

Abstract
Entorhinal cortex lesioning (ECL) causes an extensive deafferentation of the hippocampus that is classically followed by a compensatory reinnervation, where apolipoprotein E, the main extracellular lipid-carrier in the CNS, has been shown to play a crucial role by shuttling cholesterol to reconstructing neurons terminals. Hence, we investigated whether the ATP-binding cassette (ABC) transporters -A1 and -G1, known to regulate cellular cholesterol efflux and lipidation of the apolipoprotein E-containing lipoprotein complex are actively involved in this context of brain's plastic response to neurodegeneration and deafferentation. We assessed ABCA1 and ABCG1 mRNA and protein levels throughout the degenerative phase and the reinnervation process and evaluated the associated cholinergic sprouting following ECL in the adult mouse brain. We subsequently tested the effect of the pharmacological activation of the nuclear receptor LXR, prior to versus after ECL, on hippocampal ABCA1 and G1 expression and on reinnervation. ECL induced a time-dependent up-regulation of ABCA1, but not G1, that coincided with a significant increase in acetylcholine esterase (AChE) activity in the ipsilateral hippocampus. Pre-ECL, but not post-ECL i.p. treatment with the LXR agonist TO901317 also led to a significant increase solely in hippocampal ABCA1 expression, paralleled by increases in both AchE and synaptophysin protein levels in the deafferented hippocampus. Thus, ABCA1 and -G1
Keywords: ATP-binding cassette transporter, Apolipoprotein E, Cholesterol, Reinnervation, LXR, TO901317
1. Introduction
The brain is the most cholesterol-rich organ of the body as it contains ~25% of total cholesterol in 2% body weight (Dietschy and Turley, 2001). Regulation of cholesterol homeostasis in the central nervous system is maintained independently from the periphery and relies heavily on de novo cholesterol synthesis and efficient lipid transport and recycling mechanisms (Dietschy, 2009; Poirier et al., 1993). Glia and neurons provide the sterol necessary for brain development and once achieved, it is postulated that mature neurons reduce their cholesterol production in favor of lipids synthesized by astrocytes and transported by lipoproteins (Mauch et al., 2001). This derivation of glial cholesterol and other lipids to neurons is particularly necessary to support the high lipid demands imposed by synthesis of new membranes during reactive sprouting of terminals and synaptic remodeling following brain damage and neurodegenerative disease (Fig. 1) (De Chaves et al., 1997; Mauch et al., 2001; Poirier, 1994; Posse De Chaves et al., 2000). Transport of cholesterol and lipids between cells of the CNS is facilitated primarily through the formation of lipoparticles of similar size and constitution as peripheral high-density lipoproteins (HDL) (Illingworth and Glover, 1971; LaDu et al., 2001). Predominantly synthesized by astrocytes, apolipoprotein E (apoE) functions as the primary protein moiety of HDL within the CNS and has been shown to mediate the binding and internalization of glial-derived HDL by low-density lipoprotein receptor family members on regenerating nerve terminals (Pfrieger, 2003; Poirier et al., 1993). Although there is evidence to suggest that glia-derived apoE-containing lipoparticles are essential for brain development and reinnervation following injury, the mechanisms through which the assembly of HDL in the CNS is regulated are not clearly understood.
Fig. 1.

Schematic representation of hypothesized cholesterol/phospholipid recycling mechanisms in the injured CNS. Degenerating terminals are initially internalized and degraded by surrounding astrocytes. Inside astrocytes, free cholesterol (FC) is used for the assembly of an apoE/cholesterol/lipoprotein complex via the ABCA1 (1) or converted into cholesterol esters (CE) for storage purposes. The newly formed apoE/cholesterol/lipoprotein complexes are then directed (a) toward the circulation presumably through the ependymal cells surrounding the ventricles and/or (b) to specific brain cells requiring lipids. ApoE complexes are apparently internalized by the neuronal LDLR pathway (2) and cholesterol is released for dendritic proliferation and/or synaptogenesis (4). Within neurons, this free cholesterol can also be stored as CE and serves as a lipid pool for eventual needs or be hydroxylated and excreted for elimination (7). As a consequence of the internalization process, cholesterol synthesis in neurons (via the HMGCoAR Pathway) becomes progressively repressed (5). ABCA1/G1: ATP-binding cassette transporter A1/G1; APOER2: ApoE Receptor 2; BBB: Blood–Brain Barrier; E: ApoE; FC: free cholesterol; HDL: high-density lipoprotein; PL: Phospholipids; J: ApoJ; LDLR: low-density lipoprotein receptor; LRP: LDLR-related protein; and VLDLR: very low-density lipoprotein receptor.
Work in the periphery and in vitro using different cell types initially identified ATP-binding cassette (ABC) transporters family members ABCA1 and G1 as regulators of cholesterol and lipoprotein metabolism as they regulate cholesterol efflux. ABCG1 has been associated with intracellular cholesterol trafficking and efflux (Abildayeva et al., 2006; Burgess et al., 2008a, 2008b; Karten et al., 2006; Klucken et al., 2000; Vaughan and Oram, 2005), whereas ABCA1 has been linked to secretion and lipidation of apoA1 with cholesterol and HDL production in the plasma, CSF and brain (Hirsch-Reinshagen et al., 2004; Koldamova et al., 2003; Wahrle et al., 2004). Furthermore, in vitro work demonstrated that the coordinated action of ABCA1 and G1 is required for optimal removal of cellular cholesterol (Gelissen et al., 2006; Vaughan and Oram, 2006). In vivo work by Kennedy et al. (2005) showed that ABCG1 deficiency in mice compromises cholesterol efflux to surface-bound HDLs whereas ABCA1 deficiency markedly impairs cholesterol efflux to apoA1 particles.
A newly revised model for cholesterol efflux and formation of mature HDL has been proposed to explain the synergistic regulation of ABCA1 and ABCG1 during cholesterol mobilization. Briefly, ABCA1 is believed to catalyze the initial cholesterol loading of lipid-free apolipoproteins after which, ABCG1 moves in and completes lipidation and mediates cholesterol, phospholipid and sphingosine-1 phosphate export ((Vaughan and Oram, 2006), see also review (Hirsch-Reinshagen and Wellington, 2007)). Two independent teams (Hirsch-Reinshagen et al., 2004; Wahrle et al., 2004) demonstrated that very similar mechanism is regulating cholesterol and lipid mobilization in the brain. Glial ABCA1 was shown to facilitate cholesterol efflux onto apoE while lack of ABCA1 in ABCA1-deficient mice decreased cholesterol efflux and apoE levels in the hippocampus and striatum by 75–80% (Hirsch-Reinshagen et al., 2004). ABCA1 deficient mice also presented decreased cerebrospinal fluid cholesterol levels and smaller apoE lipoparticles (Wahrle et al., 2004). Finally, structural and functional deficits in neurons are also observed in mice lacking ABCA1 in the CNS (Karasinska et al., 2009). In situ hybridization studies performed in the embryo and adult showed that both ABCA1 and G1 are expressed throughout development and maturity in the brain and that they are both enriched in white and gray matters (Tachikawa et al., 2005). Notably, it was shown that ABCA1 was highly expressed in the adult hippocampus and cholinergic basal forebrain among other subcortical midbrain structures (Koldamova et al., 2005).
ABCA1, ABCG1, apoE and the LDLRs are all target genes regulated by the Liver X receptors (LXRs: schematized in Fig. 1) (Ishimoto et al., 2006; Kennedy et al., 2001; Nelissen et al., 2012; Sparrow et al., 2002; Vanmierlo et al., 2011; Wójcicka et al., 2007). LXRs are nuclear transcription factors found in glial cells and neurons in the brain (Gabbi et al., 2009; Riddell et al., 2007; Wójcicka et al., 2007). Most genes targeted by the LXR activation are involved in cholesterol transport and homeostasis in the brain. Natural agonists of LXRs are derivatives of cholesterol and when activated, LXRs promote reverse cholesterol transport, a pathway that exports cholesterol from the CNS to the liver for excretion (Koldamova et al., 2010; Wójcicka et al., 2007). As such, LXRs are considered cholesterol sensors (for review (Wójcicka et al., 2007)) and viewed as another key regulator of brain cholesterol homeostasis.
Therefore, we tested the hypothesis that ABCA1 and G1 transporters are actively recruited during the reinnervation phase that follows brain injury and neurodegeneration, in an attempt to facilitate the dynamic regulation of cholesterol recycling which is required during terminal proliferation and synaptic remodeling. Secondly, we postulated that the pharmacological activation of the nuclear receptor LXR by specific agonists could govern the ABC's local gene expression so that it enhances compensatory reinnervation. We used the well-established entorhinal cortex lesion (ECL) paradigm in adult mice and examined (a) the endogenous hippocampal expression of ABCA1 and ABCG1 time course during reactive sprouting and compensatory synaptogenesis following ECL and (b) the quantitative effects of the experimental administration of a potent LXR agonist on hippocampal ABCA1 and G1 expression and on the reinnervation process extent.
Here, we show that as could be expected ABCA1 is involved during the early phase of reinnervation, a period characterized by the intense mobilization of lipids such as cholesterol and phospholipids and their transport via apoE-HDL toward neurons undergoing active remodeling but also that ABCA1 and G1 are differentially regulated following ECL and in response to LXR agonist treatment.
