

Abstract
Rapid changes in microtubule (MT) polymerization dynamics affect regional activity of small GTPases RhoA and Rac1, which play a key role in the regulation of actin cytoskeleton and endothelial cell (EC) permeability. This study tested the role of End Binding protein-1 (EB1) in the mechanisms of increased and decreased EC permeability caused by thrombin and hepatocyte growth factor (HGF) and mediated by RhoA and Rac1 GTPases, respectively. Stimulation of human lung EC with thrombin inhibited peripheral MT growth, which was monitored by morphological and biochemical evaluation of peripheral MT and the levels of stabilized MT. In contrast, stimulation of EC with HGF promoted peripheral MT growth and protrusion of EB1-positive MT plus ends to the EC peripheral submembrane area. EB1 knockdown by small interfering RNA did not affect partial MT depolymerization, activation of Rho signaling, and permeability response to thrombin, but suppressed the HGF-induced endothelial barrier enhancement. EB1 knockdown suppressed HGF-induced activation of Rac1 and Rac1 cytoskeletal effectors cortactin and PAK1, impaired HGF-induced assembly of cortical cytoskeleton regulatory complex (WAVE-p21Arc-IQGAP1), and blocked HGF-induced enhancement of peripheral actin cytoskeleton and VE-cadherin-positive adherens junctions. Altogether, these data demonstrate a role for EB1 in coordination of MT-dependent barrier enhancement response to HGF, but show no involvement of EB1 in acute increase of EC permeability caused by the barrier disruptive agonist. The results suggest that increased peripheral EB1 distribution is a critical component of the Rac1-mediated pathway and peripheral cytoskeletal remodeling essential for agonist-induced EC barrier enhancement.
Keywords: pulmonary endothelium, permeability, microtubule dynamics, cytoskeleton, GTPase
1. INTRODUCTION
Preservation of the endothelial cell (EC) peripheral actin cytoskeleton and cell adhesive complexes is important for the maintenance of vascular barrier integrity. In turn, activation of additional barrier enhancing mechanisms is crucial for prevention of catastrophic consequences of uncontrolled vascular leakiness in the lung or other organs caused by bacterial pathogens, excessive mechanical forces, or cytokine storm during sepsis or trauma [1–4]. Activation of small GTPase RhoA and its downstream target Rho-associated kinase (Rho-kinase) may be induced by various agonists, pathologic mechanical forces, or inflammatory mediators and leads to vascular EC barrier dysfunction (see [5, 6] for review). In contrast to RhoA-mediated mechanism of EC cytoskeletal contraction and increased permeability, the enhancement of EC barrier by agonists such as prostacyclin, sphingosine 1-phosphate, or hepatocyte growth factor (HGF) involves activation of Rac1 and Rap1 GTPases which stimulate cortical actin polymerization, peripheral cytoskeletal remodeling, assembly of endothelial VE-cadherin-based adherens junction complexes and strengthening of cell-cell junctions (reviewed in [7, 8].
Although microtubules (MT) are not directly involved in the physical maintenance of EC barrier, increasing evidence suggests that dynamic changes in MT polymerization dynamics play an important signaling role in control of EC permeability. Complete or partial disassembly of MT by plant-derived alkaloids [9] or inflammatory mediators [10] leads to release of MT-associated Rho-specific guanine nucleotide exchange factor GEF-H1 [11, 12], activation of Rho signaling, Rho-kinase dependent microfilament reorganization, actomyosin contraction, and EC permeability [12, 13]. This mechanism represents a signaling crosstalk between MT and actin cytoskeleton involved in the regulation of EC barrier. Barrier disruptive agonists prevent MT growth and destabilize MT by targeting the proteins-regulators of MT polymerization and MT stability. For example, thrombin causes Rho-dependent phosphorylation of tau and dephosphorylation of stathmin, leading to disassembly of peripheral MT network [13, 14].
In contrast to EC barrier disruptive mechanisms activated by MT depolymerization, recent studies revealed a new mechanism of agonist-induced EC barrier enhancement, which required increased MT peripheral growth. Using HGF as a model agonist, which enhances basal EC barrier properties by activating Rac1 pathway [15, 16], we found that HGF promoted peripheral MT growth and MT protrusion to the EC peripheral compartment [17]. HGF-induced MT growth led to translocation of the MT-associated Rac1-specific GEF Asef to the cell periphery and local stimulation of Rac1 signaling [18].
The microtubule tip-tracking End Binding protein-1 (EB1) binds to growing plus ends of microtubules and promotes MT polymerization. EB1 does not track MT plus ends processively and instead, is exchanged rapidly at the MT tips [19]. EB1, along with another end-binding protein, CLASP2, provides a regional linkage of microtubules to actin filaments through IQGAP1, which is important for cell migration [20]. However, the role of EB1 as a potential transmitter of RhoA and Rac1 mediated signaling in vascular endothelium remains unclear. This study used cell imaging, molecular and biochemical approaches to test the involvement of EB1 in agonist-induced alterations of peripheral MT network and investigated specific role of EB1 in the control of permeability response to both, barrier enhancing and barrier disruptive agonists.
