

Abstract
We describe a child with dyserythropoietic anemia; thrombocytosis; functional platelet defect and megakaryocyte dysplasia. We show that (i) this constellation of hematopoietic abnormalities was due to a germline mutation within the 5′UTR of GATA1; (ii) the mutation impaired a 5′UTR GATA1 splicing site, promoting production of the shortened GATA1s isoform lacking the N-terminus; (iii) expression of the GATA1 N-terminus is restricted to erythroblasts and megakaryocytes in normal marrow, consistent with the patient’s abnormal erythropoiesis and megakaryopoiesis. Our findings provide insights into the clinically relevant in vivo function of the N-terminal domain of GATA1 in human hematopoiesis.
Keywords: Dyserythropoietic anemia, megakaryocyte dysplasia, GATA1
Introduction
Germline mutations within the X-linked transcription factor GATA1 occur in a spectrum of childhood blood disorders (reviewed in [1]). Alternative splicing produces two GATA1 isoforms [2]: full-length GATA1 (flGATA1) and shortened GATA1 (GATA1s), which lacks the N-terminal domain.
Rare defects known as “GATA1s mutations” [3] reduce synthesis of full-length GATA1 while GATA1s is still produced. Acquired GATA1s mutations drive transient myeloproliferative disorder and AML in Down syndrome patients (reviewed in [4–6]). Germline GATA1s mutations in individuals without trisomy 21 have been associated with Diamond-Blackfan anemia (DBA) [7,8], a syndrome of RBC aplasia and otherwise normal hematopoiesis [9]. DBA-associated ribosomopathy impairs GATA1 synthesis [10], perhaps because the complex structure of the GATA1 5′UTR demands error-free ribosomes for efficient translation. Thus, ribosomopathy has been mechanistically linked to GATA1 and erythropoiesis failure in DBA. However, thrombocytopenia with structural platelet abnormalities and dyserythropoiesis [11] as well as childhood myelodysplastic syndrome (MDS) [12] were reported in other GATA1s families. Since GATA1s patients are rare and clinically heterogeneous, the clinical impact of inherited GATA1s mutations on human hematopoiesis remains to be fully elucidated.
Results
We describe a child with dyserythropoietic anemia, megakaryocyte dysplasia and platelet malfunction due to a disease-causing mutation within the 5′UTR of GATA1. We explored the impact of this novel mutation on the expression of GATA1 splice variants in human bone marrow. Our findings support the role for the N-terminus of GATA1 in in vivo megakaryocyte function and erythropoiesis.
A 4-year old male developed fatigue and pallor secondary to anemia (hemoglobin: 4.4 g/dL). His growth and development were normal with no congenital malformations. His past blood counts revealed progressive macrocytic anemia with reticulocytopenia and persistent fetal hemoglobin first noted at 3 months of age, as well as chronic thrombocytosis (platelets:387-947,000/mm3) and occasional neutropenia with no frequent infections (lowest absolute neutrophil count:495/mm3) (Figure 1A). The folate and vitamin B12 levels were normal. Normal DEB chromosome-breakage test and lymphocyte telomere length excluded Fanconi anemia and dyskeratosis congenita, respectively.
Figure 1. Hypoplastic anemia, megakaryocyte dysplasia and thrombocytosis due to mutation within the 5′UTR of GATA1.

(A) Chronic anemia and thrombocytosis over the course of 5 years. (B) Decreased erythropoiesis and megakaryocyte dysplasia seen on bone marrow aspirate. (C) Immunohistochemistry with CD71 (erythroblast marker) and CD61 (megakaryocyte marker) reveals decreased erythropoiesis and accumulation of megakaryocytes in the patient’s bone marrow compared to a healthy individual. Right panel shows dysplastic megakaryocytes (black arrows) in the patient’s marrow (Wright-Giemsa stain). (D) GATA1 sequencing reveals a novel mutation in the affected child. RT-PCR and Western blotting demonstrate decreased full-length GATA1 transcript (E) and protein (F) in the patient. (G) Schematic representation of GATA1 alternative splicing (only first three exons are shown for simplicity). The GATA1c-.21A>G mutation produces GATA1s phenotype by disrupting full-length GATA1 splicing.