2. Results
2.1. Time course of cholinergic sprouting
The well-described unilateral entorhinal cortex lesion (ECL) model is characterized by a marked deafferentation of the outer molecular layer of the dentate gyrus followed by intensive terminal sprouting and synaptic replacement. In response to entorhinal cortex injury, nearly 60% of the synaptic inputs to the granule cell layer of the DG degenerate. This synaptic loss is transient. Within few weeks, more than 80% of entorhinal synapses are replaced by fiber connections that originate from cholinergic septal neurons, glutamatergic commissural–associational cells of the CA3/hilar areas and to a lesser extent (~5%), from the contralateral entorhinal cortex (Matthews et al., 1976; Phinney et al., 2004; Poirier et al., 1993; Steward et al., 1988).
Fig. 2A illustrates the pattern of AChE-staining observed in the ipsilateral and contralateral hippocampus of animals following ECL and shows that AChE-staining density increased over time in the ipsilateral side (see arrows). Analysis of the relative AChE optical density (OD) of the staining, which correlates with the number of newly formed cholinergic synapses, reveals a significant increase at 14 DPL and a peak at 21 DPL (both p<0.05; Fig. 2B).
Fig. 2.
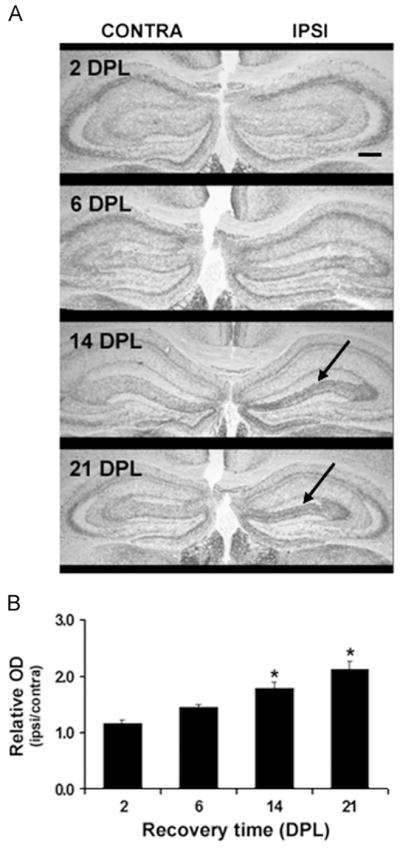
Pattern of acetylcholinesterase (AChE) activity histochemical staining of hippocampal cholinergic terminals in response to entorhinal cortex lesioning. (A) Representative photomicrographs of the AChE staining density in the dorsal region of the hippocampal formation, ispilateral (IPSI) and contralateral (CONTRA) to the lesion site at 2, 6, 14 and 21 days post-lesion (DPL). As indicated by the arrows, a significant increase in AChE staining in the outer portion of the molecular layer of the dentate gyrus is observed at 14 and 21 DPL; Coinciding with the replacement of entorhinal cortex projections by septal-hippocampal ones. The black line represents a scale bar of 20 μm in a 2.5 × magnitude photomicrographs. (B) Quantification of AChE staining density corrected for laminar shrinkage; Values are expressed as relative optical density (ipsilateral: contralateral OD) measures of AChE staining with respect to dentate molecular width. Bars correspond to the mean of 5 animals/group±SEM; *p<0.05 as compared to 2 DPL.
2.2. ABCA1 and ABCG1 mRNA expressions following ECL
Analysis of the relative ipsilateral:contralateral gene expression ratio of each ABC transporter in the HPC and FCx yielded a marked, significant 3-fold increase in ABCA1 mRNA ratio in the hippocampus of lesioned mice compared to sham-operated mice ratio at 14 DPL (p<0.001), with transcript levels returning to control levels by 21 DPL (Fig. 3A). ABCA1 up-regulation is specific to the deafferented hippocampus as no differences of mRNA ratios were detected in the frontal cortex of lesioned mice which serves as negative control in this study (Fig. 3B). In contrast, ABCG1 mRNA prevalence remains relatively unchanged in the deafferented hippocampus (Fig. 4A) as well as in the frontal cortex (Fig. 4B) of the ECL-mice.
Fig. 3.

Time course of ABCA1 mRNA expression in the hippocampus and frontal cortex of SHAM-operated and lesioned mice following entorhinal cortex lesion (ECL). The ABCA1 mRNA levels normalized to β-actin mRNA levels, are expressed as ipsilateral:contralateral ratios (mean of 5 mice/group±SEM). (A) In the hippocampus, a significant increase in the relative ABCA1 mRNA levels is observed at 14 days post-lesion (DPL) when compared to SHAM-operated mice levels (***p<0.001). (B) In the frontal cortex, relative ABCA1 mRNA expression is not observed to significantly deviate at any of the time points studied following ECL when compared to SHAM-operated relative expression levels.
Fig. 4.

Time course of ABCG1 mRNA expression in the hippocampus and frontal cortex of SHAM-operated and lesioned mice following entorhinal cortex lesion (ECL). The ABCG1 mRNA levels normalized to β-actin mRNA levels, are expressed as ipsilateral:contralateral ratios (mean of 5 mice/group±SEM). The relative expression of ABCG1 mRNA in the hippocampus (A) and frontal cortex (B) of lesioned mice are not significantly different from those observed in SHAM-operated mice at any of the observed time points (DPL: days post-lesion).
2.3. Hippocampal ABCA1 protein levels following ECL
ABCA1 protein levels were analyzed by ELISA in the ipsilateral and contralateral HPC and normalization of the data relative to the contralateral side reveals a marked ~2.8 fold increase at 14 DPL in the hippocampus of lesioned mice when compared to SHAM-operated animals (p<0.05, Fig. 5). Specificity of the signal was confirmed by Western blotting analyses where a similar pattern of expression was observed for the protein at the expected ~220 kD band (data not shown). As for the ABCA1 mRNA ratio in this region, the protein levels ratio of this transporter was transiently elevated and was back to control levels by 40 DPL (Fig. 5).
Fig. 5.

Time course analysis of ABCA1 protein levels in the hippocampus of SHAM-operated and entorhinal cortex lesioned mice. ABCA1 protein levels were determined by ELISA assays in the hippocampus ipsilateral and contralateral to the lesion and normalized to the total protein amount. Indicated are the mean ipsilateral: contralateral ratios of ABCA1 protein levels of 5 mice/group (bars)±SEM at different time points following the surgery (DPL: days post-lesion). The mean ABCA1 protein levels ratios show a significant increase that peaks at 14 DPL (*p = 0.05) and decrease subsequently compared to SHAM-operated animals.
2.4. Effect of LXR agonist treatment on ABCA1 and ABCG1 mRNA expression in ECL-mice
The LXR agonist treatment (TO901317) induces an increase of relative ABCA1 expression in the hippocampus during the remodeling phase at 25 DPL when given 7 days prior to the lesion (pre-ECL, Fig. 6A); post-hoc analyses revealed that the up-regulation (1.7 fold) was significant in the contralateral side as compared to the corresponding vehicle group (p = 0.02). In contrast, administration of the LXR agonist 7 days after the lesion (post-ECL) tends to decrease the gene expression of ABCA1 in the deafferented hippocampus (Fig. 6A). The relative mRNA levels were significantly lower in the ipsilateral and contralateral sides of post-ECL treated mice (p = 0.04 /p = 0.001) when compared to the respective pre-ECL treated groups.
Fig. 6.

Quantification of ABCA1 and ABCG1 gene expressions in the hippocampus of entorhinal cortex-lesioned mice following LXR agonist treatments: Prevention versus Rescue. Target mRNA expression levels relative to actin were determined by real time-PCR in the ipsilateral (ipsi) and contralateral (contra) hippocampus (HPC) of entorhinal cortex-lesioned (ECL) mice, treated with an LXR agonist prior to (pre-ECL) or after (post-ECL) the lesion. Bars indicate the mean of 5 mice/group±SEM at 25 days post-lesion (DPL). (A) ABCA1 expression is significantly increased in the contra-lateral (*p = 0.02) HPC of the LXR pre-lesion treatment group (prevention) compared to the corresponding vehicle group when assessed during the active synaptic remodeling process at 25 DPL. In contrast, administration of the LXR agonist after the lesion (rescue) decreased ABCA1 expression (ipsi: *p = 0.004; contra: ***p = 0.001) compared to the corresponding pre-ECL administration group during the remodeling process. (B) ABCG1 expression is not modified in the HPC of any group at 25 DPL. (C) Synaptophysin protein levels ratios in the HPC of LXR pre-ECL and post-ECL treated mice at 25 DPL. Synaptophysin protein levels were assessed by Western blot in duplicate analysis of pools of 5 hippocampi in the ipsi- and contra-lateral HPC and the ratio ipsilateral:contralateral was calculated for both pre- and post-ECL groups. This assessment served as an indicator of synaptic plasticity in order to determine whether the LXR treatment effects on ABC transporters expression coincide with reinnervation following the ECL as measured by AChE staining. An increase in synaptophysin protein levels ratio is observed in the pre-ECL but not post-ECL treated mice compared to vehicle treated mice while AChE staining in the outer molecular layer of the dentate gyrus was found to be significantly increased also in the pre-ECL (prevention) as opposed to the post-ECL (rescue) agonist treatment group (*p<0.02).
