

Abstract
The corpus callosum (CC) is the largest fiber tract in the mammalian brain, linking the bilateral cerebral hemispheres. CC development depends on the proper balance of axon growth cone attractive and repellent cues leading axons to the midline then directing them to the contralateral hemisphere. Imbalance of these cues results in CC agenesis or dysgenesis. Nogo receptors (NgR1, NgR2, NgR3) are growth cone directive molecules known for inhibiting axon regeneration after injury. We report that mice that lack Nogo receptors (NgR123-null mice) display complete CC agenesis due to axon misdirection evidenced by ectopic axons including cortical Probst bundles. Since glia and glial-derived growth cone repellent factors (especially the diffusible factor Slit2) are required for CC development, their distribution was studied. Compared to wild type mice, NgR123-null mice had a sharp increase in the glial marker GFAP and in Slit2 at the glial wedge and indusium griseum, midline structures required for CC formation. NgR123-null mice displayed reduced motor coordination and hyperactivity. These data are consistent with the hypotheses that Nogo receptors are membrane-bound growth cone repellent factors required for migration of axons across the midline at the CC, and that their absence results directly or indirectly in midline gliosis, increased Slit2, and complete CC agenesis.
INDEXING TERMS: axon, corpus callosum, glia, Probst bundle, NgR, Slit2
Graphical Abstract
Nogo receptors NgR1, NgR2 and NgR3 are cell surface molecules on neurons and glia that regulate axon outgrowth among other functions. Mice engineered to lack all Nogo receptors displayed completely penetrant agenesis of the corpus callosum, relating Nogo receptor expression to generation of the largest axon tract in the brain.
The corpus callosum (CC) is the largest fiber tract in the mammalian brain, connecting the two hemispheres to provide bilaterally coordinated neuronal activity (Paul et al., 2007; Donahoo and Richards, 2009). Development of this major axonal pathway requires a host of cells and factors that first build the appropriate midline structures and then support and guide pioneer axons from the developing cingulate cortex across the midline, which are then followed by additional axons from the neocortex (Donahoo and Richards, 2009). Failure in any one of these steps results in agenesis (complete absence) or dysgenesis (incomplete formation) of the corpus callosum in humans and in experimental animals where many genes associated with CC development have been discovered (Richards et al., 2004). Gene mutations resulting in CC agenesis or dysgenesis include those for guidance factors (both attractive and repulsive), cell adhesion molecules, growth factors, intracellular signaling molecules, and transcription factors (Richards et al., 2004; Donahoo and Richards, 2009).
The appropriate balance of attractive growth cone guidance cues, such as Netrin1 in the floor plate, and strategically placed growth cone repellant cues such as Slit2, cooperate to guide pioneer axons across the midline (Fothergill et al., 2014). Among cells responsible for CC development, important directive roles are played by midline glia including midline zipper glia, thought to aid in hemispheric fusion, the bilateral glial wedge cells that sit at key turning points for midline-crossing axons, and the glia of the indusium griseum that lie just above the CC (Richards et al., 2004). Midline glia secrete diffusible growth cone repellent cues including Slit2 and Draxin the disruption of which results in CC agenesis or dysgenesis (Bagri et al., 2002; Shu et al., 2003a; Shu et al., 2003b; Islam et al., 2009).
In addition to diffusible repulsive cues, there are membrane-bound inhibitors of axon outgrowth in the mammalian nervous system, several of which were first identified on myelin including Nogo-A, myelin-associated glycoprotein (MAG), and oligodendrocyte-myelin glycoprotein (OMgp) (Sandvig et al., 2004; Yiu and He, 2006). Chondroitin sulfate proteoglycans (CSPGs) in the extracellular matrix also inhibit axon outgrowth (Carulli et al., 2005). Receptors for axon outgrowth inhibitors include the glycophosphatidylinositol-anchored Nogo receptors NgR1, NgR2, and NgR3. NgR1 is a functional inhibitory receptor for Nogo-A, OMgp, MAG, and CSPGs, NgR2 for MAG, and NgR3 for CSPGs (Dickendesher et al., 2012; Borrie et al., 2012). Although Nogo, MAG and OMgp are prominent on myelin and NgRs are expressed by many neurons (Wang et al., 2002; Zhang et al., 2014), expression of these molecules varies among cell types in the brain (Table 1), suggesting functions in addition to axon outgrowth inhibition (Seiler et al., 2016).
TABLE 1.