DBA was initially suspected due to progressive macrocytic anemia beginning in infancy [9]. However, the patient’s bone marrow analysis revealed not only paucity of RBC precursors (Figure 1B–C) and dyserythropoiesis (Supplementary Figure 1), but also prominent megakaryocytosis (Figure 1B–C) with megakaryocyte dysplasia (Figure 1B; Supplementary Figure 2), which is not seen in classic DBA [9]. Accordingly, sequencing of the patient’s nine DBA-associated ribosome genes (RPL11, RPL35a, RPL5, RPS10, RPS17, RPS19, RPS24, RPS26, and RPS7) and deletion-duplication analysis of RPS19, RPL5, RPL11, RPL35A, RPS17 and RPS26 produced normal results. Normal cytogenetics, MDS-FISH and blast count excluded MDS. Thus, we asked whether his anemia mimicking DBA [7,8] but associated with megakaryocyte dysplasia [11] reflected a germline GATA1 defect. Indeed, Sanger sequencing revealed a novel mutation within the 5′UTR of GATA1 (c.-21A>G or c.-19-2A>G) at position 48,791,089 on the X-chromosome (GRCh38.p2 primary assembly), which affects the absolutely conserved A in the -2 position of the splice acceptor site (Figure 1D). Consistent with an X-linked recessive inheritance, mother was an asymptomatic carrier, and all healthy male siblings had wild-type GATA1 (Figure 1D).
We hypothesized the GATA1c-.21A>G transcript splicing is abnormal as our in silico analysis [13] suggested disruption of a 5′UTR consensus splice site. Accordingly, the GATA1c-.21A>G mutation decreased in vivo flGATA1 expression (Figure 1E–F), which is consistent with the GATA1s phenotype [7,8,11,12]. The patient’s anemia improved on corticosteroids similar to other GATA1s individuals [7,8,12]. Thus, the GATA1c.-21A>G mutation produced the GATA1s phenotype through destruction of a splice site within the 5′UTR of GATA1 (Figure 1G).
Past clinical reports have generated conflicting in vivo data regarding the impact of inherited GATA1s mutations on megakaryopoiesis in non-Down syndrome patients [7,8,11]. However, accumulation of dysplastic megakaryocytes in the GATA1c.-21A>G marrow with peripheral thrombocytosis strongly suggests that flGATA1 regulates human megakaryocyte proliferation and function in vivo in individuals without trisomy 21. Moreover, the GATA1c.-21A>G platelets displayed subclinical aggregation deficiencies (Supplementary Figure 3), further implicating flGATA1 in physiological megakaryopoiesis. Thus, we methodically examined GATA1 splicing in human marrow, hypothesizing that flGATA1 expression correlates with the role of flGATA1 in distinct lineages. To that end, we developed an immunohistochemistry assay specific for the GATA1 N-terminus (Figure 2A). Full-length GATA1 was absent from the GATA1c.-21A>G marrow, confirming that the mutation confers GATA1s phenotype (Figure 2B–C). Double-immunohistochemistry detected flGATA1 in healthy megakaryocytes and erythroblasts but not in other hematopoietic lineages (Figure 2C), consistent with the observation that loss of flGATA1 disrupted RBC and platelet production in our patient (Figure 1).
Figure 2. Full-length GATA1 expression is restricted to erythrocyte and megakaryocyte precursors during hematopoiesis.