As for the untreated lesioned mice, the relative ABCG1 expression was not altered by the LXR agonist treatment, when given prior to or, subsequent to lesioning (Fig. 6B).
2.5. Effect of LXR agonist treatment on synaptic density following an ECL
Analysis of the synaptic density following an LXR agonist treatment in ECL-mice using synaptophysin levels in the hippocampus reveals significant changes at 25 DPL. Synaptophysin protein levels ratios are increased by 30% at 25 DPL in the pre-ECL treatment group whereas they are relatively unchanged in the post-ECL group when compared to vehicle-treated ECL-mice (Fig. 6C: solid bars). Similarly, in situ AChE activity measured in the outer molecular layer of the dentate was shown to be significantly (p = 0.01) increased in the pre-ECL treated group at 25 DPL as opposed to the post-ECL treatment group which is not different for the vehicle-treated cohort.
3. Discussion
The present studies were designed to investigate whether ABCA1 and G1 transporters which are known to mediate cholesterol efflux are actively recruited following brain lesion to participate in the intense lipids trafficking that ensues. Using the well-established entorhinal cortex lesion paradigm, we demonstrated that ABCA1, but not ABCG1, expression is significantly (but transiently) up-regulated in the deafferented hippocampus during the early phase of the reinnervation process and the ongoing synaptic replacement. ABCA1 protein levels are also increased in the deafferented hippocampus following a similar time course profile. Both, mRNA and protein levels, were found to return to control values between 3 and 6 weeks following injury.
Several studies have demonstrated that upon experimental injury, CNS apoE protein levels as well as the neuronal binding capacity for apoE-containing lipoparticles increase sharply during the early phase of the reinnervation process (first two weeks post-injury) (Blain et al., 2004; Ignatius et al., 1987; Poirier, 1994; Poirier et al., 1991). The apoE induction time frame (Blain et al., 2004; Petit-Turcotte et al., 2005) is certainly consistent with ABCA1 expression time course observed in the present study (with a peak at 14 days post-lesion, Fig. 3) and, supports previous works showing a functional biological interaction between glial ABCA1 and apoE secretion during lipoprotein lipidation (Hirsch-Reinshagen et al., 2004; Wahrle et al., 2008, 2004). These findings are also consistent with the work of Fukumoto et al. (2002) who reported an induction of ABCA1, as measured by ABCA1 mRNA in situ hybridization at day 3 and 7 post-lesion, in response to excitotoxic lesions of the hippocampus.
We also examined the compensatory reinnervation taking place in the hippocampus after the lesion and showed increased cholinergic sprouting in the ipsilateral dentate gyrus consistent with the previous reports in the deafferented mice (Champagne et al., 2005; Veinbergs et al., 1999). The time course of the apoE and ABCA1 inductions was found to match perfectly the extent of the cholinergic remodeling process in the hippocampal areas (Figs. 2, 3, and 5). Together, these results support an active role for ABCA1 in the biochemical cascade regulating cholesterol mobilization and recycling from astrocytes to neurons undergoing synaptic remodeling and terminal sprouting in response to injury or neurodegenerative conditions (Fig. 1) (Poirier et al., 2008) by serving as a catalyst of apoE lipidation and cholesterol efflux from glial cell during the early phase of reinnervation. Mice lacking ABCA1 in the CNS were shown to present lower apoE levels accompanied with behavioral impairment and synaptic loss (Karasinska et al., 2009). APP23/ABCA1+/− transgenic mice exhibit reduced brain apoE levels and display marked memory deficits with age (Lefterov et al., 2009). The negative impact of ABCA1 deficiency on behavior and synaptic plasticity and integrity is certainly consistent with independent results obtained from apoE- and LDLR-deficient mice models (Cao et al., 2006; Champagne et al., 2005, 2002; Krugers et al., 1997; Masliah et al., 1995; Mulder et al., 2007, 2004; Oitzl et al., 1997). In all these models, authors attributed the pathophysiological deficits to the dysregulation of cholesterol trafficking and, the poor uptake of lipoprotein by brain cells, most notably neurons, during the synaptogenesis process.
Hence, to further investigate whether ABCA1 and G1 are involved in the compensatory response to brain lesion and whether they could serve as potential targets to promote reinnervation, we used a LXR agonist previously shown to induce ABCA1 and G1 expression in vivo in the CNS, i.e. TO901317. The LXR agonist administration was used in a prevention design, i.e. daily injection 7 days prior to the lesion and, in a rescue design, where daily injection of the agonist were administered 7 days after the lesion. Interestingly, only hippocampal ABCA1 expression (and not ABCG1) was found to be up-regulated in response to TO901317 treatment, and exclusively in the prevention design group (Fig. 6).
Furthermore, ABCA1 up-regulation was found to be maximal in the contralateral side of the hippocampal area. This could be explained, at least in part, by the fact that the agonist agent was injected intraperitoneally and acted on the whole brain structure. Although ipsilateral ABCA1 is normally increased as a consequence of the deafferentation process (previously shown in Figs. 3 and 6), the LXR agonist did not significantly potentiate this induction, whereas the LXR agonist effect on ABCA1 expression on the contralateral side was more exacerbated and quite significant. It is conceivable that the deafferented ipsilateral side is already at its peak physiological induction and cannot be further potentiated by pharmacological manipulations. Unexpectedly, the LXR agonist tended to decrease ABCA1 mRNA levels on both sides of the hippocampus when administered 7 days after the lesion (recovery design).
ABCA1 has been repetitively found to be increased after (6–7 days) in vivo injection of the TO901317 agonist in wild-type mice (Burns et al., 2006) and APP mice (Koldamova et al., 2005; Riddell et al., 2007). Repa et al. (2007) treated Niemann–Pick disease model mice with oral TO901317 to a regimen corresponding to 50 mg/kg for 4 weeks and still found significant increases in ABCA1 and G1 mRNA levels. Loane et al. (2011) treated mice in a pre-/post-traumatic brain injury paradigm (controlled cortical impact in a traumatic brain injury model in mice) with a single oral 25 mg/kg of TO901317 dose and found an increase in ABCA1 expression which remained elevated at least 7 days after treatment. Interestingly, results from post-injury treatments were clearly less beneficial than the ones observed from pre-injury treatments for some of the measures. Both Repa et al. study in Niemann–Pick disease model mice and the Riddell et al. study in a transgenic model of amyloidosis show an induction of one or more ABC transporters in specific brain regions under TO901317 treatment in different pathological conditions linked to cholesterol regulations in vivo. Furthermore, both teams reported a marked improvement of behavioral parameters and pathological markers with LXR agonist treatments. The results presented here on increased hippocampal AChE and synaptophysin protein levels in the pre-lesion TO901317-treated mice are certainly consistent with these observations. They are also supported by the work of Chen et al. (2010) showing that TO901317 administration stimulates synaptic remodeling and axonal regeneration in an experimental model of stroke in the mouse.
In contrast to ABCA1, ABCG1 was neither increased endogenously following the ECL nor after pharmacological TO901317 treatments. This result is counterintuitive in view of the common acceptation that LXR agonists induce ABCA1 and G1 expressions, and intriguing considering that ABCA1 is indeed upregulated and that ABCA1 and G1 were proposed to act synergistically to regulate intracellular cholesterol trafficking. However, in vivo TO901317-agonist studies classically show an increase in ABCA1 protein levels and do not report on ABCG1 alterations. In studies that examine in vivo ABCG1 expression after TO901317 treatment, it appears that ABCG1 is much less a reliable marker of the LXR agonist effectiveness than ABCA1. Comparable to our study and result, Burns et al. (2006) treated mice with i.p. doses of 25 and 50 mg/kg (same as here) for 7 days and also found an marked increase in ABCA1 protein levels in whole brain homogenates, whereas ABCG1 levels were found unchanged. Further, Repa et al. (2007), cited above, specifically examined the cerebellum of Niemann–Pick disease model mice fed with a diet containing TO901317 for 4 weeks and reported modest increase in ABCG1 mRNA levels but marked alteration in ABCA1 mRNA prevalence. In another mouse model of cholesterol homeostasis dysfunction, Vanmierlo et al. (2011) yielded results consistent with Repa et al. and findings presented here. While ABCA1 as well as ABCG1 mRNA levels were up-regulated in all brain structures considered in TO901317-mixed diet fed wild-type mice, the mutant mice displayed differential regulation of ABCA1 and G1 transcripts. ABCA1 mRNA levels were found to be increased after TO901317 treatment in cerebellum and hippocampus but not in cortical areas. In contrast, ABCG1 expression was slightly induced in cerebellum, but not in other brain structures. Finally, considering the fact that we had a limited numbers of time points post-lesion in our study, it is conceivable, although unlikely, that the expected increase in ABCG1 expression might have occur between two time points, either during the deafferentation phase or later.