2. MATERIALS AND METHODS
2.1. Cell culture and reagents
Human HGF was obtained from R&D Systems (Minneapolis, MN). Texas Red-conjugated phalloidin, Alexa Fluor 594, and Alexa Fluor 488 were purchased from Molecular Probes (Eugene, OR). End-Binding protein-1 (EB1) p21Arc, Wave, and Rac1 antibodies were purchased from BD Transduction Laboratories (San Diego, CA); Rho, VE-cadherin, IQGAP1 antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA); phospho-cortactin and p-MYPT1antibodies were from Millipore (Billerica, MA); p-MLC and p-PAK antibodies was obtained from Cell Signaling (Beverly, MA). Unless otherwise specified, all biochemical reagents were obtained from Sigma (St. Louis, MO). Human pulmonary artery endothelial cells (HPAEC) were obtained from Lonza (East Rutherford, NJ) and used for experiments at passages 5–7.
2.2. Si-RNA and DNA transfections
Pre-designed EB1-specific siRNA of standard purity was ordered from Ambion (Austin, TX). Transfection of EC with siRNA was performed as previously described [21]. Non-specific, non-targeting RNA (Dharmacon, Lafayette, CO) was used as a control treatment. Seventy-two hours after transfection, cells were harvested and used for experiments. Plasmid encoding GFP-EB1 and GFP-cortactin were purchased from Addgene (Cambridge, MA). Transient transfections of HPAEC were carried out using PolyJet reagent from Signagen Laboratories (Rockville, MD) as recommended by the manufacturer [13, 22]. After 24 hr of transfection, pulmonary EC were treated with either vehicle or HGF and used for experiments.
2.3. Analysis of EC permeability
Measurements of transendothelial electrical resistance (TER) across HPAEC were performed using the electrical cell-substrate impedance sensing system (ECIS) (Applied Biophysics, Troy, NY) as described [13, 23].
2.4. Imaging studies
Endothelial monolayers plated on glass cover slips were subjected to immunofluorescence staining with Texas Red phalloidin to visualize F-actin or VE-cadherin [22, 24]. For microtubule quantification, cells were fixed with 100% methanol cooled to −20 °C, and immunostaining was carried out with α-tubulin or EB1 antibodies [25, 26]. Briefly, after the cell boundaries were outlined, the concentric outline shapes reduced to 70% were applied to the images to mark peripheral (outer 30% of diameter) and central (inner 70% of diameter) regions. The integrated fluorescence density in the peripheral area was measured using MetaMorph software and was calculated as a percentage of the integrated fluorescence density in the total cell area. The results were normalized in each experiment. Similar methods were applied to EB1 quantification in fixed cells except that EB1 immunoactivity was manually counted and results were not normalized. Minimum 10 cells per condition, in three experimental repeats were analyzed. For time-lapse microtubule plus end tracking, cells were seeded on MatTek dishes (MatTek, Ashland, MA) and transfected with GFP-EB1. Images were acquired with 100x NA 1.45 oil objective in a 3I Marianas Yokogawa-type Spinning Disk Confocal system equipped with a CO2 chamber and a heated stage. Time-lapse images were taken with 2 second intervals for 60 seconds. 20 consecutive images in each condition were used for projection analysis using ImageJ software. For tracking analysis, EB1 in the cell margin area (2–10 μm from cell border) was tracked with the Manual Tracking plug-in in ImageJ software. The median track length was calculated using Excel software. For live cell imaging of cortactin, cells were plated on MatTek dishes (MatTek, Ashland, MA) and transfected with GFP-cortactin. Images were acquired with 100x NA 1.45 oil objective in a 3I Marianas Yokogawa-type Spinning Disk Confocal system equipped with a CO2 chamber and a heated stage. Time-lapse images were taken with 15 second intervals for 10 min. Images were processed with Image J software (National Institute of Health, Washington, USA) and Adobe Photoshop CS5 (Adobe Systems, San Jose, CA) software. Quantitative analysis of HGF-induced cortactin peripheral accumulation was performed as described above at peripheral area corresponding to 10% of cell radius in control and EB1-depleted cells.
2.5. GTPase activation assays
Rac1 and RhoA activation was evaluated in pulldown assays using agarose beads with immobilized PAK1-PBD and rhotekin-RBD, respectively [23]. In brief, after stimulation, cell lysates were collected, and GTP-bound Rac1 or RhoA were captured using pull-down assays with immobilized PAK1-PBD or Rhotekin-RBD, respectively. The levels of activated small GTPases as well as total Rac1 and RhoA content were evaluated by western blot analysis.