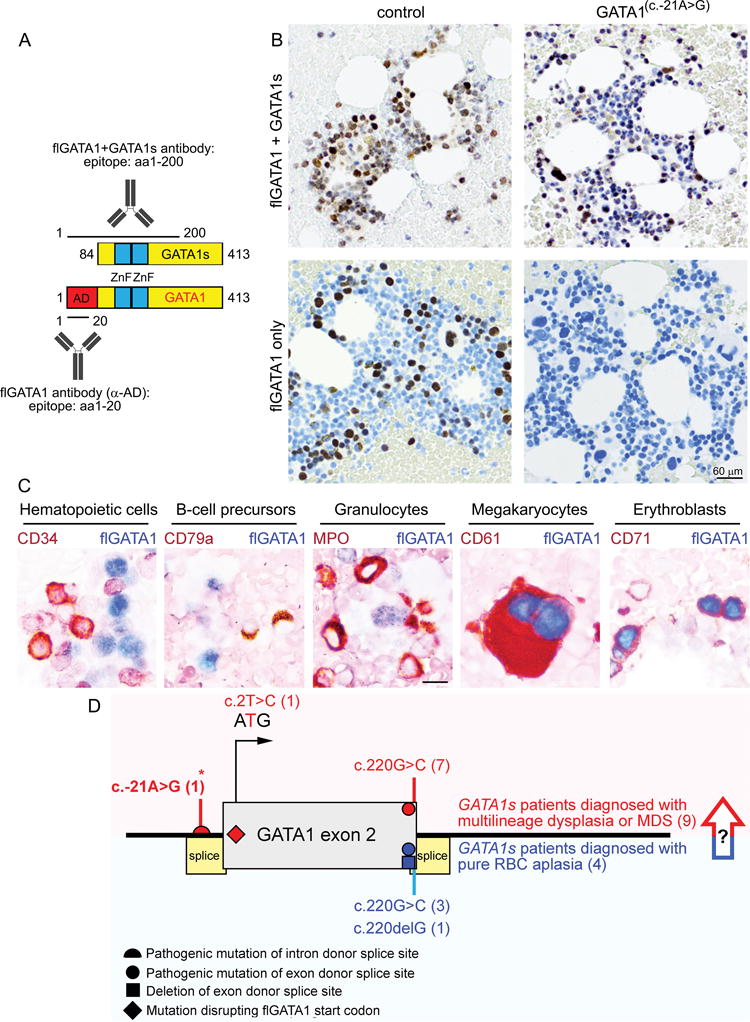
(A) Antibodies used for immunohistochemistry. Antibody against the N-terminus of GATA1 recognizes only full-length GATA1, while the C-terminal antibody recognizes both GATA1 isoforms (flGATA1 and GATA1s). (B) Loss of flGATA1 expression in the GATA1c.-21A>G patient’s bone marrow. Note that (i) flGATA1 is not expressed in all hematopoietic cells of a healthy individual, and (ii) GATA1s production is not affected by the GATA1c.-21A>G mutation. (C) Expression of N-terminal GATA1 domain (flGATA1; blue) is restricted to erythroblasts and megakaryocytes during human hematopoiesis. Appropriate hematopoietic lineage markers (brown) were co-stained as shown. (D) GATA1s mutations cause a range of phenotypes from Diamond-Blackfan anemia (red) to multilineage hematopoietic dysplasia (blue). Novel mutation described in this work is marked with asterisk. The arrow indicates that the long-term risk of MDS in GATA1s patients presenting with pure RBC aplasia remains to be determined as one GATA1s patient initially diagnosed with DBA developed MDS later in childhood [12].
Discussion
GATA1 orchestrates the production of RBCs and platelets [14–16]. Alternative splicing provides an incompletely understood pathway to fine-tune this transcription factor’s activity during hematopoiesis. Full-length GATA1 differs from the short isoform (GATA1s) by the presence of the 83 amino acid-long N-terminus, which activates GATA1-driven erythropoiesis [2,17] and recruits flGATA1 to a subset of megakaryocyte and erythroblast genes [3]. Thus, GATA1 isoforms control partially overlapping but not identical transcriptional modules of erythroblast and megakaryocyte maturation.
