and EU‐RMP groups) did not attend later PK assessments and despite returning for the follow‐up visit, were not classed as having completed the study. One subject in the MSB11022 group was lost to follow‐up. All other subjects completed the study (98.3%).
Figure 1.
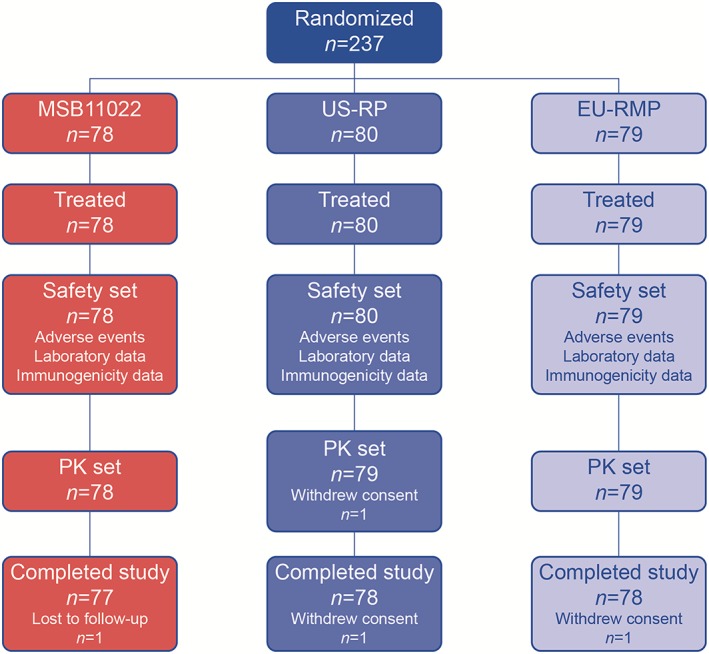
Subject disposition. EU‐RMP, EU‐reference medicinal product (adalimumab); PK, pharmacokinetic; US‐RP, US‐reference product (adalimumab)
Demographics and baseline characteristics
Baseline demographics and clinical characteristics of study participants were well balanced between treatment groups and are outlined in Table 1. Randomized subjects had a mean age of 32.7 years (range 18–56 years), mean weight 77.6 kg (60.2–94.8 kg) and mean BMI 24.9 kg m− 2 (20.1–29.8 kg m− 2). The majority of subjects were male 235/237 (99.2%) and Caucasian 163/237 (68.8%). All enrolled subjects were included in the safety and immunogenicity assessments.
Table 1.
Baseline demographics and clinical characteristics
| MSB11022 | US‐RP | EU‐RMP | ||||
|---|---|---|---|---|---|---|
| Demographic characteristic | n = 78 | n = 80 | n = 79 | |||
| Age (years) | ||||||
| Mean (SD) | 32.1 | (9.68) | 32.4 | (9.40) | 33.7 | (9.68) |
| Gender, n (%) | ||||||
| Male | 77 | (98.7) | 79 | (98.8) | 79 | (100.0) |
| Female | 1 | (1.3) | 1 | (1.3) | 0 | (0.0) |
| Race, n (%) | ||||||
| White | 51 | (65.4) | 54 | (67.5) | 58 | (73.4) |
| Black or African American | 10 | (12.8) | 10 | (12.5) | 10 | (12.7) |
| Asian | 10 | (12.8) | 10 | (12.5) | 6 | (7.6) |
| Other | 7 | (9.0) | 6 | (7.5) | 5 | (6.3) |
| Ethnicity, n (%) | ||||||
| Not Hispanic or Latino | 77 | (98.7) | 79 | (98.8) | 79 | (100.0) |
| Hispanic or Latino | 1 | (1.3) | 1 | (1.3) | 0 | (0.0) |
| Height (cm) | ||||||
| Mean (SD) | 176.7 | (7.7) | 175.8 | (6.4) | 177.0 | (5.7) |
| Weight (kg) | ||||||
| Mean (SD) | 77.4 | (7.9) | 78.3 | (8.2) | 77.1 | (8.2) |
| Body mass index (kg m−2) | ||||||
| Mean (SD) | 24.8 | (2.4) | 25.3 | (2.6) | 24.6 | (2.4) |
EU‐RMP, Europe‐approved reference medicinal product; SD, standard deviation; US‐RP, US‐licensed reference product.