Also, ABCG1's function in the regulation of CNS cholesterol homeostasis still remains quite controversial. While some studies failed to detect any effect of ABCG1 overexpression or deficiency on cholesterol efflux and brain lipid levels in apoE deficient (Burgess et al., 2008a) and PDAPP transgenic mice (Burgess et al., 2008b), others report positive correlations between ABCG1 expression and peripheral lipid tissue levels (Kennedy et al., 2005). ABCA1 has repeatedly been reported to act in concert with ABCG1, and both were shown to act sequentially in promoting phospholipids and cholesterol efflux from peripheral cell and to facilitate apoE-HDL lipidation (Gelissen et al., 2006; Karten et al., 2006; Kennedy et al., 2005; Vaughan and Oram, 2006). Cholesterol loading in vitro enhances ABCG1, but not ABCA1, expression and correlates best with cholesterol efflux from astrocytes (Karten et al., 2006). While difficult to reconcile, the in vivo and in vitro discrepancies still point to some common features: ABCG1 over-expression does not influence cognition, learning and memory, nor hippocampal synaptic plasticity (Parkinson et al., 2009), nor ABCA1 or apoE levels (Burgess et al., 2008a, 2008b) in transgenic mice; suggesting a rather modest contribution of ABCG1, if any, in lipid mobilization and in the maintenance of synaptic integrity or plasticity in the adult brain.
Alternatively, it is conceivable that neuronal cell death, as opposed to the simple deafferentation, may be required to functionally up-regulate ABCG1 in the injured CNS. Hence, ABCG1 mRNA levels might be altered at the lesion site and not in the deafferented hippocampus. Results obtained on cholinergic sprouting in the granular layer of the dentate gyrus in association with increase in ABCA1 mRNA and protein levels are in turn consistent with an active role of this ABC transporter in the maintenance of lipids and cholesterol homeostasis following injury: specifically in the regulation of glial cholesterol efflux and redistribution in response to hippocampal deafferentation from the entorhinal cortex. However, this does not exclude the possibility that the observed induction of ABCA1 in the hippocampus is the result of a retrograde process originating in the lesioned entorhinal cortex area.
Finally, considering the suggested coordinated actions of ABCA1 and G1 in the formation of mature fully lipidated apoE-HDL, the fact that ABCG1 expression is not altered by the LXR agonist treatments could explain the modest impact of the drug on local reinnervation as measured by synaptophysin and AChE levels (Fig. 6). It is of interest to note that members of the ABCG family were shown to dimerize to form functional heterogeneous transporters such as ABCG5 and 8 (Graf et al., 2003), or are functional as homotetramers like ABCG2 (Xu, 2004). In the brain, multiple evidence point at ABCG4 as a potential dimer partner to ABCG1 since they are both expressed in the brain (Oldfield et al., 2002; Savary et al., 1996), induced by LXR (Engel et al., 2001; Venkateswaran et al., 2000) and Wang et al. (2008) recently demonstrated an in vivo role for brain ABCG4 in cholesterol efflux. The latter study describes a complex interaction between ABCG1 and ABCG4 involvement in cholesterol biosynthetic pathway, LXR activation and as a consequence ABCA1 expression and apoE secretion. Therefore, the examination of other members of the ABCG family and the cholesterol biosynthetic pathway intermediates during neurodegeneration and reinnervation could certainly lead to a much more comprehensive understanding of the complex interactions regulating the intra-and extracellular transport of lipids in the adult brain.
In conclusion, results of this study show that the ABC transporter ABCA1 (but not ABCG1) plays a role in the early remodeling process that ensues brain injury. The present in vivo study adds to the growing evidence for a role of brain ABCA1 in the lipidation of apoE and supports the proposed molecular cascade that regulates glial cholesterol efflux via ABCA1 and apoE. However, these results also point at the need to decipher the role of ABCG1 and its interaction with other ABC transporters before the use of pharmacological tools like LXR synthetic agonist to enhance brain plasticity. Peripheral administration of an LXR agonist while appearing sufficient to stimulate these mechanisms in vivo and to modulate the extent of synaptic recovery, does so only when used prior to neuronal injury. It was found to completely fail when administered after the injury; suggesting little or no recovery potential.
4. Experimental procedures
4.1. Animals
A total of sixty-five male C57BL/6J mice aged 12 weeks were purchased from Jackson Laboratories (Bar Harbor, ME, USA). All animals were housed individually in an enriched environment and fed with a diet of standard laboratory chow ad libitum. A 12-h light-dark cycle was maintained with light onset at 07:00 and offset at 19:00, local time. All protocols were carried out in accordance with the Canadian Guidelines for Use and Care of Laboratory Animals, and were approved by the McGill University Animal Care Committee.
4.2. Unilateral entorhinal cortex lesions (ECL)
Unilateral electrolytic lesions to the entorhinal cortex were conducted according to the technique adapted for mice described by Blain et al. (2004). A total of forty-five fifteen-week-old anaesthetized mice (intramuscular injection of a ketamine–xylazine/acepromazine mix, 1 μL/g of body weight) were placed into a stereotaxic apparatus in a flat skull position. Four lesion coordinates were determined from Lambda: (1) [AP: 0 mm], [L: −3.0 mm], and [DV: −3.0 mm, − 4.0 mm]; (2) [AP: 0 mm], [L: −3.5 mm], and [DV: −3.0 mm, − 4.0 mm]; (3) [AP: +0.5 mm], [L: −4.0 mm], and [DV: −3.0 mm, −4.0 mm]; (4) [AP: +1.0 mm], [L: −4.0 mm], and [DV: −3.0 mm, −4.0 mm], where a 1 mA current was applied for 10 s (sec) at each coordinate. SHAM-operated animals (n = 5) were treated similarly except that the electrode was lowered only 1 mm and no current was passed. Following surgery, mice were given a subcutaneous bolster of physiological saline to prevent dehydration and nursed throughout their recovery.
4.3. LXR agonist administration
The LXR agonist selected was TO901317 (Cayman Chemicals, Ann Arbor, MI, USA) because it is a full agonist which induces binding of co-activators and disassociation of co-repressors (Wójcicka et al., 2007). Following acclimatization, a subgroup of mice were randomly assigned to receive daily intra-peritoneal injections of 30 mg/kg/day of TO901317 in 0.125% (wt/vol) carboxymethycellulose (CMC) at one of two different starting time points and continued until sacrifice in order to assess the therapeutic potential of the treatment as prevention (7 days before lesion (ECL); n = 5) or rescue (7 days after lesion (ECL); n = 5). The choice of the 30 mg/kg/day dose was guided by previous studies which reported beneficial outcomes of TO901317 in the CNS (Chen et al., 2010; Morales et al., 2008; Riddell et al., 2007). Furthermore, a prior pilot study was conducted in our laboratory using a small set of animals. The reported effect on brain ABCA1 expression levels was replicated at the chosen dose (data not shown). Control animals (n = 5) were treated daily with vehicle (0.125% (wt/vol) CMC) in the same conditions, starting 7 days prior to the ECL and continued until sacrifice.
4.4. Experimental endpoints
Mice were sacrificed at different key time points following ECL to establish a time course that covers (i) the degenerative phase (0–14 DPL) and (ii), the reactive sprouting and synaptic remodeling phases (14–30 DPL) until completion of the reinnervation process and late recovery (40–60 DPL). Thus, at 2, 14, 21, 40 and 60 days post-lesion (DPL), 14 DPL for the SHAM-operated mice and 25 DPL for the LXR-treated mice and their vehicle-treated controls, a subgroup of mice (n = 5/group) were decapitated and their brain quickly removed and dissected out on dry ice for hippocampus (HPC) and frontal cortex (FCx). Ipsilateral and contralateral side of each brain structure were stored separately at −80 °C until use.
Other subgroups of ECL-mice were allocated for histochemistry and were then administered a lethal dose of anesthetic and perfused transcardially with 30 mL of ice-cold 0.01 M phosphate buffered saline (PBS) solution at 2, 6, 14 and 21 DPL (n = 5/group). Following perfusion, whole brains were removed, flash-frozen at −40 °C in isopentane and stored at −80 °C until use.
4.5. Acetylcholinesterase (AChE) histochemistry for assessment of cholinergic sprouting
Coronal brain section of 20 μm through the region of the dorsal hippocampal formation were mounted on poly-L-lysine-coated glass slides, desiccated overnight at 4 °C, and stored at −80 °C until use. In situ AChE activity was evaluated by incubating slides at room temperature in the substrate solution (0.0072% (wt/vol) ethopropazine, 0.075% (wt/vol) glycine, 0.5% (wt/vol) cupric sulfate, 0.12% (wt/vol) acetylthiocholine iodide, 0.68% (wt/vol) sodium acetate; pH 5.0) for 4 h (h). Following this, the slides were rinsed 3 times for 5 min in H2O and then placed in the developer solution (0.38% (wt/vol) sodium sulfide; pH 7.8) for 6 min. After a second series of H2O rinses, silver intensification was performed by placing the slides into 1% silver nitrate solution for 2 min in total obscurity. Slides were rinsed a third time in H2O and post-fixed in a 4% (wt/vol) paraformaldehyde solution in 0.01 M PBS (pH 7.4) for 2 h. After a final rinse in 0.01 M PBS (pH 7.4), slides were dehydrated in a series of alcohol baths and cleared in xylene (2 min in each bath). Slides were cover-slipped with DPX mounting medium and stored in total obscurity until analyzed. All products used were purchased from Sigma (Sigma-Aldrich, St. Louis, MO).