Acquired GATA1s mutations are key drivers of transient myeloproliferation and AML in Down syndrome [4–6]. Inherited GATA1s mutations that decrease production of flGATA1 through disrupting exon 2 splice sites [7,8,11] or the initiation codon [12] have been reported in rare non-Down syndrome patients (Figure 2). GATA1s patients uniformly develop hyporegenerative anemia, confirming that the N-terminus of GATA1 is indispensable for erythropoiesis. However, since the reported clinical symptoms in GATA1s individuals range from DBA [7,8] to erythroblast and megakaryocyte dysplasia [11] and DBA progressing to MDS [12], the role of flGATA1 in other hematopoietic cell lines needs clarification.
Striking accumulation of dysplastic megakaryocytes in the GATA1c.-21A>G patient’s marrow (Figure 1) suggests that loss of flGATA1 unleashes megakaryocyte proliferation in vivo, possibly due to de-repression of the E2F cell-cycle regulator [18]. In further support of this notion, independent ex vivo studies showed that hematopoiesis in GATA1s iPS cells is skewed towards generation of abnormal megakaryocytes at the cost of erythropoiesis [3]. Indeed, expression of the GATA1 N-terminus is restricted to erythroblasts and megakaryocytes (Figure 2), supporting a physiological role of flGATA1 in these hematopoietic lineages. This assay may provide a future screening tool for GATA1s phenotype. Intriguingly, both thrombocytopenia [11] and thrombocytosis (this work) occur in GATA1s individuals, including GATA1s patients diagnosed with DBA [8]. A systematic evaluation of megakaryopoiesis in additional GATA1s patients will determine whether megakaryocyte dysplasia is universally seen in GATA1s individuals. Potential genotype-phenotype correlations will remain unknown until more GATA1s patients are identified, especially since the same GATA1c.220G>C mutation caused DBA-like phenotype in one family [8] and multilineage dysplasia in another [11].
The GATA1c.-21A>G platelets are defective (Supplementary Figure 3). This is in agreement with ultrastructural platelet abnormalities described in GATA1c.220G>C family [11], likely secondary to disrupted cytoskeletal remodeling in flGATA1-deficient megakaryocytes [19]. Future studies will determine whether GATA1s patients develop clinically significant bleeding later in life.
Prominent megakaryocyte abnormalities (this work; [11]) may provide subtle clinical clues to differentiate GATA1s mutations from “classical” DBA caused by ribosomopathy, which is defined as pure red blood cell aplasia [9]. GATA1 sequencing should be considered in males with congenital multi-lineage dysplasia and/or DBA-like clinical presentation. Interestingly, MDS-associated spliceosome mutations globally alter expression of multiple hematopoietic regulators, including GATA1 [20]. Given the potential risk of MDS in GATA1s patients [12], hematopoiesis should be closely monitored in individuals with inborn GATA1s mutations.
Supplementary Material
Supplemental Text 1. This file contains supplemental materials, methods and references, as well as three supplemental figures.
Acknowledgments
We are grateful to the patient and family who generously provided specimens used in this study. GN is supported by the NIH K12 Indiana Pediatric Scientist Award; Barth Syndrome/Bone Marrow Failure Research Fund at Riley Children’s Foundation; and the Heroes Foundation. JZ is supported by an NIH T32 Clinical Pharmacology Fellowship. DNA chromatogram was kindly provided by Dr. Michael Chicka, PhD (Prevention Genetics). We apologize to the investigators whose work has not been cited due to space limitations.