PK results
All subjects were exposed to a single 40 mg dose of their allocated treatment as a subcutaneous injection. Figure 2 shows the mean serum concentration–time profiles for the three treatment groups. The mean profiles for each treatment were similar. Mean serum concentrations of MSB11022, US‐RP and EU‐RMP appeared to decline in a monophasic manner and remained quantifiable up to 1680 h post‐dose. Geometric mean values and geometric mean ratios for the primary study endpoints (mean AUC(0,∞), AUC(0,t last) and C max) are shown by treatment group in Table 2. MSB11022 data demonstrated PK equivalence to US‐RP and EU‐RMP for all three primary endpoints, as the 90% CIs for the test : reference ratios were entirely contained within the predefined equivalence interval of 80–125%, thereby demonstrating bioequivalence. For the AUC parameters, the geometric mean ratios were in the range 89–96% for MSB11022 when compared with both the US‐RP and EU‐RMP, with the AUC(0,∞) lower 90% CI being just above 80% (Table 3). Bioequivalence was also observed between US‐RP and EU‐RMP.
Figure 2.
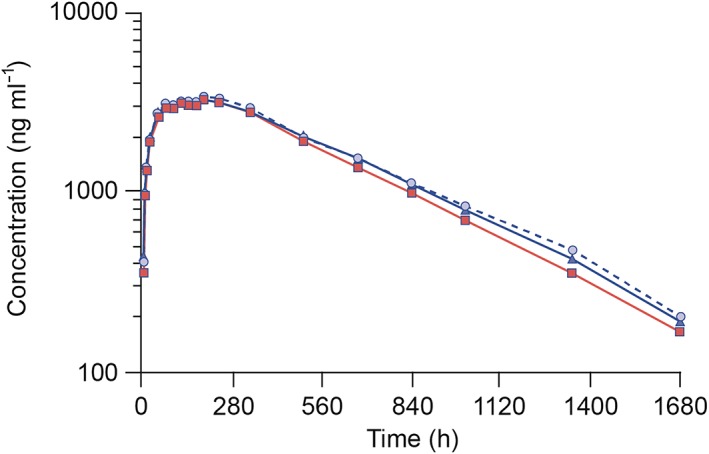
Mean serum concentration–time profiles (on semi‐logarithmic scale) by treatment following a single subcutaneous injection of 40 mg MSB11022, US‐RP and EU‐RMP. Data are presented as means. EU‐RMP, EU‐reference medicinal product (adalimumab); US‐RP, US‐reference product (adalimumab). EU‐RMP,
EU‐RMP,  MSB11022,
MSB11022,  US‐RP
US‐RP
Table 2.
Pharmacokinetic endpoints
| Treatment | ||||
|---|---|---|---|---|
| Parameter (units) | Statistic | MSB11022 | US‐RP | EU‐RMP |
| AUC(0,∞) | n | 76 | 75 | 77 |
| (μg ml−1 h) | Geometric mean | 2276.05 | 2515.98 | 2553.89 |
| GeoCV% | 44.5 | 37.5 | 41.9 | |
| AUC(0,tlast) | n | 78 | 79 | 79 |
| (μg ml−1 h) | Geometric mean | 1983.90 | 2065.99 | 2167.38 |
| GeoCV% | 43.5 | 52.6 | 45.2 | |
| Cmax | n | 78 | 79 | 79 |
| (μg ml−1) | Geometric mean | 3.44 | 3.53 | 3.60 |
| GeoCV% | 36.5 | 32.7 | 30.3 | |
| tmax | n | 78 | 79 | 79 |
| (h) | Median | 191.41 | 191.07 | 190.75 |
| Range | 24.00–506.00 | 48.00–339.90 | 48.00–503.80 | |
| t½ | n | 76 | 75 | 77 |
| (h) | Geometric mean | 295.46 | 352.50 | 348.61 |
| GeoCV% | 63.2 | 49.3 | 51.9 | |
| CL/F | n | 76 | 75 | 77 |
| (l h−1) | Geometric Mean | 0.0176 | 0.0159 | 0.0157 |
| GeoCV% | 44.5 | 37.5 | 41.9 | |
| Vz/F | n | 76 | 75 | 77 |
| (l) | Geometric Mean | 7.491 | 8.085 | 7.877 |
| GeoCV% | 40.6 | 32.9 | 31.3 | |
AUC(0,∞), area under the concentration–time curve from time 0 extrapolated to infinity; AUC(0,t last), AUC from time 0 to the last quantifiable concentration; CL/F, apparent total clearance; C max, maximum observed concentration; EU‐RMP, Europe‐approved reference medicinal product; GeoCV%, geometric coefficient of variation based on the geometric mean (sample size calculations are based on the arithmetic CV); t ½, terminal half‐life; t max, time to reach C max; US‐RP, US‐licensed reference product; V z/F, apparent volume of distribution during the terminal phase.
