

Abstract
Oncolytic viruses (OVs), like oncolytic herpes simplex virus (oHSV), are genetically engineered to selectively replicate in and kill cancer cells, while sparing normal cells. Initial OV infection, cell death, and subsequent OV propagation within the tumor microenvironment leads to a cascade of host responses (innate and adaptive), reflective of natural anti-viral immune responses. These host-virus interactions are critical to the balance between OV activities, anti-viral immune responses limiting OV, and induction of anti-tumor immunity. The host response against oHSV is complex, multifaceted, and modulated by the tumor microenvironment and immunosuppression. As a successful pathogen, HSV has multiple mechanisms to evade such host responses. In this review, we will discuss these mechanisms and HSV evasion, and how they impact oHSV therapy.
Graphical abstract
Introduction
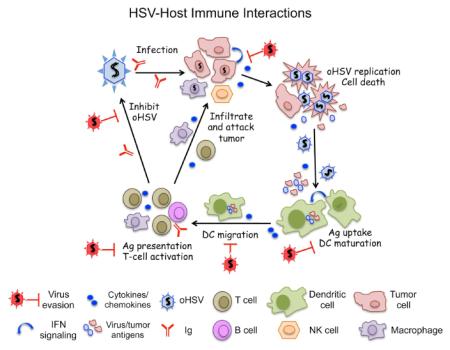
Oncolytic viruses (OVs) are a new and unique class of cancer therapeutics, with cancer cell selective replication, cytotoxicity, and spread, that is often coupled to anti-tumor immunity (immunovirotherapy) [1*,2]. Herpes simplex virus type 1 (HSV-1) was the first genetically engineered OV, originally developed for glioma [3]. An enveloped virus with a 152 kb DNA genome, HSV-1 is a human pathogen that is endowed with oncolytic activity by mutating non-essential genes involved in virus growth in post-mitotic cells (i.e., thymidine kinase, ribonucleotide reductase (ICP6)), counteracting innate anti-viral responses (i.e., γ34.5, ICP0, Us3) and pathogenicity (i.e., γ34.5, UL56, ribonucleotide reductase (ICP6)) [3]. In 2015, the FDA approved the first OV, an oncolytic HSV (oHSV), talimogene laherparepvec (T-VEC, Imlygic), for advanced melanoma [4]. Cancer treatment with oHSV, like other OVs, induces a variety of immune and inflammatory responses, both beneficial and detrimental. However, it has become increasingly recognized that OV-induced anti-tumor immunity plays a vital role in effective OV therapy of cancer [1*]. The cascade of host responses against HSV infection, which applies to oHSV, includes: serum factors (complement proteins and innate immunoglobulins); innate cytokine/chemokine cascades; recruitment and activation of innate immune cells; and induction of adaptive immune responses [5,6] (Figure 1). As a successful pathogen, HSV has multiple mechanisms to evade such host responses: inactivation of complement and immunoglobulins by viral glycoproteins; inhibition of interferon (IFN) responses and cytokine/chemokine production from infected cells [5]; blocking maturation of antigen presenting cells (APCs) [6]; escape from host immune surveillance through down-regulation of MHC class I expression [7]; and inhibition of apoptosis and cytotoxic T lymphocyte (CTL)-induced cell death [8]. A better understanding of the complex landscape of virus-host interaction is thus essential to refine oHSV strategies to improve therapeutic outcomes and understand oHSV immune vulnerabilities. In this review we will discuss different innate and adaptive anti-viral mechanisms elicited by a host in response to HSV-1 infection, and how HSV-1 overcomes the host’s anti-viral defensive mechanisms.
Figure 1.

Graphic representation of HSV-host immune interactions. [Graphical Abstract]
Host responses against HSV infection
Complement proteins and immunoglobulins
Serum complement proteins play an important role in limiting the HSV-1 infection [9]. Sera originating from HSV-1 sero-negative humans inactivate more than 88% of oHSVs, while sera from HSV-unexposed mice also showed nearly identical anti-HSV potency [10]. The effect of complement can be partially ablated by treatment with cobra venom factor (CVF), such that intravascular delivery of oHSV to intracerebral tumors was greatly enhanced by CVF treatment [11]. Innate immunoglobulins IgG and IgM present in the sera of HSV-seronegative humans, rats and mice can specifically bind to oHSV, while pretreatment with anti-IgG or -IgM antibodies significantly depleted the ability of human or rat, but not mouse serum to inactivate oHSV [10,12]. Exposure to cyclophosphamide (CPA) acutely can partially suppress this IgM effect, while at longer times CPA suppresses neutralizing antibodies [12]. CPA in combination with CVF further improves oHSV propagation [11]. HSV-IgM complexes activate C1q, a subcomponent of C1 complement complex, and the classical complement pathway, which was sufficient to neutralize HSV [13].
Cytokine/chemokine signaling pathways
Once HSV enters into tumor cells, it replicates, kills the tumor cells, and subsequently propagates within the tumor microenvironment. During this process, HSV can activate multiple signal transduction pathways, including Nuclear Factor kappa B (NF-κB), interferon (IFN) Regulatory Factors (IRFs) and Mitogen-Activated Protein Kinase (MAPK) pathways, resulting in induction of cytokine production in HSV-infected cells [14,15]. Stimulation of NF-κB and IRF pathways occur when HSV glycoprotein H/L complex interacts with the αvβ3-integrin signaling pathway, which results in induction of type I IFNs [16]. HSV infection activates pathogen recognition receptors (PRRs) like Toll-like receptor (TLR) 2 and 9 in dendritic cells (DCs), which in turn induce downstream activation of NF-κB and transcription of immunomodulatory cytokines, such as IL-1β, IL-6, IL-8, IL-10, IL-12, TNF-α, and IFN-γ [17]. HSV infection of human monocytes induces IL-15 expression due to TLR2 signaling [18]. Engagement of TLR3 and activation of NF-κB also occurs following HSV infection of neuronal cells and astrocytes, resulting in up-regulation of cytokines IL-6 and TNF-α [5]. A variety of other cytokines have also been detected in HSV-infected tissues of both mouse and human, including; IFN-α, IFN-β, IL-1, IL-2, IL-4, IL-5, and IL-23 [19].