2.6. Microtubule fractionation, coimmunoprecipitation, and analysis og protein phosphorylation
Microtubule fractionation: MT-enriched fractions were isolated as previously described [13]. Briefly, cells were incubated with buffer containing PEM (100 mM Pipes pH 6.75, 1 mM EGTA, 1 mM MgSO4, pH 6.75), 0.5% NP-40 (10 min, RT). Cytosolic fraction containing soluble tubulin was collected by centrifugation (12000 rpm, 15 min, RT). The attached cells containing polymerized MT were incubated on ice for 30 min to induce microtubule depolymerization and tubulin release into the soluble fraction. Cells were scraped in PEM; the cell debris was removed by centrifugation (2000g, 2 min, 4°C). Protein extracts were separated by SDS-PAGE. Coimmunioprecipitation: After agonist stimulation, cells were washed in cold phosphate buffered saline (PBS) and lysed on ice with cold TBS-NP40 lysis buffer (20 mM Tris pH 7.4, 150 mM NaCl, 1% NP40) supplemented with protease and phosphatase inhibitor cocktails (Roche, Indianapolis, IN). Clarified lysates were then incubated with antibodies to IQGAP1 (BD Transduction Laboratories, San Diego, CA) overnight at 4°C, washed 3–4 times with TBS-NP40 lysis buffer, and the complexes were analyzed by Western blotting using appropriate antibodies. Protein phosphorylation: For analysis of protein phosphorylation profile, cells were stimulated, then lysed, and protein extracts were separated by SDS-PAGE, transferred to polyvinylidene fluoride (PVDF) membrane, and probed with specific antibodies. Equal protein loading was verified by reprobing membranes with antibody to β-tubulin or specific protein of interest. The relative intensities of immunoreactive protein bands (RDU, relative density units) were analyzed and quantified by scanning densitometry using Image Quant software (Molecular Dynamics, Sunnyvale, CA).
2.7. Statistical analysis
Results are expressed as means ± SD of three to five independent experiments. Stimulated samples were compared to controls by unpaired Student’s t-tests. For multiple-group comparisons, a one-way variance analysis (ANOVA), followed by the post hoc Fisher’s test, were used. P<0.05 was considered statistically significant.
3. RESULTS
3.1. Contrasting effects of HGF and thrombin on microtubule arrangement, stability, growth dynamics, and peripheral pool of EB1
To assess effects of HGF and thrombin on peripheral MT density, pulmonary EC were treated with vehicle, thrombin or HGF, then fixed with methanol and subjected to immunofluorescence staining with antibody to α-tubulin (Figure 1A). Quantitative analysis of MT structure showed increased fraction of peripheral MT in HGF-treated EC and significantly decreased fraction of peripheral MT in thrombin-treated EC (Figure 1B). HGF treatment increased the fraction of polymerized MT (Figure 1C) and the levels of acetylated tubulin representing a pool of stabilized MT (Figure 1D), while thrombin stimulation significantly decreased both, the fraction of polymerized MT and acetylated tubulin (Figure 1CD). To assess effects of HGF and thrombin on the density of peripheral EB1 positive MT, methanol-fixed pulmonary EC were stained with antibody to EB1, the protein that tracks the growing plus end of microtubules. HGF treatment increased the pool of peripheral EB1-positive MT (Figure 2A, see also insets with higher magnification images). In contrast, thrombin challenge significantly decreased the fraction of peripheral EB1 positive MT. Quantitative analysis of immunofluorescence data is presented in Figure 2B. Staining with VE-cadherin antibody was used to visualize the cell borders. Effects of HGF and thrombin on MT dynamics were further examined using a live imaging approach. For this purpose, EC were transfected with GFP-tagged EB1. EB1 tracking in live cells was performed by live videomicroscopy, and projection images were generated as described in the Methods section. HGF stimulation increased, while thrombin treatment decreased the length of EB1 tracks, which represent episodes of uninterrupted microtubule growth (Figure 3A). Quantitative analysis of median track length measured in several single cells before and after agonist challenge is presented in Figure 3B.
Figure 1. Effects of thrombin and HGF on microtubule dynamics and stability.

A and B - HPAEC grown on coverslips were stimulated with vehicle, thrombin (0.3 U/ml, 10 min), or HGF (50 ng/ml, 10 min) followed by immunofluorescence staining with an antibody against α-tubulin (A). Bar graph depicts results of quantitative analysis of peripheral microtubules in methanol-fixed EC; *P<0.05; n=4; 6 images from each experiment (B). C - HPAEC were stimulated with thrombin or HGF followed by fractionation assay. Content of polymerized tubulin in MT-enriched fraction and depolymerized tubulin in cytosolic fraction was determined by western blotting with α-tubulin antibodies. Results are represented as mean ± SD; *P<0.05; n=4. D – Effect of HGF and thrombin stimulation on the pool of stable MT was evaluated by western blot of whole cell lysates with antibody against acetylated tubulin. Equal tubulin content was confirmed by probing of membranes for α-tubulin. Results are representative of three independent experiments. Results are represented as mean ± SD; *P<0.05; n=4.
Figure 2. Effects of thrombin and HGF on EB1-positive MT tips distribution.

A and B - HPAEC grown on coverslips were stimulated with vehicle, thrombin (0.3 U/ml, 10 min), or HGF (50 ng/ml, 10 min) followed by fixation with methanol and double immunofluorescence staining for EB1 (green) and Ve-cadherin (red). Insets show high magnification images of cell periphery areas with EB1-positive microtubule tips. VE-cadherin staining outlines cell borders. Results are representative of five independent experiments (A). Bar graph depicts results of quantitative analysis of peripheral EB1; *P<0.05; n=3; 10 images from each experiment (B).
Figure 3. Effects of thrombin and HGF on MT growth.