Note: λz was not estimable for all subjects and therefore for the λz dependent parameters the n is reduced to 76, 75 and 77 subjects, respectively, for MSB11022, US‐RP and EU‐RMP.
Table 3.
Statistical comparison of primary pharmacokinetic endpoints between MSB11022 and reference products
| Parameter (units) | Treatment | n | Geometric LS mean | Comparison | Ratio (%) | 90% CI of ratio |
|---|---|---|---|---|---|---|
| AUC(0,∞) (μg ml−1 h) | MSB11022 | 76 | 2276.05 | MSB11022/US‐RP | 90.46 | (81.29, 100.67) |
| US‐RP | 75 | 2515.98 | MSB11022/EU‐RMP | 89.12 | (80.14, 99.10) | |
| EU‐RMP | 77 | 2553.89 | US‐RP/EU‐RMP | 98.52 | (88.56, 109.59) | |
| AUC(0,tlast) (μg ml−1 h) | MSB11022 | 78 | 1983.90 | MSB11022/US‐RP | 96.03 | (85.32, 108.08) |
| US‐RP | 79 | 2065.99 | MSB11022/EU‐RMP | 91.53 | (81.33, 103.02) | |
| EU‐RMP | 79 | 2167.38 | US‐RP/EU‐RMP | 95.32 | (84.72, 107.25) | |
| Cmax (μg ml−1) | MSB11022 | 78 | 3.44 | MSB11022/US‐RP | 97.22 | (89.27, 105.88) |
| US‐RP | 79 | 3.53 | MSB11022/EU‐RMP | 95.38 | (87.58, 103.87) | |
| EU‐RMP | 79 | 3.60 | US‐RP/EU‐RMP | 98.10 | (90.11, 106.81) |
AUC(0,∞), area under the concentration–time curve from time 0 extrapolated to infinity; AUC(0,t last), AUC from time 0 to the last quantifiable concentration; CI, confidence interval; C max, maximum observed concentration; EU‐RMP, Europe‐approved reference medicinal product; LS, least‐squares; US‐RP, US‐licensed reference product.
All other PK endpoints, including t max, t ½, CL/F and V z/F were also similar among the three treatment groups (Table 2). The median t max was 191 h for each treatment group. The geometric mean t ½ was 295.5 h for MSB11022 and approximately 350 h for the US‐RP and EU‐RMP. However, given the observed inter‐individual variability in t 1/2 these were considered to be similar (Table 2).
Immunogenicity
Antibodies to adalimumab were detected in 14.1% (n = 11), 20.5% (n = 16) and 12.7% (n = 10) of subjects for MSB11022, US‐RP and EU‐RMP up to and including day 15 and 82.1% (n = 64), 81.3% (n = 65) and 83.5% (n = 66) of subjects for MSB11022, US‐RP and EU‐RMP, respectively, up to and including day 71.
To assess the influence of immunogenicity on the PK of each allocated treatment, subanalyses based on the post‐dose ADA results were performed. Geometric mean AUC(0,∞), AUC(0,t last) and C max were comparable between treatments, regardless of ADA status, although mean exposure to all allocated treatments in ADA‐positive subjects appeared to be lower than in ADA‐negative subjects (Figure 3, Table S3).
Figure 3.