The time course and extent of cholinergic sprouting in the dentate gyrus (DG) following ECL was assessed as the relative optical density (OD) of AChE activity staining defined as the ratio between ipsilateral and contralateral OD, thereby providing a within-section control for variations in histochemical processing. AChE staining OD was evaluated using the MCDI-II image analysis system on digital microphotographs of each brain section captured using a Zeiss Axioskop 2 Plus microscope under a 2.5 × /0.075 objective (Plan-Neofluar, Zeiss, Germany) and the Northern Eclipse Version 6.0 Image Analysis Software. Per side, six different OD measures were made along the dorsal blade of the outer molecular layer (OML) of the DG on 5 sections per animal and averaged. Since the lamina-specific denervation of the DG following ECL has been shown to result in atrophy localized to the OML that may confound the histochemical estimation of AChE activity (Phinney et al., 2004; Steward et al., 1973), measures of OML width were also assessed at 6 positions along the dorsal blade of the DG and incorporated into the equation according to the model of Fagan and Gage (1994):
4.6. Real-time polymerase chain reaction (RT-PCR)
ABCA1 and ABCG1 gene expression was assessed with the SYBR® Green technique, using actin as an internal control. Total RNA was extracted from ipsilateral and contralateral hippocampus and frontal cortex using the QIAGEN RNeasy Mini Kit (QIAGEN Inc., Mississauga, ON) according to the manufacturer's guidelines. Following extraction, 2 μg of total RNA from each sample was reverse-transcribed in the Gen-eAmp 5700 sequence detection system (PE Applied Biosystems) to generate cDNA in the following reaction mixture: 1 × RT buffer; 5.5 mM MgCl2; 500 μM dNTPs; 2.5 μM Oligo DT; 0.4 U/μL RNase inhibitor; 1.25 U/μL Multiscribe Reverse Transcriptase, in a final reaction volume of 100 μL. The reverse transcriptase program included the following thermal cycle: 10 min at 25 °C, 30 min at 48 °C, 5 min at 95 °C to stop the reaction. Real-time PCR was conducted in the GeneAmp 5700 sequence detection system on each sample of cDNA in triplicate. Primer pairs (forward: fwd and reverse: rev) used for PCR amplification were as follows: mABCA1-fwd 5′-GACCGTACTCTC-GCAGGG-3′ with mABCA1-rev 5′-GCGGCCTTGCCGGTAT-3′; mABCG1-fwd 5′CCGATGTGAACCCGTTTCTT-3′ with mABCG1-rev 5′-AGGCGGAGTCCTCTTCAGC-3′; and mACTIN-fwd 5′-TGACCGAGCGTGGCTACA-3′ with mACTIN-rev 5′-TCTCT-TTGATGTCACGCACGAT-3′. Primer pairs were generated using the Primer Express PE Biosystems software. Primer specificity was confirmed through dissociation curve analysis which demonstrated single product specific melting temperatures. No primer-dimers were observed during the 40 PCR cycles. The master-mix solution for each 35 μL PCR reaction was prepared as follows: 17.5 μL of SYBR Green PCR Master Mix, 3.5 μL of 10 μM stocks of forward and reverse primers, 7.5 μL RNase-free H2O, 3 μL RT product. All reagents used were purchased from PE Biosystems (Perkin-Elmer, Foster City, CA). The program applied for real-time PCR cycling consisted of 2 min at 50 °C, 10 min at 95 °C and 40 cycles of 15 s at 95 °C and 1 min at 60 °C. Relative gene expression was calculated using the 2−ΔδCt method with β-actin as reference gene and results are expressed as ratios between ipsilateral and con-tralateral sides to the lesion±SEM.
4.7. ELISA bioassay
ABCA1 protein levels were determined by indirect ELISA in the ipsilateral and contralateral hippocampus. Homogenates were prepared by sonication in 0.01 M PBS containing pro-tease inhibitors (Boehringer Mannheim, Germany) and total protein concentration was measured with the BCA protein dosage kit (Pierce, Rockford, IL). Each sample were applied in triplicate on Costar 96-well EIA/RIA plates (Fisher Scientific) as well as a rat brain homogenate (sonicated in PBS 0.01 M) diluted in bicarbonate/carbonate buffer (100 mM, pH 9.6) to serve as standards (375–2812.5 μg/mL) and incubated overnight at 4 °C. The next day, the primary antibody (rabbit polyclonal ABCA1 antibody, ab14146; Abcam, Cambridge, USA) diluted in a solution of PBS 0.01 M with 1% (wt/vol) bovine serum albumin (BSA) was added for 2 h at room temperature. Following three washes with Tris buffered saline-Tween 20 (TBS-T) solution, the biotinylated detection antibody (anti-rabbit antibody, ab6720; Abcam, Cambridge, USA) was added for 2 h. After a second series of washes, detection of bound secondary antibodies was realized by incubation with an alkaline-phosphatase-conjugated strepta-vidin solution (Invitrogen Canada Inc., Burlington, ON) for 1 h at room temperature. Finally, plates were washed four times with TBS-T and once with H2O and incubated with an alkaline phosphatase fluorescent substrate (AttoPhos, Promega, San Luis Obispo, USA) for 30 min at 37 °C. Fluorescence was measured with the microplate fluorescent reader (FL600, Bio-Tek Instruments) at a 450 nm/20 nm excitation and 560 nm/20 nm emission. Relative protein levels of ABCA1 are expressed as ratios between ipsi- and contra-lateral fluorescence unit±SEM.
4.8. Western blot immunodetection
Synaptophysin protein levels were evaluated by Western blot in the ipsilateral and contralateral hippocampi. Homogenates were prepared and their total protein concentration was measured as described above. Twelve micrograms of pools of five HPC protein homogenates were applied in duplicate and separated by SDS-PAGE gel electrophoresis under reducing conditions (NuPAGE Novex 4–12% gradient tris-glycine precast gels) and transferred to a nitrocellulose membrane with the iBlot Dry Blotting System according to the manufacturer's protocol (Invitrogen, Carlsbad, CA). Membranes were blocked in 5% (wt/vol) non-fat dry milk in TBS-T for 1 h at room temperature, as for all subsequent incubations. First, a mouse monoclonal antibody against synaptophysin (5768, Sigma-Aldrich, MO, USA) diluted 1/1000 in TBS-T, then a secondary horseradish peroxidase (HRP)-linked anti-mouse antibody (Amersham, Oakville, Canada) diluted 1/5000 in TBS-T after TBS-T washing. On the same membranes, detection of α-tubulin was carried out as described above using a mouse monoclonal anti-tubulin antibody (M61409M, BioDesign Int., Saco, USA) and a HRP-linked sheep anti-mouse antibody (Amersham) to control for the amount of protein loaded onto the gel. Detected proteins were revealed with an Immun-Star™ WesternC™ Chemiluminescence Kit (Bio-Rad, CA, USA) or protein chemiluminescence reagents (ZmTech Scientifique, QC, CA) and visualized by exposition on a Kodak Image Station 440CF (Kodak). Each band was analyzed with the Carestream Molecular Imaging Software (Carestream). The synaptophysin-result for each pool was normalized with the corresponding tubulin-result and expressed as the ratio between ipsi- and contra-lateral HPC.
4.9. Statistical analyses
One-way ANOVA analyses were applied from the SPSS version 15.0 software and followed by Tukey's HSD post-hoc tests. Results were considered statistically significant when p ≤ 0.05 (non-significant: n.s.).
Acknowledgments
This work was supported by grants from the Canadian Institutes of Health Research (JP) and the Natural Sciences and Engineering Research Council of Canada (JP) and the Alzheimer Society of Canada (SB).
Abbreviations
- ABCA1/G1
ATP-binding cassette transporter A1/G1
- AChE
acetylcholinesterase
- apoE
apolipoprotein E
- BBB
Blood–Brain Barrier
- CMC
carboxymethycellulose
- DPL
days post-lesion
- ECL
entorhinal cortex lesion
- FC
frontal cortex
- HDL
high-density lipoprotein
- HMGCoAR
3-hydroxy-3-methylglutaryl-coenzyme A reductase
- HPC
hippocampus
- HRP
horseradish peroxidase
- LDLR
low-density lipoprotein receptor
- LXR
liver X receptor
- OD
optical density
- OML
outer molecular layer
- PBS
phosphate buffered saline
- TBS-T
Tris Bufferd Saline-Tween 20
Footnotes
The authors have no confiicts of interest to disclose.