A and B - Live cell imaging of HPAEC transfected with GFP-EB1 and stimulated with thrombin (0.3 U/ml, 10 min) or HGF (50 ng/ml, 10 min). Projection analysis of 20 consecutive images before and after treatment is shown (A). Graphs depict quantification of GFP-EB1 track length. Each pair of dots represents the median track length in a cell before and after thrombin or HGF stimulation (B). Insets: Bar graphs represent agonist-induced changes in EB1 tracks. Data are expressed as mean ± SD of four independent experiments, 6–8 cells for each experiment; *p<0.05.
3.2. Role of EB1 in agonist-induced EC permeability response
The role of EB1 in the agonist-induced EC barrier control was tested in cell monolayers with siRNA-induced EB1 knockdown. EB1 protein depletion was verified by immunoblotting of total cell lysates with antibody to EB1 (Figure 4). The results were compared to control cells transfected with non-specific RNA. Measurements of transendothelial electrical resistance (TER) were performed to monitor agonist-induced changes in EC permeability over time.
Figure 4. Effect of EB1 knockdown on EC permeability response by thrombin and HGF.

HPAEC grown on microelectrodes were transfected with 150 nM EB1-specific siRNA or non-specific RNA (ns-RNA) for 72 hrs. A - C - At the time point indicated by arrow, EC were stimulated with thrombin (0.3 U/ml) (A), TNFα (10 ng/ml) (B), or HGF (50 ng/ml) (C), and measurements of transendothelial resistance (TER) were performed over time. Presented TER measurements are normalized average resistance values +/− SE from three independent readings in one experiment, the data are representative of four independent experiments. D - Bar graphs depict results of quantitative analysis of permeability data; n=4, *P<0.05. Inset: western blot analysis of siRNA-induced EB1 protein depletion in permeability assays.
Experiments with measurements of TER in EC monolayers showed that siRNA-induced depletion of EB1 did not significantly affect the basal EC barrier properties when compared with control monolayers transfected with non-specific RNA (1217+/−108 Ohm vs 1189+/−144 Ohm, respectively). EB1 depletion also did not affect the acute increase in EC permeability at the peak of response to thrombin (Figure 4A). EC treatment with another physiological barrier-compromising mediator showed that similar to thrombin stimulation EB1 knockdown did not affect the barrier disruptive effect of TNFα (Figure 4B). In contrast, EB1 knockdown attenuated the HGF-induced TER increase (Figure 4C). Quantitative analysis of permeability experiments summarizes effects of EB1 depletion on EC permeability response to barrier-protective and barrier-disruptive agonists (Figure 4D).
3.3. Effect of EB1 knockdown on thrombin-induced F-actin remodeling, disruption of adherens junctions, and Rho signaling
EC treated with non-targeting RNA or EB1-specific siRNA were treated with thrombin, and thrombin-induced actin stress fiber formation and disruption of adherens junctions was monitored by immunofluorescence staining with Texas Red-labeled phalloidin and antibody to VE-cadherin, respectively. EB1 knockdown did not affect the thrombin-induced F-actin remodeling and disruption of VE-cadherin-positive adherens junctions (Figure 5AB). EB1 protein depletion was verified by double-immunofluorescence staining with EB1 and F-actin or EB1 and VE-cadherin antibodies (Figure 5AB, insets). In agreement with the absence of effect on thrombin-induced EC hyper-permeability, EB1 knockdown did not influence the thrombin-induced activation of RhoA measured in Rho-GTP pulldown assay and phosphorylation of RhoA downstream targets, myosin light chain phosphatase (MYPT1) and myosin light chain (MLC) (Figure 5C).
Figure 5. Effect of EB1 knockdown on thrombin-induced EC barrier disruption and Rho pathway activation.


Human pulmonary EC were transfected with EB1-specific or non-specific siRNA. A and B - Cells plated onto glass coverslips were treated with thrombin (0.3 U/ml, 10 min). Effects of EB1 depletion on thrombin-induced actin cytoskeleton (A) and cell junction (B) remodeling was monitored by immunofluorescence staining with Texas-Red phalloidin and VE-cadherin antibody, respectively. Insets: control and EB1-depleted EC were stained for EB1 and F-actin (A) or EB1 and VE-cadherin actibodies (B). C - Thrombin-induced Rho activation was assessed by RhoGTP pulldown assay. The content of activated Rho was normalized to the total Rho content in cell lysates. SiRNA-induced EB1 protein depletion was confirmed by western blotting. D - Western blot analysis of thrombin-induced MYPT1 and MLC phosphorylation. Probing for β-actin was used as a normalization control. Bar graphs represent the results of quantitative densitometry of western blot panels in control and treated HPAEC monolayers. Data are expressed as mean ± SD of three independent experiments; *P<0.05.
3.4. Effect of EB1 knockdown on HGF-induced remodeling of peripheral actin cytoskeleton, adherens junctions, and Rac signaling
Morphological analysis of EC monolayers with siRNA-induced protein depletion of EB1 showed significant attenuation of HGF-induced peripheral F-actin enhancement (Figure 6A) and decreased VE-cadherin-positive staining of adherens junctions (Figure 6B). EB1 knockdown also attenuated the HGF-induced activation of Rac1 (Figure 6C) and its downstream targets, PAK1 and cortactin (Figure 6D). Impaired activation of Rac1 signaling in the HGF-stimulated EC with EB1 knockdown was accompanied by suppressed translocation of cortactin and VE-cadherin to the cell membrane fraction (Figure 6D), the events essential for activation of subcortical actin cytoskeletal remodeling and strengthening of endothelial adherens junctions [27, 28]. EB1 involvement in the cortical actin cytoskeletal dynamics induced by HGF was further evaluated in live microscopy studies using endothelial cells expressing GFP-cortactin. HGF induced expansion of peripheral cortactin positive cell areas and formation of cortactin-rich lamellipodia in control cells treated with non-specific RNA (Figure 6F, top panels). In contrast, HGF-induced peripheral remodeling was abolished in the cells with EB1 knockdown (Figure 6F, bottom panels).