Cytokines and chemokines derived from oHSV-infected tumor cells or cells of the tumor microenvironment limit oHSV replication and spread within the tumor and further induce stronger innate immune responses [20,21]. Chemokines CXCL9 (MIG) and CXCL10 (IP-10) are important in limiting HSV infections, recruiting natural killer (NK) and CD8+ T cells to the CNS [21]. OHSV 1716 infection of ovarian carcinomas induced CXCL9 and CXCL10 from monocytes and DCs, which led to the infiltration of NK and CD8+ T cells and the inhibition of tumor growth [22]. HSV-1 infection of macrophages and fibroblasts induces chemokine RANTES/CCL5 expression through a PKR-dependent mechanism [23]. Aside from its chemokine activity, RANTES binds directly to HSV virions via glycoprotein B (gB), damaging the virions [24]. The type I IFN (IFN-α/β) signaling pathway is critical in suppressing HSV replication and pathogenicity [25**], and orchestrating innate immune responses [15]. STAT1 activation in infected malignant peripheral nerve sheath tumor (MPNST) cells, with induction of IFN-stimulated genes (ISGs), was associated with reduced oHSV R3616 (γ34.5Δ) replication, while JAK inhibitor ruxolitinib increased virus yield [26]. STAT3, as opposed to STAT1 and STAT2, is an important negative regulator of type I IFN signaling and is activated in many tumors [27]. Activation of STAT3 by HDAC inhibitor VPA or IL-6 enhanced oHSV replication in glioma cells, through inhibition of type I IFN signaling, whereas inhibition of STAT3 reduced replication [20].
Innate cellular immune responses
Cytokines and chemokines recruit innate immune cells (macrophages, NK, NKT, γδ T cells) to the site of HSV infection, and activate them for further production of cytokines and chemokines, leading to a toxic cycle for HSV [28,29]. For example, IL-15 produced from peripheral blood mononuclear cells up-regulates the cytotoxic activity of NK cells against HSV infections in vitro [30]. CPA treatment decreased innate cytokine expression (IFNα/β, TNFα, IL-15) in mononuclear cells and increased oHSV spread in brain tumors and extended survival [31]. IL-18 produced from activated macrophages induced IFN-γ production by NK cells and protected mice from HSV infections [32]. IFN-γ is also produced from activated macrophages and γδ T cells in response to HSV infections, which further stimulates macrophages to produce TNF-α, which activates NK cells for HSV control [33]. Depletion of macrophages with clodronate liposomes or modulation of the tumor microenvironment with CPA (reducing IFN-γ expression and infiltration of macrophages) improves oHSV intratumoral spread and efficacy in brain tumors [34]. OHSV infections lead to increased infiltration of M1 type macrophages into the tumor, which produce significant amounts of TNF-α, resulting in TNF-α-induced apoptosis in infected tumor cells and inhibition of viral replication [35]. IL-6 produced from macrophages, has also been shown to control HSV-1 infection and immunopathology [36], in contrast to the effects of IL-6 in enhancing oHSV replication in glioma cells [20]. Microglial IL-6 expression was protective to neural progenitor cells after HSV-1 infection in vitro, while contrarily IL-6 and phospho-STAT3 were found to be elevated in HSV encephalitic brain, illustrating dependence on cell context [37].
HSV can trigger NK recognition of infected fibroblasts due to up-regulation of ligands for natural cytotoxicity receptors (NCR; NKp30, NKp46) [38]. Interestingly, this ligand up-regulation also occurs in oHSV-infected glioblastoma cells, which mediates NK lysis of oHSV-infected cells and decreased oHSV efficacy [29]. Identification of the ligands could provide a target to enhance anti-tumor activity. TGF-β, an immunosuppressive cytokine, has been shown reverse NK cell and macrophage mediated inhibition of oHSV replication in glioblastoma cells in vitro and to improve oHSV efficacy in vivo [39*]. Transgenic mice expressing dominant negative TGF-β receptor in CD11c innate cells, but not CD4+ T-cells, had reduced virus replication in the eye, while expression in CD4+ T-cells resulted in reduced leukocyte infiltration [40].
Adaptive immune responses
DCs are professional APCs that coordinate innate and adaptive immune responses. They are key to protecting against HSV; immature DCs sense HSV and efficiently take-up and process antigens leading to their maturation and migration to lymph nodes where they prime and activate T-cells [6]. DCs express a broad array of PRRs that recognize HSV pathogen-associated molecular patterns (PAMPs) [14]. HSV gH/gL and gB bind to TLR2 and activate NF-κB [41]. Activated DCs stimulate IFN-γ secretion from HSV-specific CD4+ and CD8+ T-cells [42]. OHSV infection of tumor cells leads to immunogenic cell death and the release of viral and tumor-associated antigens (TAAs), which can be cross-presented by DCs [1*]. Both CD4+ Th1 cells and CD8+ effector T-cells play a role in limiting HSV disease. HSV-specific CD8+ T-cells can clear established lytic infections, but are less effective at blocking the initial spread of virus [43]. HSV has also been shown to activate T regulatory cells (Tregs), including upregulation of OX40 and CTLA-4, which suppress effector and memory CD8+ cells [44]. Cancer cell specific CTL activity and anti-tumor efficacy is increased by CPA after oHSV treatment of syngeneic tumors [45]. B cells are also important in controlling HSV infection, through both innate (IgM) and adaptive (APCs activating CD4+ T-cell responses) immunity [46]. B cells cooperated with DCs in Th1 memory CD4+ T-cell recall responses, with immunized mice succumbing to HSV-2 pathogenicity in their absence [47]. A schematic overview of host responses against HSV infection is provided in Figure 2.
Figure 2.

Schematic overview of host immune responses to oHSV infection.
HSV evasion of host antiviral responses
While HSV induces anti-viral innate and adaptive immune responses, it also expresses proteins that inhibit both the induction and activity of those responses, allowing the virus to evade the immune system and establish both a productive and latent infection [5,15] (Figure 1). In the context of oHSV immunovirotherapy, some of these viral genes can be detrimental or beneficial. The immune evasion functions of HSV genes are listed in Table 1. In addition, pharmacological manipulation of different immune pathways or cell types can enhance oHSV efficacy [2].
Table 1.