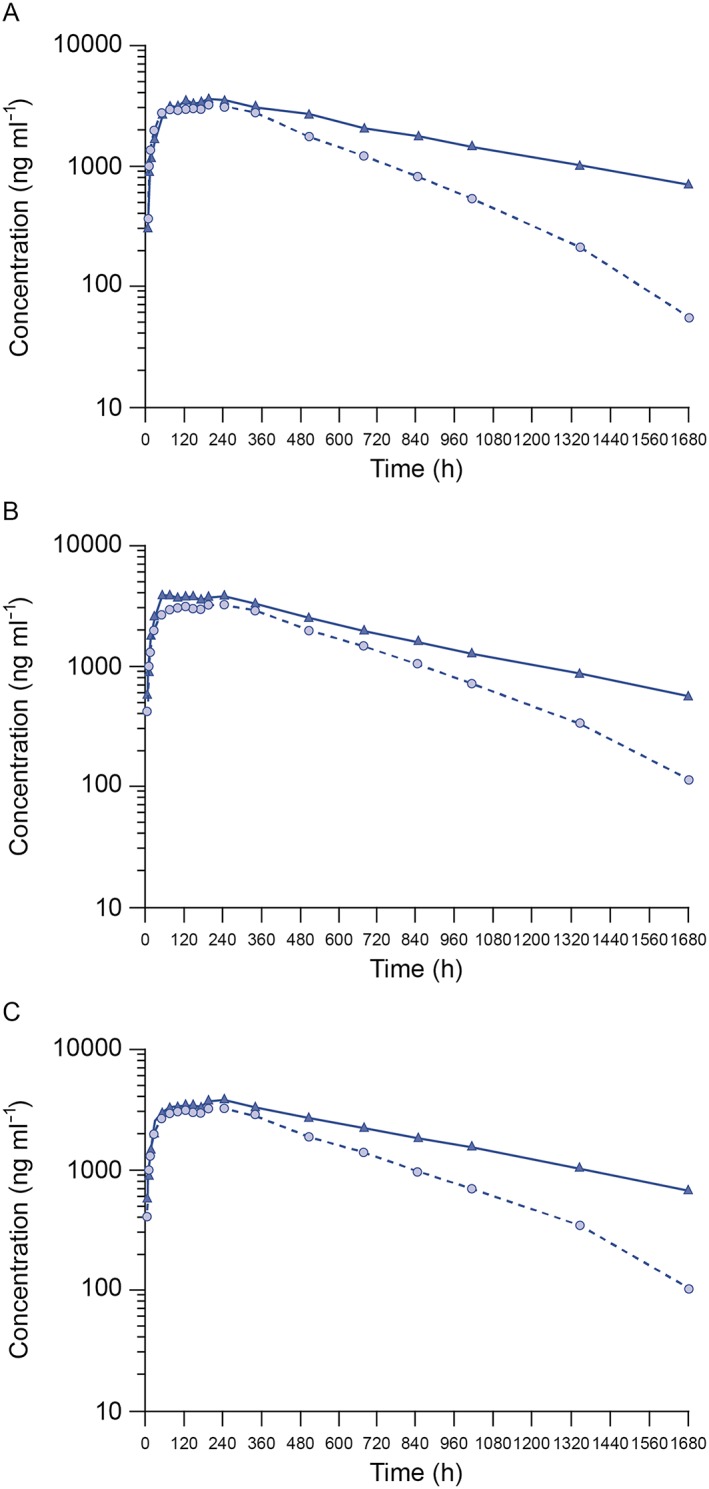
Mean serum concentration–time profiles (on semi‐logarithmic scale) following a single subcutaneous injection of 40 mg MSB11022, US‐RP, and EU‐RMP according to ADA status for (A) MSB11022, (B) US‐RP, and (C) EU‐RMP. Data are presented as means. ADA, anti‐drug antibody; EU‐RMP, EU‐reference medicinal product (adalimumab); US‐RP, US‐reference product (adalimumab).  ADA negative,
ADA negative,  ADA positive
ADA positive
Safety and tolerability
Overall, 263 TEAEs were reported in 143/237 (60.3%) subjects. The most common TEAEs encountered are outlined in Table 4. Forty‐nine subjects receiving MSB110022 reported at least one TEAE (62.8%, 95 TEAEs), as did 45 subjects receiving US‐RP (56.3%, 82 TEAEs) and 49 subjects receiving EU‐RMP (62.0%, 86 TEAEs). Most of the TEAEs were considered mild and unrelated to study drug. None of the TEAEs led to study withdrawal. In terms of TEAEs considered to be related to study drug, headache was the most frequently reported complaint, followed by injection site pain and oropharyngeal pain.
Table 4.