Figure 6. Effect of EB1 knockdown on HGF-induced EC barrier enhancement and activation of Rac1 signaling.


Human pulmonary EC were transfected with EB1-specific or non-specific siRNA. A and B - Cells grown on glass coverslips were treated with HGF (50 ng/ml, 10 min). Effects of EB1 depletion on HGF-induced actin cytoskeleton (A) and cell junction (B) remodeling was monitored by immunofluorescence staining with Texas-Red phalloidin and VE-cadherin antibody, respectively. C - Rac1 activation was determined by Rac GTPase pulldown assay. The content of activated Rac1 was normalized to the total protein content in EC lysates. SiRNA-induced EB1 protein depletion was confirmed by western blotting. D - HGF-induced PAK and cortactin phosphorylation in control and EB1-depleted EC was evaluated by western blot. Equal protein loading was confirmed by probing with β-actin antibody. E - Cells were treated with HGF for 5 or 10 min, and membrane translocation of cortactin and VE-cadherin was analyzed by western blot analysis of EC membrane fractions. Protein content in corresponding total cell lysates was used as a normalization control. Bar graphs represent the results of quantitative densitometry of western blot panels in control and treated HPAEC monolayers. Data are expressed as mean ± SD of three independent experiments; *p<0.05. F - Live cell imaging of the cells expressing GFP-cortactin. Snapshots depict HGF-induced cortical dynamics at the cell periphery of control and EB1-depleted cells. Arrows and higher magnification insets show peripheral cortactin accumulation and lamellipodia formation upon HGF treatment. Insets show cortactin accumulation at cell periphery after 3 min of HGF stimulation in control, but not EB1-depleted cells. The bar graph depicts quantitative image analysis of GFP-cortactin immunoreactivity at the cell cortical compartment. Results are average +/− SD of six microscopy fields per condition.
3.5. EB1 knockdown attenuates HGF-induced assembly of WAVE-p21Arc-IQGAP1 cytoskeletal complex
The results described above suggest that HGF-induced enhancement of cortical actin cytoskeleton involves cortactin activation and its accumulation at the cell periphery. Our published studies also show HGF-induced interactions between cortactin, EB1, and multifunctional scaffold protein IQGAP1 [17]. Next experiments tested the role of EB1 in the HGF-induced peripheral accumulation of IQGAP1-dependent activators of actin polymerization, WAVE and p21Arc [29, 30], and assembly of WAVE-p21Arc-IQGAP1 complex. Imaging studies performed in human pulmonary endothelium revealed increased p21-Arc accumulation at the cell periphery upon HGF stimulation, which was abolished in EC with EB1 knockdown Figure 7A). Next, interactions between endogenous IQGAP1, p21Arc, and WAVE in the lung EC were evaluated by coimmunoprecipitation assays. HGF induced association of IQGAP1 with p21Arc and WAVE (Figure 7B). These effects were inhibited in EB1-depleted cells. Collectively, these results suggest that EB1 is critical for the recruitment of regulators of cytoskeletal remodeling, IQGAP1, p21Arc and WAVE in response to HGF treatment.
Figure 7. Effect of EB1 knockdown on HGF-mediated regulation of actin remodeling.
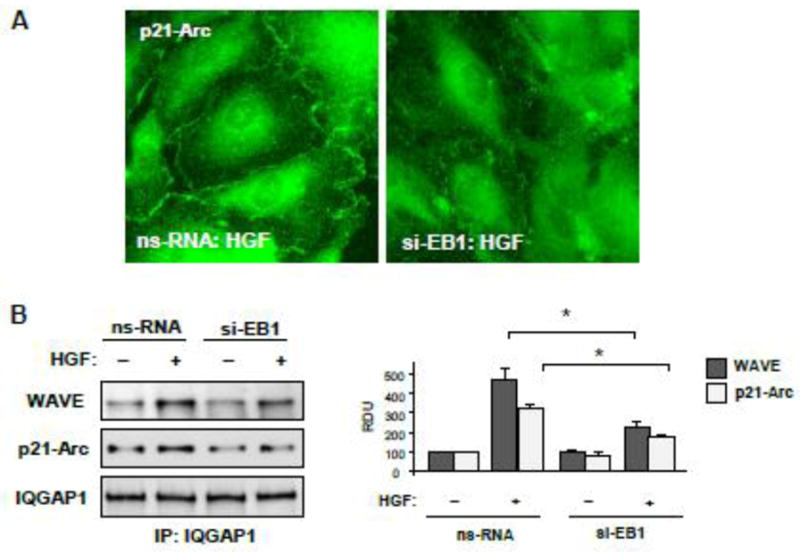
Human pulmonary EC were transfected with EB1-specific or non-specific siRNA. A - Cells grown on glass coverslips were stimulated with HGF (50 ng/ml, 10 min). Intracellular redistribution of p21-Arc was examined by immunofluorescence staining with corresponding antibody. Shown are representative results of three independent experiments. B – Control and EB1-depleted EC were used for co-immunoprecipitation assays with IQGAP1 antibody followed by western blot detection of WAVE and p21-Arc. Bar graphs depict quantitative densitometry analysis of immunoblotting data. Results are represented as mean ± SD; *P<0.05; n=4.