Abbreviations key
- CD
Cluster of differentiation
- DEB
Diepoxybutane
- DBA
Diamond-Blackfan anemia
- FISH
Fluorescent in situ hybridization
- flGATA1
Full-length GATA1
- GATA1
Globin transcription factor 1
- GATA1s
Shortened GATA1
- iPS cells
Induced pluripotent stem cells
- IRB
Institutional Review Board
- MDS
Myelodysplastic syndrome
- NIH
National Institutes of Health
- RBC
Red blood cell(s)
- RNA
Ribonucleic acid
- RPL
Ribosomal protein L
- RPS
Ribosomal protein S
- RT-PCR
Reverse transcription-polymerase chain reaction
- 5′UTR
5′ untranslated region
Footnotes
Authorship
G.N. and J.Z. directed patient’s diagnostics and clinical management; J.Z. designed and performed Western blotting and RT-PCR experiments; C.T. and M.C. designed, performed and analyzed immunohistochemistry experiments; G.N. acquired microscopy images and analyzed data; G.N. wrote and all authors edited the paper.
Conflict of interest disclosure
The authors declare no competing financial interests.
References
- 1.Crispino JD, Weiss MJ. Erythro-megakaryocytic transcription factors associated with hereditary anemia. Blood. 2014;123(20):3080–3088. doi: 10.1182/blood-2014-01-453167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Calligaris R, Bottardi S, Cogoi S, Apezteguia I, Santoro C. Alternative translation initiation site usage results in two functionally distinct forms of the GATA-1 transcription factor. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(25):11598–11602. doi: 10.1073/pnas.92.25.11598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Byrska-Bishop M, VanDorn D, Campbell AE, Betensky M, Arca PR, Yao Y, Gadue P, Costa FF, Nemiroff RL, Blobel GA, French DL, Hardison RC, Weiss MJ, Chou ST. Pluripotent stem cells reveal erythroid-specific activities of the GATA1 N-terminus. The Journal of clinical investigation. 2015;125(3):993–1005. doi: 10.1172/JCI75714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roberts I, Izraeli S. Haematopoietic development and leukaemia in Down syndrome. British journal of haematology. 2014;167(5):587–599. doi: 10.1111/bjh.13096. [DOI] [PubMed] [Google Scholar]
- 5.Malinge S, Izraeli S, Crispino JD. Insights into the manifestations, outcomes, and mechanisms of leukemogenesis in Down syndrome. Blood. 2009;113(12):2619–2628. doi: 10.1182/blood-2008-11-163501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shimizu R, Engel JD, Yamamoto M. GATA1-related leukaemias. Nature reviews Cancer. 2008;8(4):279–287. doi: 10.1038/nrc2348. [DOI] [PubMed] [Google Scholar]
- 7.Klar J, Khalfallah A, Arzoo PS, Gazda HT, Dahl N. Recurrent GATA1 mutations in Diamond-Blackfan anaemia. British journal of haematology. 2014;166(6):949–951. doi: 10.1111/bjh.12919. [DOI] [PubMed] [Google Scholar]
- 8.Sankaran VG, Ghazvinian R, Do R, Thiru P, Vergilio JA, Beggs AH, Sieff CA, Orkin SH, Nathan DG, Lander ES, Gazda HT. Exome sequencing identifies GATA1 mutations resulting in Diamond-Blackfan anemia. The Journal of clinical investigation. 2012;122(7):2439–2443. doi: 10.1172/JCI63597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vlachos A, Ball S, Dahl N, Alter BP, Sheth S, Ramenghi U, Meerpohl J, Karlsson S, Liu JM, Leblanc T, Paley C, Kang EM, Leder EJ, Atsidaftos E, Shimamura A, Bessler M, Glader B, Lipton JM, Participants of Sixth Annual Daniella Maria Arturi International Consensus C Diagnosing and treating Diamond Blackfan anaemia: results of an international clinical consensus conference. British journal of haematology. 