Immune evasion mechanisms of HSV.
|
HSV genes or
proteins |
Functions | References |
|---|---|---|
| Glycoprotein B | Down-regulation of MHC II processing pathway in CD4+
cells |
[73] |
| Glycoprotein C | Inactivates serum complement proteins | [13] |
| Glycoprotein D | Down-regulates NK receptor ligand and NK-mediated lysis; inhibition of apoptosis |
[8,51] |
| Glycoproteins E and I |
Inactivate circulating immunoglobulins | [49] |
| Us3 | Inhibits NF-κB activation and reduces cytokine expression, such as IL-8; inhibits induction of apoptosis; hyperphosphorylates IRF3 to block activation of RLR signaling pathway |
[8,56,62] |
| Us5 (gJ) | Antagonizes Fas ligand- and granzyme B-mediated CTL- induced apoptosis; inhibits perforin- or UV-induced cell death and apoptosis |
[86,87] |
| VP16 | Inhibits NF-κB activation and blocks IRF3 pathway and IFN-β production |
[15] |
| VP24 | Inhibits IRF3 phosphorylation | [61] |
| Us11 | Inhibits RLR signaling, PKR-induced eIF-2α phosphorylation and autophagy; blocks OAS |
[58,59*,66,67,82*] |
| ICP0 | Blocks NF-κB-mediated transcription of immunomodulatory cytokines, and IRF3- and IRF7- induced anti-viral signaling pathways; inhibits IRF3 translocation to the nucleus; inhibits IFI16; degradation of mature DC marker (CD83) |
[54,63,64,71] |
| ICP6 | Blocks TNF-α- and Fas ligand-mediated apoptosis through interacting with caspase 8 and necroptosis |
[88,89] |
| ICP27 | Blocks NF-κB and IRF3 signaling pathways; blocks STAT1 activation and its translocation to the nucleus |
[68] |
| γ34.5 | Suppression of PKR/eIF-2α signaling pathway and IFN- induced anti-viral mechanisms; inhibits DC maturation and antigen presentation; blocks MHC class II accumulation on the cell surface; autophagy inhibition |
[14,69,80**] |
| UL36 | Inhibits TRAF3 of TLR pathway | [15] |
| UL41 (vhs) | Inhibits IFN-stimulated genes viperin, ZAP, and tetherin; blocks DC activation and maturation; blocks MHC class II accumulation on the cell surface |
[14,65,74] |
| UL42 | Inhibits nuclear translocation of NF-κB | [55] |
| ICP47 | Down-regulates MHC class I by inhibiting TAP | [7] |
Evasion of host initial defense mechanisms
HSV envelope glycoproteins not only function for virus entry, but also to escape humoral defense mechanisms. HSV gC binds and inactivates complement protein molecules (C3b, C5, and properdin), protecting HSV from complement-mediated virus neutralization induced by natural IgM [13] and antibody-independent complement neutralization [48]. Glycoprotein E and gI encode Fc receptors (FcγR) that bind IgG, with gE/gI binding monomeric IgG and gE binding IgG complexes [49]. This binding blocks antibody-mediated complement activation and antibody-dependent cellular cytotoxicity and contributes to pathogenicity in vivo [50]. Glycoprotein D (gD) down-regulates CD112, a ligand for the DNAM-1 (DNAX accessory molecule 1) receptor of activated NK cells, resulting in NK inability to bind and lyse HSV-infected or gD-transfected cells [51]. HSV reduces the expression of CD1d molecules on the surface of APCs, impairing their stimulation of NKT cells [52].
Inhibition of IFN signaling and cytokine/chemokine signaling pathways
HSV expresses several immune evasion genes to escape the host’s anti-viral immune surveillance mechanisms [5,15] (Table 1). Because of defects in those pathways in cancer cells, mutations in many of these genes endow oncolytic activity to HSV [3]. Type 1 IFN production and its signaling pathway is key to antiviral innate immunity and thus HSV targets multiple steps in the pathway: (i) TLR signaling pathway; TLR3 and TLR2 signaling to TRAF6 are inhibited by Us3 [53], TRAF3 is inhibited by UL36 deubiquitinase [15], MyD88 and NF-kB subunits P50 and P65 are inhibited by ICP0 [54], nuclear translocation of NF-kB is inhibited by UL42 and Us3 [55,56], and ICP27 blocks NF-kB by binding to IkBα [57]. (ii) RIG-I-like receptor (RLR) signaling pathway; Us11 binds to PACT, RIG-I and MDA-5 to inhibit interaction with MAVS [58,59*], γ34.5 binds to TBK1 and inhibits IRF3 phosphorylation [60*], as does VP24 [61], Us3 hyper-phosphorylation of IRF3 blocks activation [62], ICP0 inhibits IRF3 translocation to the nucleus [63], and VP16 interferes with IRF3-CBP complex to inhibit transcription [15]. (iii) DNA sensor signaling pathway; IFI16 is inhibited by ICP0 in a dynamic fashion in the nucleus [64]. (iv) IFN-stimulated genes: UL41 (vhs) inhibits viperin, ZAP, and tetherin through mRNA degradation [15,65], and Us11 inhibits OAS [66] and PKR [67]. PKR phosphorylates eIF2α, which shuts down protein synthesis. γ34.5 activates protein phosphatase 1α to dephosphorylate p-eIF2α and restore protein synthesis [14], and overcomes IFN-induced anti-viral mechanisms [14]. ICP27 prevents STAT-1 phosphorylation and activation [68]. Several of these immune evasion genes (γ34.5, ICP0, Us3) have been deleted/mutated in oHSV [3], because cancer cells tend to be defective in IFN signaling [1*].
Inhibition of adaptive immune responses
HSV has evolved numerous mechanisms to block DC function [6]. HSV efficiently infects both immature and mature human DCs, which leads to an inability to mature or down-regulation of maturation, but not cytokine secretion, and induces apoptosis early after infection [6]. γ34.5, through targeting TBK1 and IKKα/β, inhibits DC maturation and interferes with antigen presentation through inhibition of autophagy [14,69]. HSV activates β2 integrins in infected mature human monocyte-derived DCs, which enhances adhesion, and down-regulates expression of CCR7, which mediates CCL19 chemotaxis to lymph nodes, thereby impeding migration and T-cell activation [70]. ICP0 induces degradation of CD83, a marker of DC maturation, resulting in reduced T-cell stimulation [71]. ICP47 down-regulates MHC I-peptide presentation by blocking transporter associated with antigen presentation (TAP) in infected human cells, including cancer cells [7]. This results in no MHC I-mediated antigen presentation to CD8+ T-cells and escape from immune surveillance [7]. Inhibition of mouse TAP with oHSV expressing bovine herpes virus UL49.5 improved anti-tumor efficacy in syngeneic tumor models, dependent on CD8+ T-cells, and with increased virus [72*]. The MHC II processing pathway in CD4+ T cells is down-regulated by HSV-1 through reduction in invariant chain (Ii) expression and binding of gB to HLA-DM and -DR [73]. Both γ34.5 and UL41 block accumulation of MHC class II proteins on the cell surface of glioblastoma cells, resulting in interference in antigen presentation to CD4+ T cells [74].