4. DISCUSSION
The major finding of this study is a demonstration of EB1 involvement in the control of agonist-induced EC permeability. This study uncovered specific role of EB1 in control of MT extension and activation of Rac-specific peripheral actin and cell junction remodeling, leading to enhancement of endothelial barrier. In contrast, EB1 was not involved in acute permeability increase caused by barrier-disruptive agonists such as thrombin or TNFα. Both agonists induce destabilization and disassembly of peripheral MT [13, 24, 31], which per se further stimulates RhoA-dependent mechanisms of EC barrier dysfunction. As the results of this study show, these barrier-disruptive effects were not influenced by EB1 knockdown.
EB1-mediated interactions with growing MTs are important to coordinate cell shape changes and directed migration of epithelial cells placed in three-dimensional environment [32]. However, precise mechanisms of EB1-dependent regulation of cell motility and other functional responses by vascular endothelium, such as agonist-induced vascular endothelial permeability remain to be investigated. We recently found that HGF stimulation caused pronounced peripheral MT growth in pulmonary EC. Growing MT tips reached the cell cortex, where they became captured by multifunctional scaffold IQGAP1 [17]. These findings suggest that EB1 is required for peripheral targeting of signaling molecules, which leads to activation of cytoskeletal and cell junction remodeling.
We observed the striking inhibitory effect of EB1 knockdown on HGF-induced increase in transendothelial electrical resistance, which was linked to suppression of HGF-induced Rac1 activation and Rac1-dependent cytoskeletal responses. Morphological analysis of EC cultures using immunofluorescence staining showed that EB1 knockdown attenuated HGF-induced cortical actin cytoskeletal remodeling, peripheral accumulation of cytoskeletal Rac effectors cortactin and p21Arc, and impaired enhancement of VE-cadherin positive adherens junctions. Control experiments showed that EB1 knockdown did not noticeably affect the basal EC barrier function, F-actin and VE-cadherin distribution. Furthermore, we found that HGF-induced association of IQGAP1 with its cytoskeletal partners WAVE and p21Arc is regulated by EB1. Knockdown of EB1 abolishes accumulation of a member of Arp2/3 complex, p21-Arc at the periphery of HGF-stimulated EC and assembly of p21-Arc/WAVE/IQGAP1 complex. These effects are consistent with the known Rac1- mechanism of Arc/WAVE complex activation and formation of branched F-actin network [33, 34]. This mechanism is critical for the enhancement of peripheral actin cytoskeleton and EC barrier function, and our results show additional regulation of this mechanism by EB1.
Our results show that in contrast to HGF-stimulated conditions, EB1 is not involved in the acute phase of thrombin-induced EC permeability mediated by Rho mechanism. EB1 knockdown did not affect the acute phase of EC barrier dysfunction and was without effect on thrombin-induced activation of RhoA and its downstream signaling. This observation may be explained by thrombin-induced inhibition of MT growth and partial depolymerization of peripheral MT network, the process which apparently is not controlled by EB1. On the other hand, MT dynamics is intimately involved in control of Rho signaling by binding the RhoA-specific guanine nucleotide exchange factor GEF-H1 [11, 12, 35]. Accordingly, alterations in the pool of MT-bound GEF-H1 are directly linked with Rho activation and have been shown to regulate barrier properties of epithelial [36, 37] and endothelial [22, 24, 38] monolayers.
Interestingly, EB1 presence becomes increasingly important during EC monolayer recovery after thrombin-induced barrier disruption, which is associated with activation of Rac1/Cdc42 dependent mechanisms [28, 39, 40]. Indeed, recovery of EC barrier properties after thrombin challenge was delayed in the cells with EB1 knockdown (Figure 4A). These results may be explained by EB1 involvement in re-activation of MT growth and elongation of the MT to the cell cortical compartment. We speculate that the role of such MT elongation is similar to the MT remodeling caused by HGF: growing MT tips reaching the cell cortex become captured by linker proteins and may unload signaling molecules, such as Rac-specific GEFs shown to stimulate Rac-dependent mechanisms of cytoskeletal remodeling and EC barrier restoration.
In conclusion, this study demonstrates for the first time the differential role of EB1 in the mediation of pulmonary EC permeability responses to barrier-enhancing and barrier-disruptive agonists. We speculate that EB1 involvement in HGF-induced EC barrier enhancement includes EB1-dependent capturing of microtubules to the cell cortex via EB1-IQGAP1-cortactin interaction, which is required for local activation of Rac signaling, increased interaction of Rac-dependent actin-binding proteins - activators of cortical actin polymerization with IQGAP1, and enhancement of EC monolayer barrier. This mechanism may be also involved in the EC barrier recovery after stimulation with barrier disruptive agonists such as thrombin. Therefore, pharmacological control of MT dynamics during EC barrier recovery may be an important aspect of future strategies aimed at the improved restoration of vascular function in variety of syndromes with increased vascular leak.