References
- Abildayeva K, Jansen PJ, Hirsch-Reinshagen V, Bloks VW, Bakker AHF, Ramaekers FCS, de Vente J, Groen AK, Wellington CL, Kuipers F, Mulder M. 24(S)-hydroxycholesterol participates in a liver X receptor-controlled pathway in astrocytes that regulates apolipoprotein E-mediated cholesterol efflux. J Biol Chem. 2006;281:12799–12808. doi: 10.1074/jbc.M601019200. http://dx.doi.org/10.1074/jbc.M601019200. [DOI] [PubMed] [Google Scholar]
- Blain JF, Paradis E, Gaudreault SB, Champagne D, Richard D, Poirier J. A role for lipoprotein lipase during synaptic remodeling in the adult mouse brain. Neurobiol Dis. 2004;15:510–519. doi: 10.1016/j.nbd.2003.12.001. [DOI] [PubMed] [Google Scholar]
- Burgess B, Naus K, Chan J, Hirsch-Reinshagen V, Tansley G, Matzke L, Chan B, Wilkinson A, Fan J, Donkin J, Balik D, Tanaka T, Ou G, Dyer R, Innis S, McManus B, Lütjohann D, Wellington C. Overexpression of human ABCG1 does not affect atherosclerosis in fat-fed ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 2008a;28:1731–1737. doi: 10.1161/ATVBAHA.108.168542. http://dx.doi.org/10.1161/ATVBAHA.108.168542. [DOI] [PubMed] [Google Scholar]
- Burgess BL, Parkinson PF, Racke MM, Hirsch-Reinshagen V, Fan J, Wong C, Stukas S, Theroux L, Chan JY, Donkin J, Wilkinson A, Balik D, Christie B, Poirier J, Lütjohann D, Demattos RB, Wellington CL. ABCG1 influences the brain cholesterol biosynthetic pathway but does not affect amyloid precursor protein or apolipoprotein E metabolism in vivo. J Lipid Res. 2008b;49:1254–1267. doi: 10.1194/jlr.M700481-JLR200. http://dx.doi.org/10.1194/jlr.M700481-JLR200. [DOI] [PubMed] [Google Scholar]
- Burns MP, Vardanian L, Pajoohesh-Ganji A, Wang L, Cooper M, Harris DC, Duff K, Rebeck GW. The effects of ABCA1 on cholesterol efflux and Abeta levels in vitro and in vivo. J Neurochem. 2006;98:792–800. doi: 10.1111/j.1471-4159.2006.03925.x. http://dx.doi.org/10.1111/j.1471-4159.2006.03925.x. [DOI] [PubMed] [Google Scholar]
- Cao D, Fukuchi K, Wan H, Kim H, Li L. Lack of LDL receptor aggravates learning deficits and amyloid deposits in Alzheimer transgenic mice. Neurobiol Aging. 2006;27:1632–1643. doi: 10.1016/j.neurobiolaging.2005.09.011. http://dx.doi.org/10.1016/j.neurobiolaging.2005.09.011. [DOI] [PubMed] [Google Scholar]
- Champagne D, Dupuy JB, Rochford J, Poirier J. Apolipoprotein E knockout mice display procedural deficits in the Morris water maze: analysis of learning strategies in three versions of the task. Neuroscience. 2002;114:641–654. doi: 10.1016/s0306-4522(02)00313-5. [DOI] [PubMed] [Google Scholar]
- Champagne D, Rochford J, Poirier J. Effect of apolipoprotein E deficiency on reactive sprouting in the dentate gyrus of the hippocampus following entorhinal cortex lesion: role of the astroglial response. Exp Neurol. 2005;194:31–42. doi: 10.1016/j.expneurol.2005.01.016. http://dx.doi.org/10.1016/j.expneurol.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Chen J, Zacharek A, Cui X, Shehadah A, Jiang H, Roberts C, Lu M, Chopp M. Treatment of stroke with a synthetic liver X receptor agonist, TO901317, promotes synaptic plasticity and axonal regeneration in mice. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab. 2010;30:102–109. doi: 10.1038/jcbfm.2009.187. http://dx.doi.org/10.1038/jcbfm.2009.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Chaves Posse, Vance EI, Campenot DE, Kiss RB, Vance JE, RS Uptake of lipoproteins for axonal growth of sympathetic neurons. J Biol Chem. 2000;275:19883–19890. doi: 10.1074/jbc.275.26.19883. [DOI] [PubMed] [Google Scholar]
- De Chaves EI, Rusiñol AE, Vance DE, Campenot RB, Vance JE. Role of lipoproteins in the delivery of lipids to axons during axonal regeneration. J Biol Chem. 1997;272:30766–30773. doi: 10.1074/jbc.272.49.30766. [DOI] [PubMed] [Google Scholar]
- Dietschy JM. Central nervous system: cholesterol turnover, brain development and neurodegeneration. Biol Chem. 2009;390:287–293. doi: 10.1515/BC.2009.035. http://dx.doi.org/10.1515/BC.2009.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietschy JM, Turley SD. Cholesterol metabolism in the brain. Curr Opin Lipidol. 2001;12:105–112. doi: 10.1097/00041433-200104000-00003. [DOI] [PubMed] [Google Scholar]
- Engel T, Lorkowski S, Lueken A, Rust S, Schlüter B, Berger G, Cullen P, Assmann G. The human ABCG4 gene is regulated by oxysterols and retinoids in monocyte-derived macrophages. Biochem Biophys Res Commun. 2001;288:483–488. doi: 10.1006/bbrc.2001.5756. http://dx.doi.org/10.1006/bbrc.2001.5756. [DOI] [PubMed] [Google Scholar]
- Fagan AM, Gage FH. Mechanisms of sprouting in the adult central nervous system: cellular responses in areas of terminal degeneration and reinnervation in the rat hippocampus. Neuroscience. 1994;58:705–725. doi: 10.1016/0306-4522(94)90449-9. [DOI] [PubMed] [Google Scholar]
- Fukumoto H, Deng A, Irizarry MC, Fitzgerald ML, Rebeck GW. Induction of the cholesterol transporter ABCA1 in central nervous system cells by liver X receptor agonists increases secreted Abeta levels. J Biol Chem. 2002;277:48508–48513. doi: 10.1074/jbc.M209085200. http://dx.doi.org/10.1074/jbc.M209085200. [DOI] [PubMed] [Google Scholar]
- Gabbi C, Warner M, Gustafsson JA. Minireview: liver X receptor beta: emerging roles in physiology and diseases. Mol Endocrinol Baltimore Md. 2009;23:129–136. doi: 10.1210/me.2008-0398. http://dx.doi.org/10.1210/me.2008-0398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelissen IC, Harris M, Rye KA, Quinn C, Brown AJ, Kockx M, Cartland S, Packianathan M, Kritharides L, Jessup W. ABCA1 and ABCG1 synergize to mediate cholesterol export to apoA-I. Arterioscler Thromb Vasc Biol. 2006;26:534–540. doi: 10.1161/01.ATV.0000200082.58536.e1. http://dx.doi.org/10.1161/01.ATV.0000200082.58536.e1. [DOI] [PubMed] [Google Scholar]
- Graf GA, Yu L, Li WP, Gerard R, Tuma PL, Cohen JC, Hobbs HH. ABCG5 and ABCG8 are obligate heterodimers for protein trafficking and biliary cholesterol excretion. J Biol Chem. 2003;278:48275–48282. doi: 10.1074/jbc.M310223200. http://dx.doi.org/10.1074/jbc.M310223200. [DOI] [PubMed] [Google Scholar]
- Hirsch-Reinshagen V, Wellington CL. Cholesterol metabolism, apolipoprotein E, adenosine triphosphate-binding cassette transporters, and Alzheimer’s disease. Curr Opin Lipidol. 2007;18:325–332. doi: 10.1097/MOL.0b013e32813aeabf. http://dx.doi.org/10.1097/MOL.0b013e32813aeabf. [DOI] [PubMed] [Google Scholar]
- Hirsch-Reinshagen V, Zhou S, Burgess BL, Bernier L, McIsaac SA, Chan JY, Tansley GH, Cohn JS, Hayden MR, Wellington CL. Deficiency of ABCA1 impairs apolipoprotein E metabolism in brain. J Biol Chem. 2004;279:41197–41207. doi: 10.1074/jbc.M407962200. http://dx.doi.org/10.1074/jbc.M407962200. [DOI] [PubMed] [Google Scholar]
- Ignatius MJ, Shooter EM, Pitas RE, Mahley RW. Lipoprotein uptake by neuronal growth cones in vitro. Science. 1987;236:959–962. doi: 10.1126/science.3576212. [DOI] [PubMed] [Google Scholar]
- Illingworth DR, Glover J. The composition of lipids in cerebrospinal fluid of children and adults. J Neurochem. 1971;18:769–776. doi: 10.1111/j.1471-4159.1971.tb12006.x. [DOI] [PubMed] [Google Scholar]
- Ishimoto K, Tachibana K, Sumitomo M, Omote S, Hanano I, Yamasaki D, Watanabe Y, Tanaka T, Hamakubo T, Sakai J, Kodama T, Doi T. Identification of human low-density lipoprotein receptor as a novel target gene regulated by liver X receptor alpha. FEBS Lett. 2006;580:4929–4933. doi: 10.1016/j.febslet.2006.08.010. http://dx.doi.org/10.1016/j.febslet.2006.08.010. [DOI] [PubMed] [Google Scholar]