HSV infection also modulates T cells. Us3 interrupts TCR signaling by inhibiting the linker for activation of T cells (LAT) [53]. In T cells, HSV inhibits NF-κB activation and synthesis of pro-inflammatory cytokines like IL-2 and TNF-α, while activating STAT3 and the MAPK p38 and JNK pathways, and IL-10 synthesis [75]. Co-culture of T cells with HSV-infected fibroblasts or direct infection induced apoptosis [76]. For cancer therapy in immmunocompetent models, oHSV acts as an in situ vaccine, with T-cells playing a critical role as immune cell mediators of efficacy [2,77]. Both in mice and T-Vec melanoma responders, oHSV treatment led to a reduction in Tregs [77,78]. In the T-Vec phase III clinical trial durable responses were observed not only in injected lesions, but also non-injected, with 11% of patients having complete responses, strongly suggestive of robust immune responses [1,78*]. Augmenting this with immune modulators like immune checkpoint inhibitors or expression of immune modulatory transgenes like IL-12 is actively being pursued [3,77,78].
Autophagy is a major cellular degradative pathway, including for cellular pathogens like HSV, as well as for MHC I and II presentation in APCs [79]. γ34.5 binds beclin 1, preventing autophagosome formation [80**]. HSV-1 with γ34.5 deleted for the beclin 1 binding domain has increased autophagy in culture and is attenuated for neuropathogenicity, but is able to dephosphorylate eIF2α and replicate similar to wild-type virus in culture [80**,81]. As opposed to γ34.5-deleted oHSV, oHSV lacking the beclin 1 binding domain, Δ68H-6, is able to replicate in glioblastoma stem cells [81]. Us11 is also able to block autophagy through its interaction with PKR [82*].
Inhibition of cell death
Several HSV genes, such as Us3, Us5 (gJ), gD, and ICP6 are involved in the inhibition of apoptosis of HSV-infected cells, thus allowing HSV-infected cells to survive long enough to complete viral replication [8] (Table 1). Us3 inhibits apoptosis through multiple interactions; inhibiting cytochrome c and caspase-3 activation, programmed cell death protein 4 (PDCD4) [83], and pro-apoptotic proteins Bad and Bid [84]. Because of this, Us3-deleted HSV has oncolytic properties [85]. Glycoprotein J antagonizes the Fas ligand- and granzyme B-mediated pathways of CTL-induced apoptosis [86]. HSV deleted for Us12 or Us3 were defective in preventing T cell mediated lysis but not apoptosis, while gJ-deleted HSV was defective in both [87]. Necroptosis is another form of regulated cell death that occurs in the absence of caspases. HSV-1 ICP6 and HSV-2 ICP10 suppress apoptosis by blocking TNF-α- and Fas ligand-mediated caspase 8 [88], as well as blocking TNF-induced necroptosis in human cells by inhibiting the interactions between RIP1 and RIP3 [89]. In mice, ICP6 has the opposite effect, binding to RIP3 to activate necroptosis [8,90*].
Conclusions
HSV-host interactions are complex and accumulating evidence suggests that the interaction impacts the therapeutic outcome of oHSV therapy of cancer. Successful use of HSV as an oncolytic agent requires molecular and cellular understanding of HSV-host immune interactions, involving an array of signaling pathways and host cell types. HSV utilizes various immune evasion mechanisms to evade host anti-viral responses and promote virus replication. To minimize pathogenicity and ensure tumor selectivity, oHSV have deletions of genes with immune-modulating functions such as γ34.5, ICP6, and Us3. Such genetic alterations may elicit increased innate immune responses and attenuate viral replication and spread within the tumor, which in turn could reduce the magnitude of viral oncolysis and release of tumor antigens and other danger signals. Achieving a robust and smooth transition from innate to adaptive tumor-specific immunity is key to optimizing oHSV therapy of cancer. The immune responses can be further modulated pharmacologically (i.e., CPA, immune checkpoint inhibitors) and by arming oHSV with immunomodulatory transgenes that affect the tumor microenvironment, host immune defense mechanisms, and anti-tumor immunity to enhance anti-tumor efficacy. Further elucidation of mechanisms relating to HSV-host interactions will facilitate the design of next generation oHSV for improved cancer therapy.
Highlights.
oHSV infection induces anti-viral responses that limit efficacy
HSV encodes multiple proteins that inhibit innate and adaptive immune responses.
oHSV antitumor immunity is a balance between immune stimulation and evasion
Acknowledgements
This work was supported in part by a grant from NIH, RO1CA160762, and the Thomas A. Pappas Chair in Neurosciences.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and recommended reading
• of special interest
•• of outstanding interest
- 1.Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov. 2015;14:642–662. doi: 10.1038/nrd4663. * A thorough review describing oncolytic viruses, including oHSV, mechanisms of action and interactions with the immune system and how these can be modulated for anti-tumor immunotherapeutic efficacy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chiocca EA, Rabkin SD. Oncolytic viruses and their application to cancer immunotherapy. Cancer Immunol Res. 2014;2:295–300. doi: 10.1158/2326-6066.CIR-14-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peters C, Rabkin SD. Designing Herpes Viruses as Oncolytics. Mol Ther Oncolytics. 2015;2:15010. doi: 10.1038/mto.2015.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ott PA, Hodi FS. Talimogene Laherparepvec for the Treatment of Advanced Melanoma. Clin Cancer Res. 2016;22:3127–3131. doi: 10.1158/1078-0432.CCR-15-2709. [DOI] [PubMed] [Google Scholar]
- 5.Suazo PA, Ibanez FJ, Retamal-Diaz AR, Paz-Fiblas MV, Bueno SM, Kalergis AM, Gonzalez PA. Evasion of early antiviral responses by herpes simplex viruses. Mediators Inflamm. 2015;2015:593757. doi: 10.1155/2015/593757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bedoui S, Greyer M. The role of dendritic cells in immunity against primary herpes simplex virus infections. Front Microbiol. 2014;5:533. doi: 10.3389/fmicb.2014.00533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eggensperger S, Tampe R. The transporter associated with antigen processing: a key player in adaptive immunity. Biol Chem. 2015;396:1059–1072. doi: 10.1515/hsz-2014-0320. [DOI] [PubMed] [Google Scholar]