Highlights.
Thrombin caused disassembly of peripheral MT, while HGF promoted MT growth
EB-1 knockdown did not affect thrombin-induced Rho activation and permeability
EB-1 knockdown suppressed HGF-induced Rac1 activation and cytoskeletal remodeling
Increased EB-1 peripheral distribution is essential for EC barrier enhancement
Acknowledgments
This work was supported by the grants: HL107920 and HL130431 from the National Heart, Lung, and Blood Institute, and GM114171 from the National Institute of General Medical Sciences.
Non-standard Abbreviations
- EB1
End-Binding protein-1
- EC
endothelial cells
- GEF
guanine nucleotide exchange factor
- HPAEC
human pulmonary artery endothelial cells
- MLC
myosin light chain
- MT
microtubules
- MYPT1
myosin light chain phosphatase
- ns-RNA
non-specific RNA
- TER
transendothelial electrical resistance
- XPerT
express permeability testing assay
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Frank JA, Matthay MA. Crit Care. 2003;7(3):233–241. doi: 10.1186/cc1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tremblay LN, Slutsky AS. Intensive Care Med. 2006;32(1):24–33. doi: 10.1007/s00134-005-2817-8. [DOI] [PubMed] [Google Scholar]
- 3.Maniatis NA, Kotanidou A, Catravas JD, Orfanos SE. Vascul Pharmacol. 2008;49(4–6):119–133. doi: 10.1016/j.vph.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deng JC, Standiford TJ. Compr Physiol. 2011;1(1):81–104. doi: 10.1002/cphy.c090011. [DOI] [PubMed] [Google Scholar]
- 5.Beckers CM, van Hinsbergh VW, van Nieuw Amerongen GP. Thromb Haemost. 2010;103(1):40–55. doi: 10.1160/TH09-06-0403. [DOI] [PubMed] [Google Scholar]
- 6.Spindler V, Schlegel N, Waschke J. Cardiovasc Res. 2010;87(2):243–253. doi: 10.1093/cvr/cvq086. [DOI] [PubMed] [Google Scholar]
- 7.Pannekoek WJ, Kooistra MR, Zwartkruis FJ, Bos JL. Biochim Biophys Acta. 2009;1788(4):790–796. doi: 10.1016/j.bbamem.2008.12.010. [DOI] [PubMed] [Google Scholar]
- 8.Birukov KG, Zebda N, Birukova AA. Compr Physiol. 2013;3(1):429–484. doi: 10.1002/cphy.c100066. [DOI] [PubMed] [Google Scholar]
- 9.Verin AD, Birukova A, Wang P, Liu F, Becker P, Birukov K, Garcia JG. Am J Physiol Lung Cell Mol Physiol. 2001;281(3):L565–574. doi: 10.1152/ajplung.2001.281.3.L565. [DOI] [PubMed] [Google Scholar]
- 10.Shivanna M, Srinivas SP. Exp Eye Res. 2009;89(6):950–959. doi: 10.1016/j.exer.2009.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ren Y, Li R, Zheng Y, Busch H. J Biol Chem. 1998;273(52):34954–34960. doi: 10.1074/jbc.273.52.34954. [DOI] [PubMed] [Google Scholar]
- 12.Krendel M, Zenke FT, Bokoch GM. Nat Cell Biol. 2002;4(4):294–301. doi: 10.1038/ncb773. [DOI] [PubMed] [Google Scholar]
- 13.Birukova AA, Birukov KG, Smurova K, Adyshev DM, Kaibuchi K, Alieva I, Garcia JG, Verin AD. FASEB J. 2004;18(15):1879–1890. doi: 10.1096/fj.04-2328com. [DOI] [PubMed] [Google Scholar]
- 14.Song Y, Wong C, Chang DD. J Cell Biochem. 2000;80(2):229–240. doi: 10.1002/1097-4644(20010201)80:2<229::aid-jcb120>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 15.Birukova AA, Alekseeva E, Mikaelyan A, Birukov KG. FASEB J. 2007;21(11):2776–2786. doi: 10.1096/fj.06-7660com. [DOI] [PubMed] [Google Scholar]
- 16.Singleton PA, Salgia R, Moreno-Vinasco L, Moitra J, Sammani S, Mirzapoiazova T, Garcia JG. J Biol Chem. 2007;282(42):30643–30657. doi: 10.1074/jbc.M702573200. [DOI] [PubMed] [Google Scholar]
- 17.Tian Y, Tian X, Gawlak G, O’Donnell JJ, 3rd, Sacks DB, Birukova AA. Mol Cell Biol. 2014;34(18):3546–3558. doi: 10.1128/MCB.00248-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tian Y, Gawlak G, Shah AS, Higginbotham K, Tian X, Kawasaki Y, Akiyama T, Sacks DB, Birukova AA. J Biol Chem. 2015;290(7):4097–4109. doi: 10.1074/jbc.M114.620377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Akhmanova A, Steinmetz MO. Nat Rev Mol Cell Biol. 2008;9(4):309–322. doi: 10.1038/nrm2369. [DOI] [PubMed] [Google Scholar]