- Karasinska JM, Rinninger F, Lütjohann D, Ruddle P, Franciosi S, Kruit JK, Singaraja RR, Hirsch-Reinshagen V, Fan J, Brunham LR, Bissada N, Ramakrishnan R, Wellington MR, Parks JS, Hayden MR. Specific loss of brain ABCA1 increases brain cholesterol uptake and influences neuronal structure and function. J Neurosci Off J Soc Neurosci. 2009;29:3579–3589. doi: 10.1523/JNEUROSCI.4741-08.2009. http://dx.doi.org/10.1523/JNEUROSCI.4741-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karten B, Campenot RB, Vance DE, Vance JE. Expression of ABCG1, but not ABCA1, correlates with cholesterol release by cerebellar astroglia. J Biol Chem. 2006;281:4049–4057. doi: 10.1074/jbc.M508915200. http://dx.doi.org/10.1074/jbc.M508915200. [DOI] [PubMed] [Google Scholar]
- Kennedy MA, Barrera GC, Nakamura K, Baldán A, Tarr P, Fishbein MC, Frank J, Francone OL, Edwards PA. ABCG1 has a critical role in mediating cholesterol efflux to HDL and preventing cellular lipid accumulation. Cell Metab. 2005;1:121–131. doi: 10.1016/j.cmet.2005.01.002. http://dx.doi.org/10.1016/j.cmet.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Kennedy MA, Venkateswaran A, Tarr PT, Xenarios I, Kudoh J, Shimizu N, Edwards PA. Characterization of the human ABCG1 gene: liver X receptor activates an internal promoter that produces a novel transcript encoding an alternative form of the protein. J Biol Chem. 2001;276:39438–39447. doi: 10.1074/jbc.M105863200. http://dx.doi.org/10.1074/jbc.M105863200. [DOI] [PubMed] [Google Scholar]
- Klucken J, Büchler C, Orsó E, Kaminski WE, Porsch-Ozcürümez M, Liebisch G, Kapinsky M, Diederich W, Drobnik W, Dean M, Allikmets R, Schmitz G. ABCG1 (ABC8), the human homolog of the Drosophila white gene, is a regulator of macrophage cholesterol and phospholipid transport. Proc Natl Acad Sci USA. 2000;97:817–822. doi: 10.1073/pnas.97.2.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koldamova R, Fitz NF, Lefterov I. The role of ATP-binding cassette transporter A1 in Alzheimer’s disease and neurodegeneration. Biochim Biophys Acta. 2010;1801:824–830. doi: 10.1016/j.bbalip.2010.02.010. http://dx.doi.org/10.1016/j.bbalip.2010.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koldamova RP, Lefterov IM, Ikonomovic MD, Skoko J, Lefterov PI, Isanski BA, DeKosky ST, Lazo JS. 22R-hydroxycholesterol and 9-cis-retinoic acid induce ATP-binding cassette transporter A1 expression and cholesterol efflux in brain cells and decrease amyloid beta secretion. J Biol Chem. 2003;278:13244–13256. doi: 10.1074/jbc.M300044200. http://dx.doi.org/10.1074/jbc.M300044200. [DOI] [PubMed] [Google Scholar]
- Koldamova RP, Lefterov IM, Staufenbiel M, Wolfe D, Huang S, Glorioso JC, Walter M, Roth MG, Lazo JS. The liver X receptor ligand T0901317 decreases amyloid beta production in vitro and in a mouse model of Alzheimer’s disease. J Biol Chem. 2005;280:4079–4088. doi: 10.1074/jbc.M411420200. http://dx.doi.org/10.1074/jbc.M411420200. [DOI] [PubMed] [Google Scholar]
- Krugers HJ, Mulder M, Korf J, Havekes L, de Kloet ER, Joëls M. Altered synaptic plasticity in hippocampal CA1 area of apolipoprotein E deficient mice. Neuroreport. 1997;8:2505–2510. doi: 10.1097/00001756-199707280-00018. [DOI] [PubMed] [Google Scholar]
- LaDu MJ, Shah JA, Reardon CA, Getz GS, Bu G, Hu J, Guo L, Van Eldik LJ. Apolipoprotein E and apolipoprotein E receptors modulate A beta-induced glial neuroinflammatory responses. Neurochem Int. 2001;39:427–434. doi: 10.1016/s0197-0186(01)00050-x. ( http://dx.doi.org/11578778) [DOI] [PubMed] [Google Scholar]
- Lefterov I, Fitz NF, Cronican A, Lefterov P, Staufenbiel M, Koldamova R. Memory deficits in APP23/Abca1+/− mice correlate with the level of Aβ oligomers. ASN Neuro. 2009:1. doi: 10.1042/AN20090015. http://dx.doi.org/10.1042/AN20090015. [DOI] [PMC free article] [PubMed]
- Loane DJ, Washington PM, Vardanian L, Pocivavsek A, Hoe HS, Duff KE, Cernak I, Rebeck GW, Faden AI, Burns MP. Modulation of ABCA1 by an LXR agonist reduces β-amyloid levels and improves outcome after traumatic brain injury. J Neurotrauma. 2011;28:225–236. doi: 10.1089/neu.2010.1595. http://dx.doi.org/10.1089/neu.2010.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Mallory M, Ge N, Alford M, Veinbergs I, Roses AD. Neurodegeneration in the central nervous system of apoE-deficient mice. Exp Neurol. 1995;136:107–122. doi: 10.1006/exnr.1995.1088. http://dx.doi.org/10.1006/exnr.1995.1088. [DOI] [PubMed] [Google Scholar]
- Matthews DA, Cotman C, Lynch G. An electron microscopic study of lesion-induced synaptogenesis in the dentate gyrus of the adult rat. I Magnitude and time course of degeneration. Brain Res. 1976;115:1–21. doi: 10.1016/0006-8993(76)90819-2. [DOI] [PubMed] [Google Scholar]
- Mauch DH, Nägler K, Schumacher S, Göritz C, Müller EC, Otto A, Pfrieger FW. CNS synaptogenesis promoted by glia-derived cholesterol. Science. 2001;294:1354–1357. doi: 10.1126/science.294.5545.1354. http://dx.doi.org/10.1126/science.294.5545.1354. [DOI] [PubMed] [Google Scholar]
- Morales JR, Ballesteros I, Deniz JM, Hurtado O, Vivancos J, Nombela F, Lizasoain I, Castrillo A, Moro MA. Activation of liver X receptors promotes neuroprotection and reduces brain inflammation in experimental stroke. Circulation. 2008;118:1450–1459. doi: 10.1161/CIRCULATIONAHA.108.782300. http://dx.doi.org/10.1161/CIRCULATIONAHA.108.782300. [DOI] [PubMed] [Google Scholar]
- Mulder M, Jansen PJ, Janssen BJA, van de Berg WDJ, van der Boom H, Havekes LM, de Kloet RE, Ramaekers FCS, Blokland A. Low-density lipoprotein receptor-knockout mice display impaired spatial memory associated with a decreased synaptic density in the hippocampus. Neurobiol Dis. 2004;16:212–219. doi: 10.1016/j.nbd.2004.01.015. http://dx.doi.org/10.1016/j.nbd.2004.01.015. [DOI] [PubMed] [Google Scholar]
- Mulder M, Koopmans G, Wassink G, Al Mansouri G, Simard ML, Havekes LM, Prickaerts J, Blokland A. LDL receptor deficiency results in decreased cell proliferation and presynaptic bouton density in the murine hippocampus. Neurosci Res. 2007;59:251–256. doi: 10.1016/j.neures.2007.07.004. http://dx.doi.org/10.1016/j.neures.2007.07.004. [DOI] [PubMed] [Google Scholar]
- Nelissen K, Mulder M, Smets I, Timmermans S, Smeets K, Ameloot M, Hendriks JJA. Liver X receptors regulate cholesterol homeostasis in oligodendrocytes. J Neurosci Res. 2012;90:60–71. doi: 10.1002/jnr.22743. http://dx.doi.org/10.1002/jnr.22743. [DOI] [PubMed] [Google Scholar]
- Oitzl MS, Mulder M, Lucassen PJ, Havekes LM, Grootendorst J, de Kloet ER. Severe learning deficits in apolipoprotein E-knockout mice in a water maze task. Brain Res. 1997;752:189–196. doi: 10.1016/s0006-8993(96)01448-5. [DOI] [PubMed] [Google Scholar]
- Oldfield S, Lowry C, Ruddick J, Lightman S. ABCG4: a novel human white family ABC-transporter expressed in the brain and eye. Biochim Biophys Acta. 2002;1591:175–179. doi: 10.1016/s0167-4889(02)00269-0. [DOI] [PubMed] [Google Scholar]
- Parkinson PF, Kannangara TS, Eadie BD, Burgess BL, Wellington CL, Christie BR. Cognition, learning behavior and hippocampal synaptic plasticity are not disrupted in mice over-expressing the cholesterol transporter ABCG1. Lipids Health Dis. 2009;8:5. doi: 10.1186/1476-511X-8-5. http://dx.doi.org/10.1186/1476-511X-8-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit-Turcotte C, Aumont N, Beffert U, Dea D, Herz J, Poirier J. The apoE receptor apoER2 is involved in the maintenance of efficient synaptic plasticity. Neurobiol Aging. 2005;26:195–206. doi: 10.1016/j.neurobiolaging.2004.04.007. http://dx.doi.org/10.1016/j.neurobiolaging.2004.04.007. [DOI] [PubMed] [Google Scholar]