Summary of Treatment‐Emergent Adverse Events by System Organ Class and Preferred Term
| MSB11022 n = 78 | US‐RP n = 80 | EU‐RMP n = 79 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| System organ class preferred term | n | (%) | Events | n | (%) | Events | n | (%) | Events |
| Subjects with at least one TEAE | 49 | (62.8) | 95 | 45 | (56.3) | 82 | 49 | (62.0) | 86 |
| Nervous system disorders | 15 | (19.2) | 18 | 18 | (22.5) | 24 | 9 | (11.4) | 12 |
| Headache | 13 | (16.7) | 16 | 15 | (18.8) | 18 | 7 | (8.9) | 10 |
| Infections and infestations | 18 | (23.1) | 22 | 15 | (18.8) | 15 | 15 | (19.0) | 17 |
| Rhinitis | 6 | (7.7) | 6 | 8 | (10.0) | 8 | 3 | (3.8) | 4 |
| Nasopharyngitis | 3 | (3.8) | 4 | 4 | (5.0) | 4 | 4 | (5.1) | 5 |
| Upper respiratory tract infection | 2 | (2.6) | 2 | 2 | (2.5) | 2 | 3 | (3.8) | 3 |
| General disorders and administration site conditions | 12 | (15.4) | 14 | 8 | (10.0) | 9 | 10 | (12.7) | 12 |
| Injection site pain | 3 | (3.8) | 4 | 2 | (2.5) | 2 | 2 | (2.5) | 2 |
| Injection site erythema | 3 | (3.8) | 3 | 1 | (1.3) | 1 | 2 | (2.5) | 2 |
| Injection site pruritus | 1 | (1.3) | 1 | 1 | (1.3) | 1 | 1 | (1.3) | 1 |
| Injection site bruising | 1 | (1.3) | 1 | 1 | (1.3) | 1 | 0 | (0.0) | 0 |
| Injection site rash | 2 | (2.6) | 2 | 0 | (0.0) | 0 | 0 | (0.0) | 0 |
| Fatigue | 1 | (1.3) | 1 | 0 | (0.0) | 0 | 4 | (5.1) | 4 |
| Gastrointestinal disorders | 12 | (15.4) | 13 | 9 | (11.3) | 9 | 5 | (6.3) | 6 |
| Toothache | 1 | (1.3) | 1 | 1 | (1.3) | 1 | 2 | (2.5) | 2 |
| Abdominal discomfort | 3 | (3.8) | 3 | 1 | (1.3) | 1 | 0 | (0.0) | 0 |
| Musculoskeletal and connective tissue disorders | 7 | (9.0) | 9 | 5 | (6.3) | 6 | 9 | (11.4) | 10 |
| Arthralgia | 0 | (0.0) | 0 | 3 | (3.8) | 3 | 2 | (2.5) | 2 |
| Musculoskeletal pain | 0 | (0.0) | 0 | 1 | (1.3) | 1 | 3 | (3.8) | 3 |
| Respiratory, thoracic and mediastinal disorders | 9 | (11.5) | 9 | 7 | (8.8) | 8 | 7 | (8.9) | 8 |
| Oropharyngeal pain | 7 | (9.0) | 7 | 4 | (5.0) | 4 | 3 | (3.8) | 3 |
| Skin and subcutaneous tissue disorders | 3 | (3.8) | 3 | 7 | (8.8) | 7 | 8 | (10.1) | 10 |
| Injury, poisoning and procedural complications | 2 | (2.6) | 2 | 2 | (2.5) | 2 | 4 | (5.1) | 4 |
| Eye disorders | 0 | (0.0) | 0 | 1 | (1.3) | 1 | 3 | (3.8) | 3 |
EU‐RMP, Europe‐approved reference medicinal product; TEAE, treatment‐emergent adverse event; US‐RP, US‐licensed reference product.
Immunogenicity did not influence TEAE frequency. At least one TEAE was reported for 37/64 ADA‐positive subjects (57.8%, 75 events) in the MSB11022 group. A similar distribution of TEAEs was also reported for 36/65 ADA‐positive subjects (55.4%, 64 events) in the US‐RP group and for 41/66 ADA‐positive subjects (62.1%, 72 events) in the EU‐RMP group. Likewise, a similar number of TEAEs was also reported in ADA‐negative subjects. At least one TEAE was reported for 12/14 ADA‐negative subjects (85.7%, 20 events) in the MSB11022 group, 9/15 ADA‐negative subjects (60.0%, 18 events) in the US‐RP group and 8/13 ADA‐negative subjects (61.5%, 14 events) in the EU‐RMP group.