- 8.Yu X, He S. The interplay between human herpes simplex virus infection and the apoptosis and necroptosis cell death pathways. Virol J. 2016;13:77. doi: 10.1186/s12985-016-0528-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wakimoto H, Johnson PR, Knipe DM, Chiocca EA. Effects of innate immunity on herpes simplex virus and its ability to kill tumor cells. Gene Ther. 2003;10:983–990. doi: 10.1038/sj.gt.3302038. [DOI] [PubMed] [Google Scholar]
- 10.Wakimoto H, Ikeda K, Abe T, Ichikawa T, Hochberg FH, Ezekowitz RA, Pasternack MS, Chiocca EA. The complement response against an oncolytic virus is species-specific in its activation pathways. Mol Ther. 2002;5:275–282. doi: 10.1006/mthe.2002.0547. [DOI] [PubMed] [Google Scholar]
- 11.Ikeda K, Wakimoto H, Ichikawa T, Jhung S, Hochberg FH, Louis DN, Chiocca EA. Complement depletion facilitates the infection of multiple brain tumors by an intravascular, replication-conditional herpes simplex virus mutant. J Virol. 2000;74:4765–4775. doi: 10.1128/jvi.74.10.4765-4775.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ikeda K, Ichikawa T, Wakimoto H, Silver JS, Deisboeck TS, Finkelstein D, Harsh GRt, Louis DN, Bartus RT, Hochberg FH, et al. Oncolytic virus therapy of multiple tumors in the brain requires suppression of innate and elicited antiviral responses. Nat Med. 1999;5:881–887. doi: 10.1038/11320. [DOI] [PubMed] [Google Scholar]
- 13.Hook LM, Lubinski JM, Jiang M, Pangburn MK, Friedman HM. Herpes simplex virus type 1 and 2 glycoprotein C prevents complement-mediated neutralization induced by natural immunoglobulin M antibody. J Virol. 2006;80:4038–4046. doi: 10.1128/JVI.80.8.4038-4046.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ma Y, He B. Recognition of herpes simplex viruses: toll-like receptors and beyond. J Mol Biol. 2014;426:1133–1147. doi: 10.1016/j.jmb.2013.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Su C, Zhan G, Zheng C. Evasion of host antiviral innate immunity by HSV-1, an update. Virol J. 2016;13:38. doi: 10.1186/s12985-016-0495-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gianni T, Leoni V, Campadelli-Fiume G. Type I interferon and NF-kappaB activation elicited by herpes simplex virus gH/gL via alphavbeta3 integrin in epithelial and neuronal cell lines. J Virol. 2013;87:13911–13916. doi: 10.1128/JVI.01894-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sato A, Linehan MM, Iwasaki A. Dual recognition of herpes simplex viruses by TLR2 and TLR9 in dendritic cells. Proc Natl Acad Sci U S A. 2006;103:17343–17348. doi: 10.1073/pnas.0605102103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ahmad R, El Bassam S, Cordeiro P, Menezes J. Requirement of TLR2-mediated signaling for the induction of IL-15 gene expression in human monocytic cells by HSV-1. Blood. 2008;112:2360–2368. doi: 10.1182/blood-2008-02-137711. [DOI] [PubMed] [Google Scholar]
- 19.Hukkanen V, Broberg E, Salmi A, Eralinna JP. Cytokines in experimental herpes simplex virus infection. Int Rev Immunol. 2002;21:355–371. doi: 10.1080/08830180213276. [DOI] [PubMed] [Google Scholar]
- 20.Okemoto K, Wagner B, Meisen H, Haseley A, Kaur B, Chiocca EA. STAT3 activation promotes oncolytic HSV1 replication in glioma cells. PLoS One. 2013;8:e71932. doi: 10.1371/journal.pone.0071932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thapa M, Welner RS, Pelayo R, Carr DJ. CXCL9 and CXCL10 expression are critical for control of genital herpes simplex virus type 2 infection through mobilization of HSV-specific CTL and NK cells to the nervous system. J Immunol. 2008;180:1098–1106. doi: 10.4049/jimmunol.180.2.1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Benencia F, Courreges MC, Conejo-Garcia JR, Mohamed-Hadley A, Zhang L, Buckanovich RJ, Carroll R, Fraser N, Coukos G. HSV oncolytic therapy upregulates interferon-inducible chemokines and recruits immune effector cells in ovarian cancer. Mol Ther. 2005;12:789–802. doi: 10.1016/j.ymthe.2005.03.026. [DOI] [PubMed] [Google Scholar]
- 23.Melchjorsen J, Pedersen FS, Mogensen SC, Paludan SR. Herpes simplex virus selectively induces expression of the CC chemokine RANTES/CCL5 in macrophages through a mechanism dependent on PKR and ICP0. J Virol. 2002;76:2780–2788. doi: 10.1128/JVI.76.6.2780-2788.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakayama T, Shirane J, Hieshima K, Shibano M, Watanabe M, Jin Z, Nagakubo D, Saito T, Shimomura Y, Yoshie O. Novel antiviral activity of chemokines. Virology. 2006;350:484–492. doi: 10.1016/j.virol.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 25.Leib DA, Harrison TE, Laslo KM, Machalek MA, Moorman NJ, Virgin HW. Interferons regulate the phenotype of wild-type and mutant herpes simplex viruses in vivo. J Exp Med. 1999;189:663–672. doi: 10.1084/jem.189.4.663. ** Describes the use of IFN receptor knock-out mice to demonstrate the major role that type 1 IFNs play in HSV neuropathogenicity, especially in targeting γ34.5 mutants. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jackson JD, Markert JM, Li L, Carroll SL, Cassady KA. STAT1 and NF-kappaB Inhibitors Diminish Basal Interferon-Stimulated Gene Expression and Improve the Productive Infection of Oncolytic HSV in MPNST Cells. Mol Cancer Res. 2016;14:482–492. doi: 10.1158/1541-7786.MCR-15-0427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuchipudi SV. The Complex Role of STAT3 in Viral Infections. J Immunol Res. 2015;2015:272359. doi: 10.1155/2015/272359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wuest TR, Carr DJ. The role of chemokines during herpes simplex virus-1 infection. Front Biosci. 