2008;142(6):859–876. doi: 10.1111/j.1365-2141.2008.07269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ludwig LS, Gazda HT, Eng JC, Eichhorn SW, Thiru P, Ghazvinian R, George TI, Gotlib JR, Beggs AH, Sieff CA, Lodish HF, Lander ES, Sankaran VG. Altered translation of GATA1 in Diamond-Blackfan anemia. Nature medicine. 2014;20(7):748–753. doi: 10.1038/nm.3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hollanda LM, Lima CS, Cunha AF, Albuquerque DM, Vassallo J, Ozelo MC, Joazeiro PP, Saad ST, Costa FF. An inherited mutation leading to production of only the short isoform of GATA-1 is associated with impaired erythropoiesis. Nature genetics. 2006;38(7):807–812. doi: 10.1038/ng1825. [DOI] [PubMed] [Google Scholar]
- 12.Parrella S, Aspesi A, Quarello P, Garelli E, Pavesi E, Carando A, Nardi M, Ellis SR, Ramenghi U, Dianzani I. Loss of GATA-1 full length as a cause of Diamond-Blackfan anemia phenotype. Pediatric blood & cancer. 2014;61(7):1319–1321. doi: 10.1002/pbc.24944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reese MG, Eeckman FH, Kulp D, Haussler D. Improved splice site detection in Genie. Journal of computational biology: a journal of computational molecular cell biology. 1997;4(3):311–323. doi: 10.1089/cmb.1997.4.311. [DOI] [PubMed] [Google Scholar]
- 14.Romeo PH, Prandini MH, Joulin V, Mignotte V, Prenant M, Vainchenker W, Marguerie G, Uzan G. Megakaryocytic and erythrocytic lineages share specific transcription factors. Nature. 1990;344(6265):447–449. doi: 10.1038/344447a0. [DOI] [PubMed] [Google Scholar]
- 15.Martin DI, Zon LI, Mutter G, Orkin SH. Expression of an erythroid transcription factor in megakaryocytic and mast cell lineages. Nature. 1990;344(6265):444–447. doi: 10.1038/344444a0. [DOI] [PubMed] [Google Scholar]
- 16.Elagib KE, Racke FK, Mogass M, Khetawat R, Delehanty LL, Goldfarb AN. RUNX1 and GATA-1 coexpression and cooperation in megakaryocytic differentiation. Blood. 2003;101(11):4333–4341. doi: 10.1182/blood-2002-09-2708. [DOI] [PubMed] [Google Scholar]
- 17.Chlon TM, McNulty M, Goldenson B, Rosinski A, Crispino JD. Global transcriptome and chromatin occupancy analysis reveal the short isoform of GATA1 is deficient for erythroid specification and gene expression. Haematologica. 2015;100(5):575–584. doi: 10.3324/haematol.2014.112714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klusmann JH, Godinho FJ, Heitmann K, Maroz A, Koch ML, Reinhardt D, Orkin SH, Li Z. Developmental stage-specific interplay of GATA1 and IGF signaling in fetal megakaryopoiesis and leukemogenesis. Genes & development. 2010;24(15):1659–1672. doi: 10.1101/gad.1903410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elagib KE, Rubinstein JD, Delehanty LL, Ngoh VS, Greer PA, Li S, Lee JK, Li Z, Orkin SH, Mihaylov IS, Goldfarb AN. Calpain 2 activation of P-TEFb drives megakaryocyte morphogenesis and is disrupted by leukemogenic GATA1 mutation. Developmental cell. 2013;27(6):607–620. doi: 10.1016/j.devcel.2013.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim E, Ilagan JO, Liang Y, Daubner GM, Lee SC, Ramakrishnan A, Li Y, Chung YR, Micol JB, Murphy ME, Cho H, Kim MK, Zebari AS, Aumann S, Park CY, Buonamici S, Smith PG, Deeg HJ, Lobry C, Aifantis I, Modis Y, Allain FH, Halene S, Bradley RK, Abdel-Wahab O. SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer cell. 2015;27(5):617–630. doi: 10.1016/j.ccell.2015.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Text 1. This file contains supplemental materials, methods and references, as well as three supplemental figures.