TEAEs of infection were comparable between groups (23.1%, 22 events in MSB11022, 18.8%, 15 events in US‐RP and 19.0%, 17 events in EU‐RMP arms, respectively). No serious infections were reported. Hypersensitivity reactions were reported for 2/78 subjects (2.6%) in the MSB11022 group, 2/80 subjects (2.5%) in the US‐RP group and 6/79 subjects (7.6%) in the EU‐RMP group. The majority of the events reported as hypersensitivity reactions were mild in severity and considered to be related to the study drug. Injection site reactions were reported in a small number of subjects (16/237, 6.7%); nine subjects on MSB11022, four subjects on US‐RP and three subjects on EU‐RMP. Of the 11 injection site reactions in the MSB11022 arm, 10 were considered to be mild in severity; in the US‐licensed Humira arm, all five injection site reactions were mild in severity and of the five injections site reactions reported in the EU‐approved Humira arm, four were considered to be mild in severity. No definite time pattern was observed between the time of injection administration and the occurrence of injection site reactions. The majority of the reactions resolved within 3 days.
No deaths or SAEs related to the study drug were reported. Two SAEs, both considered to be unrelated to the study drug, were reported with the MSB11022 group (head injury and impaired glucose tolerance test). The event of head injury occurred following an assault leading to hospitalization of the subject; the subject was subsequently released without any clinical complications. An abnormal glucose tolerance test was reported for a subject with a family history of diabetes.
No safety concerns based on laboratory measurements, vital signs or 12‐lead ECG were reported.
Discussion
This phase 1 double‐blind, three arm, parallel group, single dose study was designed to evaluate and compare the PK profiles of the investigational adalimumab biosimilar MBS11022 and the RMP/RPs (Humira®; AbbVie Ltd, Maidenhead, UK/AbbVie Inc., North Chicago, IL, USA). Secondary objectives were to confirm comparable safety/tolerability and immunogenicity of the two products.
The PK profile of adalimumab has been described previously in patients with RA 1. Adalimumab demonstrates linear PK characteristics throughout its clinical dose range. After subcutaneous administration of a single 40 mg dose, absorption and distribution of adalimumab are slow, with peak serum concentrations being reached ~5 days after administration. The mean t ½ is ~2 weeks 1, 2, 3. Adalimumab biosimilar compounds would be expected to mirror these characteristics.
All 237 randomized subjects in this trial received a single dose of study drug, and demographic characteristics were similar across the three groups. A dose level of 40 mg was selected as this is the recommended therapeutic dose of the EU‐RMP/US‐RP presentations of adalimumab for subjects weighing >30 kg 2, 3. At this dose, all PK parameters were found to be equivalent across the treatment groups (i.e. MSB11022 and adalimumab as the US‐RP and EU‐RMP). Most notably, equivalent drug exposures for all three treatment groups were shown by predefined equivalence intervals for AUC parameters and C max. The median t max observed across groups was consistent and broadly concordant with the average of ~5 days reported for the US‐RP and EU‐RMP 2, 3. Geometric mean systemic clearance of the treatment groups ranged from 0.016 to 0.018 l h−1 and were comparable with 0.012 l h−1 reported by the manufacturer of adalimumab in patients with RA, while volumes of distribution approximating to 8.0 l in these healthy volunteers were also comparable in RA patients (range 4.7–6.0 l) given the observed inter‐individual variability 2, 3. The geometric mean t ½ of 295 h for MSB11022 was slightly shorter than the mean t 1/2 observed for the US‐RP and EU‐RMP of ~350 h. However, given the observed inter‐individual variability in t 1/2 this can be considered comparable and it is also similar to the accepted mean t ½ of adalimumab (~2 weeks 336 h) reported in the product literature 2, 3 and that noted specifically in patients with RA (12 days 288 h) 1.
While it is reasonable to expect some variation relative to previously published PK data as a result of differences in drug recipient populations, experimental conditions and other factors such as assay procedures used, it is important to note that three way bioequivalence of MSB11022, US‐RP and EU‐RMP was demonstrated for all primary PK parameters across all study groups in this trial. For the AUC parameters, the geometric mean ratios were in the range 89–96% for MSB11022 when compared with both the US‐RP and EU‐RMP. However the 90% CIs of geometric least‐square mean ratios were all contained within the predefined equivalence interval of 80–125% for AUC(0,∞), AUC(0,t last) and C max. This showed the bioequivalence of MSB11022 to the two reference treatments after a single subcutaneous dose of 40 mg.
Stratifying PK parameters according to ADA status also demonstrated similarity between treatment arms for AUC(0,∞), AUC(0,t last) and C max. Mean exposures appeared to be lower in subjects testing positive for antibodies. This is thought to be related to increased clearance and the design of the PK assay, which does not allow the measurement of adalimumab when bound to ADA with neutralizing capability. Overall, there were no relevant differences in ADA positivity across the three treatment groups. Of note, the proportions of ADA‐positive subjects in all three treatment groups was higher (>80%) than historically reported with US‐RP or EU‐RMP 2, 3. This phenomenon has also been observed with other compounds and reflects the use of different and/or more sensitive methods compared with historical studies 12.