2008;13:4862–4872. doi: 10.2741/3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alvarez-Breckenridge CA, Yu J, Price R, Wojton J, Pradarelli J, Mao H, Wei M, Wang Y, He S, Hardcastle J, et al. NK cells impede glioblastoma virotherapy through NKp30 and NKp46 natural cytotoxicity receptors. Nat Med. 2012;18:1827–1834. doi: 10.1038/nm.3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ahmad A, Sharif-Askari E, Fawaz L, Menezes J. Innate immune response of the human host to exposure with herpes simplex virus type 1: in vitro control of the virus infection by enhanced natural killer activity via interleukin-15 induction. J Virol. 2000;74:7196–7203. doi: 10.1128/jvi.74.16.7196-7203.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wakimoto H, Fulci G, Tyminski E, Chiocca EA. Altered expression of antiviral cytokine mRNAs associated with cyclophosphamide's enhancement of viral oncolysis. Gene Ther. 2004;11:214–223. doi: 10.1038/sj.gt.3302143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fujioka N, Akazawa R, Ohashi K, Fujii M, Ikeda M, Kurimoto M. Interleukin-18 protects mice against acute herpes simplex virus type 1 infection. J Virol. 1999;73:2401–2409. doi: 10.1128/jvi.73.3.2401-2409.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vogel K, Thomann S, Vogel B, Schuster P, Schmidt B. Both plasmacytoid dendritic cells and monocytes stimulate natural killer cells early during human herpes simplex virus type 1 infections. Immunology. 2014;143:588–600. doi: 10.1111/imm.12337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fulci G, Dmitrieva N, Gianni D, Fontana EJ, Pan X, Lu Y, Kaufman CS, Kaur B, Lawler SE, Lee RJ, et al. Depletion of peripheral macrophages and brain microglia increases brain tumor titers of oncolytic viruses. Cancer Res. 2007;67:9398–9406. doi: 10.1158/0008-5472.CAN-07-1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meisen WH, Wohleb ES, Jaime-Ramirez AC, Bolyard C, Yoo JY, Russell L, Hardcastle J, Dubin S, Muili K, Yu J, et al. The Impact of Macrophage- and Microglia-Secreted TNFalpha on Oncolytic HSV-1 Therapy in the Glioblastoma Tumor Microenvironment. Clin Cancer Res. 2015;21:3274–3285. doi: 10.1158/1078-0432.CCR-14-3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murphy EA, Davis JM, Brown AS, Carmichael MD, Ghaffar A, Mayer EP. Effect of IL-6 deficiency on susceptibility to HSV-1 respiratory infection and intrinsic macrophage antiviral resistance. J Interferon Cytokine Res. 2008;28:589–595. doi: 10.1089/jir.2007.0103. [DOI] [PubMed] [Google Scholar]
- 37.Chucair-Elliott AJ, Conrady C, Zheng M, Kroll CM, Lane TE, Carr DJ. Microglia-induced IL-6 protects against neuronal loss following HSV-1 infection of neural progenitor cells. Glia. 2014;62:1418–1434. doi: 10.1002/glia.22689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chisholm SE, Howard K, Gomez MV, Reyburn HT. Expression of ICP0 is sufficient to trigger natural killer cell recognition of herpes simplex virus-infected cells by natural cytotoxicity receptors. J Infect Dis. 2007;195:1160–1168. doi: 10.1086/512862. [DOI] [PubMed] [Google Scholar]
- 39.Han J, Chen X, Chu J, Xu B, Meisen WH, Chen L, Zhang L, Zhang J, He X, Wang QE, et al. TGFbeta Treatment Enhances Glioblastoma Virotherapy by Inhibiting the Innate Immune Response. Cancer Res. 2015;75:5273–5282. doi: 10.1158/0008-5472.CAN-15-0894. * Describes how pretreatment with TGFβ can reverse the innate cellular responses of NK cells and macrophages, including decreased NK cytotoxicity of oHSV-infection glioblastoma cells, increased virus yields, and enhanced anti-tumor efficacy in immune-deficient or -competent mice. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Allen SJ, Mott KR, Wechsler SL, Flavell RA, Town T, Ghiasi H. Adaptive and innate transforming growth factor beta signaling impact herpes simplex virus 1 latency and reactivation. J Virol. 2011;85:11448–11456. doi: 10.1128/JVI.00678-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leoni V, Gianni T, Salvioli S, Campadelli-Fiume G. Herpes simplex virus glycoproteins gH/gL and gB bind Toll-like receptor 2, and soluble gH/gL is sufficient to activate NF-kappaB. J Virol. 2012;86:6555–6562. doi: 10.1128/JVI.00295-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Macleod BL, Bedoui S, Hor JL, Mueller SN, Russell TA, Hollett NA, Heath WR, Tscharke DC, Brooks AG, Gebhardt T. Distinct APC subtypes drive spatially segregated CD4+ and CD8+ T-cell effector activity during skin infection with HSV-1. PLoS Pathog. 2014;10:e1004303. doi: 10.1371/journal.ppat.1004303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van Lint A, Ayers M, Brooks AG, Coles RM, Heath WR, Carbone FR. Herpes simplex virus-specific CD8+ T cells can clear established lytic infections from skin and nerves and can partially limit the early spread of virus after cutaneous inoculation. J Immunol. 2004;172:392–397. doi: 10.4049/jimmunol.172.1.392. [DOI] [PubMed] [Google Scholar]
- 44.Suvas S, Kumaraguru U, Pack CD, Lee S, Rouse BT. CD4+CD25+ T cells regulate virus-specific primary and memory CD8+ T cell responses. J Exp Med. 2003;198:889–901. doi: 10.1084/jem.20030171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li H, Zeng Z, Fu X, Zhang X. Coadministration of a herpes simplex virus-2 based oncolytic virus and cyclophosphamide produces a synergistic antitumor effect and enhances tumor-specific immune responses. Cancer Res. 2007;67:7850–7855. doi: 10.1158/0008-5472.CAN-07-1087. [DOI] [PubMed] [Google Scholar]
- 46.Deshpande SP, Kumaraguru U, Rouse BT. Dual role of B cells in mediating innate and acquired immunity to herpes simplex virus infections. Cell Immunol. 2000;202:79–87. doi: 10.1006/cimm.2000.1666. [DOI] [PubMed] [Google Scholar]
- 47.Iijima N, Linehan MM, Zamora M, Butkus D, Dunn R, Kehry MR, Laufer TM, Iwasaki A. Dendritic cells and B cells maximize mucosal Th1 memory response to herpes simplex virus. J Exp Med. 2008;205:3041–3052. doi: 10.1084/jem.20082039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Friedman HM, Wang L, Pangburn MK, Lambris JD, Lubinski J. Novel mechanism of antibody-independent complement neutralization of herpes simplex virus type 1. J Immunol. 2000;165:4528–4536. doi: 10.4049/jimmunol.165.8.4528. [DOI] [PubMed] [Google Scholar]