- Pfrieger FW. Cholesterol homeostasis and function in neurons of the central nervous system. Cell Mol Life Sci CMLS. 2003;60:1158–1171. doi: 10.1007/s00018-003-3018-7. http://dx.doi.org/10.1007/s00018-003-3018-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phinney AL, Calhoun ME, Woods AG, Deller T, Jucker M. Stereological analysis of the reorganization of the dentate gyrus following entorhinal cortex lesion in mice. Eur J Neurosci. 2004;19:1731–1740. doi: 10.1111/j.1460-9568.2004.03280.x. http://dx.doi.org/10.1111/j.1460-9568.2004.03280.x. [DOI] [PubMed] [Google Scholar]
- Poirier J. Apolipoprotein E in animal models of CNS injury and in Alzheimer’s disease. Trends Neurosci. 1994;17:525–530. doi: 10.1016/0166-2236(94)90156-2. ( http://dx.doi.org/7532337) [DOI] [PubMed] [Google Scholar]
- Poirier J, Hess M, May PC, Finch CE. Astrocytic apolipoprotein E mRNA and GFAP mRNA in hippocampus after entorhinal cortex lesioning. Brain Res Mol Brain Res. 1991;11:97–106. doi: 10.1016/0169-328x(91)90111-a. [DOI] [PubMed] [Google Scholar]
- Poirier J, Lamarre-Théroux L, Dea D, Aumont N, Blain JF. Cholesterol transport and production in Alzheimer’s disease. In: Fisher A, Memo M, Stocchi F, Hanin I, editors. Advances in Alzheimer’s and Parkinson’s Disease. Springer; US, Boston, MA: 2008. pp. 211–219. [Google Scholar]
- Poirier JK, Baccichet A, Dea D, Gauthier S. Cholesterol synthesis and lipoprotein reuptake during synaptic remodelling in hippocampus in adult rats. Neuroscience. 1993;55:81–90. doi: 10.1016/0306-4522(93)90456-p. ( http://dx.doi.org/8350994) [DOI] [PubMed] [Google Scholar]
- Repa JJ, Li H, Frank-Cannon TC, Valasek MA, Turley SD, Tansey MG, Dietschy JM. Liver X receptor activation enhances cholesterol loss from the brain, decreases neuroinflammation, and increases survival of the NPC1 mouse. J Neurosci Off J Soc Neurosci. 2007;27:14470–14480. doi: 10.1523/JNEUROSCI.4823-07.2007. http://dx.doi.org/10.1523/JNEUROSCI.4823-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddell DR, Zhou H, Comery TA, Kouranova E, Lo CF, Warwick HK, Ring RH, Kirksey Y, Aschmies S, Xu J, Kubek K, Hirst WD, Gonzales C, Chen Y, Murphy E, Leonard S, Vasylyev D, Oganesian A, Martone RL, Pangalos MN, Reinhart PH, Jacobsen JS. The LXR agonist TO901317 selectively lowers hippocampal Abeta42 and improves memory in the Tg2576 mouse model of Alzheimer’s disease. Mol Cell Neurosci. 2007;34:621–628. doi: 10.1016/j.mcn.2007.01.011. http://dx.doi.org/10.1016/j.mcn.2007.01.011. [DOI] [PubMed] [Google Scholar]
- Savary S, Denizot F, Luciani M, Mattei M, Chimini G. Molecular cloning of a mammalian ABC transporter homologous to Drosophila white gene. Mamm Genome Off J Int Mamm Genome Soc. 1996;7:673–676. doi: 10.1007/s003359900203. [DOI] [PubMed] [Google Scholar]
- Sparrow CP, Baffic J, Lam MH, Lund EG, Adams AD, Fu X, Hayes N, Jones AB, Macnaul KL, Ondeyka J, Singh S, Wang J, Zhou G, Moller DE, Wright SD, Menke JG. A potent synthetic LXR agonist is more effective than cholesterol loading at inducing ABCA1 mRNA and stimulating cholesterol efflux. J Biol Chem. 2002;277:10021–10027. doi: 10.1074/jbc.M108225200. http://dx.doi.org/10.1074/jbc.M108225200. [DOI] [PubMed] [Google Scholar]
- Steward O, Cotman CW, Lynch GS. Re-establishment of electrophysiologically functional entorhinal cortical input to the dentate gyrus deafferented by ipsilateral entorhinal lesions: innervation by the contralateral entorhinal cortex. Exp Brain Res. 1973;18:396–414. doi: 10.1007/BF00239108. [DOI] [PubMed] [Google Scholar]
- Steward O, Vinsant SL, Davis L. The process of reinnervation in the dentate gyrus of adult rats: an ultrastructural study of changes in presynaptic terminals as a result of sprouting. J Comp Neurol. 1988;267:203–210. doi: 10.1002/cne.902670205. http://dx.doi.org/10.1002/cne.902670205. [DOI] [PubMed] [Google Scholar]
- Tachikawa M, Watanabe M, Hori S, Fukaya M, Ohtsuki S, Asashima T, Terasaki T. Distinct spatio-temporal expression of ABCA and ABCG transporters in the developing and adult mouse brain. J Neurochem. 2005;95:294–304. doi: 10.1111/j.1471-4159.2005.03369.x. http://dx.doi.org/10.1111/j.1471-4159.2005.03369.x. [DOI] [PubMed] [Google Scholar]
- Vanmierlo T, Rutten K, Dederen J, Bloks VW, van Vark-van der Zee LC, Kuipers F, Kiliaan A, Blokland A, Sijbrands EJG, Steinbusch H, Prickaerts J, Lütjohann D, Mulder M. Liver X receptor activation restores memory in aged AD mice without reducing amyloid. Neurobiol Aging. 2011;32:1262–1272. doi: 10.1016/j.neurobiolaging.2009.07.005. http://dx.doi.org/10.1016/j.neurobiolaging.2009.07.005. [DOI] [PubMed] [Google Scholar]
- Vaughan AM, Oram JF. ABCG1 redistributes cell cholesterol to domains removable by high density lipoprotein but not by lipid-depleted apolipoproteins. J Biol Chem. 2005;280:30150–30157. doi: 10.1074/jbc.M505368200. http://dx.doi.org/10.1074/jbc.M505368200. [DOI] [PubMed] [Google Scholar]
- Vaughan AM, Oram JF. ABCA1 and ABCG1 or ABCG4 act sequentially to remove cellular cholesterol and generate cholesterol-rich HDL. J Lipid Res. 2006;47:2433–2443. doi: 10.1194/jlr.M600218-JLR200. http://dx.doi.org/10.1194/jlr.M600218-JLR200. [DOI] [PubMed] [Google Scholar]
- Veinbergs I, Mante M, Jung MW, Van Uden E, Masliah E. Synaptotagmin and synaptic transmission alterations in apolipoprotein E-deficient mice. Prog Neuropsychopharmacol Biol Psychiatry. 1999;23:519–531. doi: 10.1016/s0278-5846(99)00013-5. [DOI] [PubMed] [Google Scholar]
- Venkateswaran A, Laffitte BA, Joseph SB, Mak PA, Wilpitz DC, Edwards PA, Tontonoz P. Control of cellular cholesterol efflux by the nuclear oxysterol receptor LXRα. Proc Natl Acad Sci. 2000;97:12097–12102. doi: 10.1073/pnas.200367697. http://dx.doi.org/10.1073/pnas.200367697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahrle SE, Jiang H, Parsadanian M, Kim J, Li A, Knoten A, Jain S, Hirsch-Reinshagen V, Wellington CL, Bales KR, Paul SM, Holtzman DM. Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. J Clin Invest. 2008;118:671–682. doi: 10.1172/JCI33622. http://dx.doi.org/10.1172/JCI33622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahrle SE, Jiang H, Parsadanian M, Legleiter J, Han X, Fryer JD, Kowalewski T, Holtzman DM. ABCA1 is required for normal central nervous system ApoE levels and for lipidation of astrocyte-secreted apoE. J Biol Chem. 2004;279:40987–40993. doi: 10.1074/jbc.M407963200. http://dx.doi.org/10.1074/jbc.M407963200. [DOI] [PubMed] [Google Scholar]
- Wang N, Yvan-Charvet L, Lütjohann D, Mulder M, Vanmierlo T, Kim TW, Tall AR. ATP-binding cassette transporters G1 and G4 mediate cholesterol and desmosterol efflux to HDL and regulate sterol accumulation in the brain. FASEB J Off Publ Fed Am Soc Exp Biol. 2008;22:1073–1082. doi: 10.1096/fj.07-9944com. http://dx.doi.org/10.1096/fj.07-9944com. [DOI] [PubMed] [Google Scholar]
- Wójcicka G, Jamroz-Wiśniewska A, Horoszewicz K, Bełtowski J. Liver X receptors (LXRs). Part I: structure, function, regulation of activity, and role in lipid metabolism. Postȩpy Hig Med Dośw. 2007;61:736–759. (Online) [PubMed] [Google Scholar]
- Xu J. Characterization of oligomeric human half-ABC transporter ATP-binding cassette G2. J Biol Chem. 2004;279:19781–19789. doi: 10.1074/jbc.M310785200. http://dx.doi.org/10.1074/jbc.M310785200. [DOI] [PubMed] [Google Scholar]