The safety analysis showed similar AE profiles across the three groups, with MSB11022 having a safety/tolerability profile consistent with the labelled safety profile of adalimumab as the US‐RP or EU‐RMP 2, 3. There were no safety concerns in any group and no new safety signals with MSB11022 when compared with the US‐RP and EU‐RMP formulations of adalimumab. The two SAEs noted in the MSB11022 group, impaired glucose tolerance test and head injury, were not related to study medication.
Because anti‐TNF therapy is associated with immune suppression 13, opportunistic infections are AEs of special interest for this drug class 14, 15. However, no serious infections were reported in the present study in any group. Moreover, hypersensitivity reactions were seen in only two subjects each in the MSB11022 and US‐RP groups, compared with six subjects in the EU‐RMP group and there were no concerns relating to clinical laboratory tests or vital signs.
These phase 1 data, obtained in healthy volunteers, suggest that MSB11022 fulfils the requirements for a biosimilar compound with reference to adalimumab as required by both US 7 and European 8 authorities in terms of PK and safety. Three way bioequivalence was demonstrated, with no new safety signals for the biosimilar.
In conclusion, this study supports the further clinical evaluation of MSB11022 as a proposed biosimilar with characteristics equivalent to the established compound adalimumab (Humira®) in the treatment of autoimmune conditions in which TNF is a significant inflammatory mediator.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare EH, MU and SR are employees of Merck and VW was an employee of Merck during the planning, conduct and analysis of this study, TM is an employee of Quintiles and is also supported by the National Institute for Health Research (NIHR) Biomedical Research Centre at Guy's and St Thomas' NHS Foundation Trust and King's College London, PV is an employee of Cytel Inc. and NA is an employee of PAREXEL.
This study was sponsored by Merck Biosimilars. Medical writing assistance was provided by Sandra Cusco PhD, Bioscript Science, Macclesfield, UK and funded by Merck Biosimilars, Aubonne, Switzerland.
The study was conducted at Quintiles Drug Research Unit at Guy's Hospital, London and Hammersmith Medicines Research, HMR Ltd, London, UK. Pharmacokinetic and immunogenicity analyses were conducted by PPD, Richmond, VA, USA.
Contributors
All authors had roles in the design and conduct of the study, collection, management, analysis and interpretation of the data and preparation, review and approval of the article.
Supporting information
Figure S1 Study design for a phase 1, randomized, double‐blind, parallel‐group, single dose trial. The subject, the investigator, as well as the sponsor, were blinded to the study drug administered. Time post‐dose and day on which samples were collected for safety, pharmacokinetics and immunogenicity (shaded time periods only) analyses are outlined. Following study drug administration, subjects remained resident at the trial site until discharge on day 8 (*later modified to day 5 following an interim safety analysis). Outpatient visits occurred on days 9, 11, 15, 22, 29, 36, 43 and 57. Follow‐up assessments were performed on day 71, the last visit of the trial
Table S1 ELISA inter‐assay precision and accuracy, validation
Table S2 ELISA inter‐assay precision and accuracy, post‐validation performance: sample analysis
Table S3 Pharmacokinetic parameters according to antidrug antibody status
Supporting info item
Hyland, E. , Mant, T. , Vlachos, P. , Attkins, N. , Ullmann, M. , Roy, S. , and Wagner, V. (2016) Comparison of the pharmacokinetics, safety, and immunogenicity of MSB11022, a biosimilar of adalimumab, with Humira® in healthy subjects. Br J Clin Pharmacol, 82: 983–993. doi: 10.1111/bcp.13039.
References
- 1. den Broeder A, van de Putte L, Rau R, Schattenkirchner M, Van Riel P, Sander O, et al. A single dose, placebo‐controlled study of the fully human anti‐tumor necrosis factor‐α antibody adalimumab (D2E7) in patients with rheumatoid arthritis. J Rheumatol 2002; 29: 2288–98. [PubMed] [Google Scholar]
- 2. Humira (adalimumab) injection, for subcutaneous use. North Chicago, IL: AbbVie Inc., September 2015. [online]. http://www.rxabbvie.com/pdf/humira.pdf (last accessed 11 December 2015).