- 49.Dubin G, Frank I, Friedman HM. Herpes simplex virus type 1 encodes two Fc receptors which have different binding characteristics for monomeric immunoglobulin G (IgG) and IgG complexes. J Virol. 1990;64:2725–2731. doi: 10.1128/jvi.64.6.2725-2731.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lubinski JM, Lazear HM, Awasthi S, Wang F, Friedman HM. The herpes simplex virus 1 IgG fc receptor blocks antibody-mediated complement activation and antibody-dependent cellular cytotoxicity in vivo. J Virol. 2011;85:3239–3249. doi: 10.1128/JVI.02509-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Grauwet K, Cantoni C, Parodi M, De Maria A, Devriendt B, Pende D, Moretta L, Vitale M, Favoreel HW. Modulation of CD112 by the alphaherpesvirus gD protein suppresses DNAM-1-dependent NK cell-mediated lysis of infected cells. Proc Natl Acad Sci U S A. 2014;111:16118–16123. doi: 10.1073/pnas.1409485111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yuan W, Dasgupta A, Cresswell P. Herpes simplex virus evades natural killer T cell recognition by suppressing CD1d recycling. Nat Immunol. 2006;7:835–842. doi: 10.1038/ni1364. [DOI] [PubMed] [Google Scholar]
- 53.Yang Y, Wu S, Wang Y, Pan S, Lan B, Liu Y, Zhang L, Leng Q, Chen D, Zhang C, et al. The Us3 Protein of Herpes Simplex Virus 1 Inhibits T Cell Signaling by Confining Linker for Activation of T Cells (LAT) Activation via TRAF6 Protein. J Biol Chem. 2015;290:15670–15678. doi: 10.1074/jbc.M115.646422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van Lint AL, Murawski MR, Goodbody RE, Severa M, Fitzgerald KA, Finberg RW, Knipe DM, Kurt-Jones EA. Herpes simplex virus immediate-early ICP0 protein inhibits Toll-like receptor 2-dependent inflammatory responses and NF-kappaB signaling. J Virol. 2010;84:10802–10811. doi: 10.1128/JVI.00063-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang J, Wang S, Wang K, Zheng C. Herpes simplex virus 1 DNA polymerase processivity factor UL42 inhibits TNF-alpha-induced NF-kappaB activation by interacting with p65/RelA and p50/NF-kappaB1. Med Microbiol Immunol. 2013;202:313–325. doi: 10.1007/s00430-013-0295-0. [DOI] [PubMed] [Google Scholar]
- 56.Wang K, Ni L, Wang S, Zheng C. Herpes simplex virus 1 protein kinase US3 hyperphosphorylates p65/RelA and dampens NF-kappaB activation. J Virol. 2014;88:7941–7951. doi: 10.1128/JVI.03394-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim JC, Lee SY, Kim SY, Kim JK, Kim HJ, Lee HM, Choi MS, Min JS, Kim MJ, Choi HS, et al. HSV-1 ICP27 suppresses NF-kappaB activity by stabilizing IkappaBalpha. FEBS Lett. 2008;582:2371–2376. doi: 10.1016/j.febslet.2008.05.044. [DOI] [PubMed] [Google Scholar]
- 58.Kew C, Lui PY, Chan CP, Liu X, Au SW, Mohr I, Jin DY, Kok KH. Suppression of PACT-induced type I interferon production by herpes simplex virus 1 Us11 protein. J Virol. 2013;87:13141–13149. doi: 10.1128/JVI.02564-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xing J, Wang S, Lin R, Mossman KL, Zheng C. Herpes simplex virus 1 tegument protein US11 downmodulates the RLR signaling pathway via direct interaction with RIG-I and MDA-5. J Virol. 2012;86:3528–3540. doi: 10.1128/JVI.06713-11. * This paper reports an additional activity of Us11, inhibiting RLR signaling and innate immune responses through its RNA-binding domain. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ma Y, Jin H, Valyi-Nagy T, Cao Y, Yan Z, He B. Inhibition of TANK binding kinase 1 by herpes simplex virus 1 facilitates productive infection. J Virol. 2012;86:2188–2196. doi: 10.1128/JVI.05376-11. * Describes a new role for γ34.5 in binding to and inhibiting TBK1, and thus blocking expression of innate antiviral genes independent of PKR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang D, Su C, Zheng C. Herpes Simplex Virus 1 Serine Protease VP24 Blocks the DNA-Sensing Signal Pathway by Abrogating Activation of Interferon Regulatory Factor 3. J Virol. 2016;90:5824–5829. doi: 10.1128/JVI.00186-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang S, Wang K, Lin R, Zheng C. Herpes simplex virus 1 serine/threonine kinase US3 hyperphosphorylates IRF3 and inhibits beta interferon production. J Virol. 2013;87:12814–12827. doi: 10.1128/JVI.02355-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Paladino P, Collins SE, Mossman KL. Cellular localization of the herpes simplex virus ICP0 protein dictates its ability to block IRF3-mediated innate immune responses. PLoS One. 2010;5:e10428. doi: 10.1371/journal.pone.0010428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Everett RD. Dynamic Response of IFI16 and Promyelocytic Leukemia Nuclear Body Components to Herpes Simplex Virus 1 Infection. J Virol. 2016;90:167–179. doi: 10.1128/JVI.02249-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zenner HL, Mauricio R, Banting G, Crump CM. Herpes simplex virus 1 counteracts tetherin restriction via its virion host shutoff activity. J Virol. 2013;87:13115–13123. doi: 10.1128/JVI.02167-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sanchez R, Mohr I. Inhibition of cellular 2'-5' oligoadenylate synthetase by the herpes simplex virus type 1 Us11 protein. J Virol. 2007;81:3455–3464. doi: 10.1128/JVI.02520-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Poppers J, Mulvey M, Khoo D, Mohr I. Inhibition of PKR activation by the proline-rich RNA binding domain of the herpes simplex virus type 1 Us11 protein. J Virol. 2000;74:11215–11221. doi: 10.1128/jvi.74.23.11215-11221.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Johnson KE, Song B, Knipe DM. Role for herpes simplex virus 1 ICP27 in the inhibition of type I interferon signaling. Virology. 2008;374:487–494. doi: 10.1016/j.virol.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gobeil PA, Leib DA. Herpes simplex virus gamma34.5 interferes with autophagosome maturation and antigen presentation in dendritic cells. MBio. 2012;3:e00267–00212. doi: 10.1128/mBio.00267-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Theodoridis AA, Eich C, Figdor CG, Steinkasserer A. Infection of dendritic cells with herpes simplex virus type 1 induces rapid degradation of CYTIP, thereby modulating adhesion and migration. Blood. 2011;118:107–115. doi: 10.1182/blood-2010-07-294363. [DOI] [PubMed] [Google Scholar]