- 20.Watanabe T, Noritake J, Kakeno M, Matsui T, Harada T, Wang S, Itoh N, Sato K, Matsuzawa K, Iwamatsu A, Galjart N, Kaibuchi K. J Cell Sci. 2009;122(Pt 16):2969–2979. doi: 10.1242/jcs.046649. [DOI] [PubMed] [Google Scholar]
- 21.Singleton PA, Chatchavalvanich S, Fu P, Xing J, Birukova AA, Fortune JA, Klibanov AM, Garcia JG, Birukov KG. Circ Res. 2009;104(8):978–986. doi: 10.1161/CIRCRESAHA.108.193367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Birukova AA, Fu P, Xing J, Yakubov B, Cokic I, Birukov KG. Am J Physiol Lung Cell Mol Physiol. 2010;298(6):L837–848. doi: 10.1152/ajplung.00263.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Birukov KG, Bochkov VN, Birukova AA, Kawkitinarong K, Rios A, Leitner A, Verin AD, Bokoch GM, Leitinger N, Garcia JG. Circ Res. 2004;95(9):892–901. doi: 10.1161/01.RES.0000147310.18962.06. [DOI] [PubMed] [Google Scholar]
- 24.Birukova AA, Adyshev D, Gorshkov B, Bokoch GM, Birukov KG, Verin AA. Am J Physiol Lung Cell Mol Physiol. 2006;290(3):L540–548. doi: 10.1152/ajplung.00259.2005. [DOI] [PubMed] [Google Scholar]
- 25.Komarova Y, De Groot CO, Grigoriev I, Gouveia SM, Munteanu EL, Schober JM, Honnappa S, Buey RM, Hoogenraad CC, Dogterom M, Borisy GG, Steinmetz MO, Akhmanova A. J Cell Biol. 2009;184(5):691–706. doi: 10.1083/jcb.200807179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tian X, Tian Y, Sarich N, Wu T, Birukova AA. Faseb J. 2012;26(9):3862–3874. doi: 10.1096/fj.12-207746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee JF, Ozaki H, Zhan X, Wang E, Hla T, Lee MJ. Histochem Cell Biol. 2006;126(3):297–304. doi: 10.1007/s00418-006-0143-z. [DOI] [PubMed] [Google Scholar]
- 28.Kouklis P, Konstantoulaki M, Vogel S, Broman M, Malik AB. Circ Res. 2004;94(2):159–166. doi: 10.1161/01.RES.0000110418.38500.31. [DOI] [PubMed] [Google Scholar]
- 29.Briggs MW, Sacks DB. EMBO Rep. 2003;4(6):571–574. doi: 10.1038/sj.embor.embor867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Le Clainche C, Schlaepfer D, Ferrari A, Klingauf M, Grohmanova K, Veligodskiy A, Didry D, Le D, Egile C, Carlier MF, Kroschewski R. J Biol Chem. 2007;282(1):426–435. doi: 10.1074/jbc.M607711200. [DOI] [PubMed] [Google Scholar]
- 31.Petrache I, Birukova A, Ramirez SI, Garcia JG, Verin AD. Am J Respir Cell Mol Biol. 2003;28(5):574–581. doi: 10.1165/rcmb.2002-0075OC. [DOI] [PubMed] [Google Scholar]
- 32.Gierke S, Wittmann T. Curr Biol. 2012;22(9):753–762. doi: 10.1016/j.cub.2012.02.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miki H, Suetsugu S, Takenawa T. Embo J. 1998;17(23):6932–6941. doi: 10.1093/emboj/17.23.6932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ten Klooster JP, Evers EE, Janssen L, Machesky LM, Michiels F, Hordijk P, Collard JG. Biochem J. 2006;397(1):39–45. doi: 10.1042/BJ20051957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chang YC, Nalbant P, Birkenfeld J, Chang ZF, Bokoch GM. Mol Biol Cell. 2008;19(5):2147–2153. doi: 10.1091/mbc.E07-12-1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Birkenfeld J, Nalbant P, Yoon SH, Bokoch GM. Trends Cell Biol. 2008;18(5):210–219. doi: 10.1016/j.tcb.2008.02.006. [DOI] [PubMed] [Google Scholar]
- 37.Kakiashvili E, Speight P, Waheed F, Seth R, Lodyga M, Tanimura S, Kohno M, Rotstein OD, Kapus A, Szaszi K. J Biol Chem. 2009;284(17):11454–11466. doi: 10.1074/jbc.M805933200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tian X, Tian Y, Gawlak G, Sarich N, Wu T, Birukova AA. J Biol Chem. 2014;289(8):5168–5183. doi: 10.1074/jbc.M113.493924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Birukova AA, Tian X, Tian Y, Higginbotham K, Birukov KG. Mol Biol Cell. 2013;24(17):2678–2688. doi: 10.1091/mbc.E13-02-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tian X, Tian Y, Gawlak G, Meng F, Kawasaki Y, Akiyama T, Birukova AA. Mol Biol Cell. 2015;26(4):636–650. doi: 10.1091/mbc.E14-02-0725. [DOI] [PMC free article] [PubMed] [Google Scholar]