- 3. SPC . Humira pre‐filled pen, pre‐filled syringe and vial. Maidenhead, UK: Abbvie Ltd, November 2015. [online]. Available at https://www.medicines.org.uk/emc/medicine/21201 (last accessed 11 December 2015).
- 4. Burmester GR, Panaccione R, Gordon KB, McIlraith MJ, Lacerda AP. Adalimumab: long‐term safety in 23,458 patients from global clinical trials in rheumatoid arthritis, juvenile idiopathic arthritis, ankylosing spondylitis, psoriatic arthritis, psoriasis and Crohn's disease. Ann Rheum Dis 2013; 72: 517–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Benucci M, Li Gobbi F, Meacci F, Manfredi M, Infantino M, Severino M, et al. Antidrug antibodies against TNF‐blocking agents: correlations between disease activity, hypersensitivity reactions, and different classes of immunoglobulins. Biologics 2015; 9: 7–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bartelds GM, Wijbrandts CA, Nurmohamed MT, Stapel S, Lems WF, Aarden L, et al. Clinical response to adalimumab: relationship to anti‐adalimumab antibodies and serum adalimumab concentrations in rheumatoid arthritis. Ann Rheum Dis 2007; 66: 921–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. FDA Guidance for Industry Scientific Considerations in Demonstrating Biosimilarity to a Reference Product. Silver Spring, MD: US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER), April 2015. [online]. Available at http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291128.pdf (last accessed 11 December 2015).
- 8. European Medicines Agency Guideline on similar biological medicinal products containing biotechnology‐derived proteins as active substance: quality issues. London, UK: European Medicines Agency, May 2012. [online]. Available at http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/05/WC500127960.pdf (last accessed 11 December 2015).
- 9. Magnenat L, Palmese A, Fremaux C, D'Amici F, Terlizzese M, Rossi M, et al Demonstration of physicochemical and functional similarity between the proposed biosimilar adalimumab MSB11022 and Humira® . mAbs 2016; Manuscript submitted. [DOI] [PMC free article] [PubMed]
- 10. van Schouwenburg PA, van de Stadt LA, de Jong RN, van Buren EE, Kruithof S, de Groot E, et al. Adalimumab elicits a restricted anti‐idiotypic antibody response in autoimmune patients resulting in functional neutralisation. Ann Rheum Dis 2013; 72: 104–9. [DOI] [PubMed] [Google Scholar]
- 11. van Schouwenburg PA, Kruithof S, Votsmeier C, van Schie K, Hart MH, de Jong RN, et al. Functional analysis of the anti‐adalimumab response using patient‐derived monoclonal antibodies. J Biol Chem 2014; 289: 34482–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Thomas SS. Comparative immunogenicity of TNF inhibitors: impact on clinical efficacy and tolerability in the management of autoimmune diseases. A systematic review and meta‐analysis. BioDrugs 2015; 29: 241–58. [DOI] [PubMed] [Google Scholar]
- 13. Tracey D, Klareskog L, Sasso EH, Salfeld JG, Tak PP. Tumor necrosis factor antagonist mechanisms of action: a comprehensive review. Pharmacol Ther 2008; 117: 244–79. [DOI] [PubMed] [Google Scholar]
- 14. Hochberg MC, Lebwohl MG, Plevy SE, Hobbs KF, Yocum DE. The benefit/risk profile of TNF‐blocking agents: findings of a consensus panel. Semin Arthritis Rheum 2005; 34: 819–36. [DOI] [PubMed] [Google Scholar]
- 15. Keystone EC. Does anti‐tumor necrosis factor‐α therapy affect risk of serious infection and cancer in patients with rheumatoid arthritis? A review of long‐term data. J Rheumatol 2011; 38: 1552–62. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Study design for a phase 1, randomized, double‐blind, parallel‐group, single dose trial. The subject, the investigator, as well as the sponsor, were blinded to the study drug administered. Time post‐dose and day on which samples were collected for safety, pharmacokinetics and immunogenicity (shaded time periods only) analyses are outlined. Following study drug administration, subjects remained resident at the trial site until discharge on day 8 (*later modified to day 5 following an interim safety analysis). Outpatient visits occurred on days 9, 11, 15, 22, 29, 36, 43 and 57. Follow‐up assessments were performed on day 71, the last visit of the trial
Table S1 ELISA inter‐assay precision and accuracy, validation
Table S2 ELISA inter‐assay precision and accuracy, post‐validation performance: sample analysis
Table S3 Pharmacokinetic parameters according to antidrug antibody status
Supporting info item