- 71.Heilingloh CS, Muhl-Zurbes P, Steinkasserer A, Kummer M. Herpes simplex virus type 1 ICP0 induces CD83 degradation in mature dendritic cells independent of its E3 ubiquitin ligase function. J Gen Virol. 2014;95:1366–1375. doi: 10.1099/vir.0.062810-0. [DOI] [PubMed] [Google Scholar]
- 72.Pourchet A, Fuhrmann SR, Pilones KA, Demaria S, Frey AB, Mulvey M, Mohr I. CD8(+) T-cell Immune Evasion Enables Oncolytic Virus Immunotherapy. EBioMedicine. 2016;5:59–67. doi: 10.1016/j.ebiom.2016.01.022. * This study demonstrates that oHSV inhibition of TAP increases anti-tumor efficacy in syngeneic models, as opposed to decreasing efficacy by infected cell evasion of CD8+ T cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Neumann J, Eis-Hubinger AM, Koch N. Herpes simplex virus type 1 targets the MHC class II processing pathway for immune evasion. J Immunol. 2003;171:3075–3083. doi: 10.4049/jimmunol.171.6.3075. [DOI] [PubMed] [Google Scholar]
- 74.Trgovcich J, Johnson D, Roizman B. Cell surface major histocompatibility complex class II proteins are regulated by the products of the gamma(1)34.5 and U(L)41 genes of herpes simplex virus 1. J Virol. 2002;76:6974–6986. doi: 10.1128/JVI.76.14.6974-6986.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sloan DD, Jerome KR. Herpes simplex virus remodels T-cell receptor signaling, resulting in p38-dependent selective synthesis of interleukin-10. J Virol. 2007;81:12504–12514. doi: 10.1128/JVI.01111-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Han JY, Sloan DD, Aubert M, Miller SA, Dang CH, Jerome KR. Apoptosis and antigen receptor function in T and B cells following exposure to herpes simplex virus. Virology. 2007;359:253–263. doi: 10.1016/j.virol.2006.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cheema TA, Wakimoto H, Fecci PE, Ning J, Kuroda T, Jeyaretna DS, Martuza RL, Rabkin SD. Multifaceted oncolytic virus therapy for glioblastoma in an immunocompetent cancer stem cell model. Proc Natl Acad Sci U S A. 2013;110:12006–12011. doi: 10.1073/pnas.1307935110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ott PA, Hodi FS. Talimogene Laherparepvec for the Treatment of Advanced Melanoma. Clin Cancer Res. 2016 doi: 10.1158/1078-0432.CCR-15-2709. [DOI] [PubMed] [Google Scholar]
- 79.O'Connell D, Liang C. Autophagy interaction with herpes simplex virus type-1 infection. Autophagy. 2016;12:451–459. doi: 10.1080/15548627.2016.1139262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Orvedahl A, Alexander D, Talloczy Z, Sun Q, Wei Y, Zhang W, Burns D, Leib DA, Levine B. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe. 2007;1:23–35. doi: 10.1016/j.chom.2006.12.001. ** This paper identified the beclin 1 binding domain of γ34.5 and how it contributes to HSV antagonism of autophagy, independent of eIF2α dephosphorylation, and neuropathogenicity. [DOI] [PubMed] [Google Scholar]
- 81.Kanai R, Zaupa C, Sgubin D, Antoszczyk SJ, Martuza RL, Wakimoto H, Rabkin SD. Effect of gamma34.5 deletions on oncolytic herpes simplex virus activity in brain tumors. J Virol. 2012;86:4420–4431. doi: 10.1128/JVI.00017-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lussignol M, Queval C, Bernet-Camard MF, Cotte-Laffitte J, Beau I, Codogno P, Esclatine A. The herpes simplex virus 1 Us11 protein inhibits autophagy through its interaction with the protein kinase PKR. J Virol. 2013;87:859–871. doi: 10.1128/JVI.01158-12. * Describes a novel effect of Us11 interacting with PKR to inhibit autophagy, a second HSV protein blocking autophagy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang X, Patenode C, Roizman B. US3 protein kinase of HSV-1 cycles between the cytoplasm and nucleus and interacts with programmed cell death protein 4 (PDCD4) to block apoptosis. Proc Natl Acad Sci U S A. 2011;108:14632–14636. doi: 10.1073/pnas.1111942108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cartier A, Broberg E, Komai T, Henriksson M, Masucci MG. The herpes simplex virus-1 Us3 protein kinase blocks CD8T cell lysis by preventing the cleavage of Bid by granzyme B. Cell Death Differ. 2003;10:1320–1328. doi: 10.1038/sj.cdd.4401308. [DOI] [PubMed] [Google Scholar]
- 85.Liu TC, Wakimoto H, Martuza RL, Rabkin SD. Herpes simplex virus Us3(−) mutant as oncolytic strategy and synergizes with phosphatidylinositol 3-kinase-Akt targeting molecular therapeutics. Clin Cancer Res. 2007;13:5897–5902. doi: 10.1158/1078-0432.CCR-07-1013. [DOI] [PubMed] [Google Scholar]
- 86.Jerome KR, Chen Z, Lang R, Torres MR, Hofmeister J, Smith S, Fox R, Froelich CJ, Corey L. HSV and glycoprotein J inhibit caspase activation and apoptosis induced by granzyme B or Fas. J Immunol. 2001;167:3928–3935. doi: 10.4049/jimmunol.167.7.3928. [DOI] [PubMed] [Google Scholar]
- 87.Aubert M, Krantz EM, Jerome KR. Herpes simplex virus genes Us3, Us5, and Us12 differentially regulate cytotoxic T lymphocyte-induced cytotoxicity. Viral Immunol. 2006;19:391–408. doi: 10.1089/vim.2006.19.391. [DOI] [PubMed] [Google Scholar]
- 88.Dufour F, Sasseville AM, Chabaud S, Massie B, Siegel RM, Langelier Y. The ribonucleotide reductase R1 subunits of herpes simplex virus types 1 and 2 protect cells against TNFalpha- and FasL-induced apoptosis by interacting with caspase-8. Apoptosis. 2011;16:256–271. doi: 10.1007/s10495-010-0560-2. [DOI] [PubMed] [Google Scholar]
- 89.Guo H, Omoto S, Harris PA, Finger JN, Bertin J, Gough PJ, Kaiser WJ, Mocarski ES. Herpes simplex virus suppresses necroptosis in human cells. Cell Host Microbe. 2015;17:243–251. doi: 10.1016/j.chom.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang X, Li Y, Liu S, Yu X, Li L, Shi C, He W, Li J, Xu L, Hu Z, et al. Direct activation of RIP3/MLKL-dependent necrosis by herpes simplex virus 1 (HSV-1) protein ICP6 triggers host antiviral defense. Proc Natl Acad Sci U S A. 2014;111:15438–15443. doi: 10.1073/pnas.1412767111. * The first demonstration that HSV-1 triggers necroptosis in mice through the ICP6 RHIM-like domain and that mice lacking RIP3 have increased HSV pathogenicity. [DOI] [PMC free article] [PubMed] [Google Scholar]