Expression of Nogo Receptors, Nogo, MAG and OMgp mRNA Transcripts in Different Cell Types Isolated From the Mouse Cerebral Cortex
| gene | protein | astrocytes | neurons | oligo- dendrocytes |
microglia |
|---|---|---|---|---|---|
| Rtn4r | NgR1 | 1.0 | 11.7 | 4.6 | 2.0 |
| Rtn4rl2 | NgR2 | 0.1 | 2.3 | 1.6 | 0.5 |
| Rtn4rl1 | NgR3 | 0.1 | 1.7 | 2.6 | 38.7 |
| Rtn4 | Nogo | 27 | 87 | 277 | 49 |
| Mag | MAG | 0.3 | 0.7 | 3750 | 15 |
| Omg | OMgp | 26.3 | 6.6 | 260 | 0.8 |
Transcript levels in isolated mouse P7 astrocytes and neurons or mouse P17 oligodendrocytes and microglia were determined by RNA-Seq. Relative quantities of transcripts are reported as fragments per kilobase of transcript sequence per million mapped fragments (FPKM). Data are from the brain RNA-Seq web site (http://web.stanford.edu/group/barres_lab/brain_rnaseq.html) as described (Zhang et al., 2014).
As part of the research on the roles of Nogo-A and Nogo receptors in axon regeneration after injury, mice genetically engineered to lack all three Nogo receptors (NgR1, NgR2, and NgR3) were generated (Dickendesher et al., 2012). In histological examination of these mice, we unexpectedly observed completely penetrant agenesis of the corpus callosum. Here we report this finding, relating Nogo receptor expression to generation of the largest axon tract in the brain.
MATERIALS AND METHODS
Animals
All animal procedures were approved by the Johns Hopkins Animal Care and Use Committee and were consistent with federal law and NIH regulations.
Mice with germline deletions in the genes coding for NgR1 (Rtn4r), NgR2 (Rtn4rl2), and NgR3 (Rtn4rl1) were created and cross-bred to generate triple-null (NgR123-null) mice as described (Dickendesher et al., 2012). Founder NgR123-null mice were the kind gift of Andrew Wood, Wyeth Neuroscience, Monmouth Junction, NJ. Triple-null mice were cross bred to generate offspring which were genetically tested as detailed below.
NgR gene disruption was confirmed by RT-PCR (data not shown) using the following primers: NgR1, 5’-ggtctagggatgcatctcag-3’ (forward) and 5’-gtggtctgtgtggctcctgc-3’ (backward); NgR2, 5’-gttggtggggttcttgtctcagg-3’ (forward) and 5’-cccccctgccccagctacgc-3’ (backward); NgR3, 5’-cgcaaagggtgctgtgtggaattgc-3’ (forward) and 5’-ggaaggtgatgcgattgttctgcagg-3’ (backward). All mutant mice used in these studies were null at all three NgR genes.
Strain identification using a 384-SNP panel (Charles River Laboratories, Wilmington, MA) revealed positive results for similarity to 129S2/SvPasCrl at >99% of the sites. Therefore, control mice for these studies were 129S2/SvPasCrl founders (Charles River Laboratories) bred on site.
The NgR123 triple-null mouse line on which these studies are based, provided by Wyeth Neuroscience, contractually could not be outbred and lost fecundity. Based on the authors’ inquiries, there are no live colonies of these mice in existence as of the time of submission. Ownership of the original triple-null mice and any frozen stocks passed to Pfizer (Groton, CT), which purchased Wyeth in 2009. Nogo receptor null mice have been separately derived elsewhere (Wills et al., 2012).
Antibody Characterization
Table 2 lists the antigen, immunogen, host species, commercial source, Research Resource Identifier (RRID), and method-specific dilutions for the eleven primary antibodies used in this study. The six antibodies used for immunohistochemistry or immunofluorescent tissue staining were cross-tested by immunoblot of SDS-PAGE resolved whole brain extracts and generated unique bands that migrated appropriately for the molecular weight of the target protein (Figs. 3 and 6): MBP, CNPase (Samanta et al., 2010), synaptophysin (Belichenko et al., 2007), β-III-tubulin (Jouhilahti et al., 2008), GFAP and Slit2 (Delloye-Bourgeois et al., 2015). The four antibodies used only for SDS-PAGE immunoblotting also identified unique appropriately migrating molecular species: Nogo-A (Gil et al., 2010; Blaise et al., 2012), OMgp (Kottis et al., 2002), MAG (Nobile-Orazio et al., 1984), and the gel loading control GAPDH. NeuN (Mullen et al., 1992) was used only for immunohistological staining of nuclei of neurons, and provided highly selective staining of appropriate neuronal layers in the wild type hippocampus.
TABLE 2.
Primary Antibodies
| Antigen | Immunogen | Source1, host species, catalog number, research resource identified |
Dilution2 |
|---|---|---|---|
| Neuron specific nuclear protein (NeuN) |
Purified cell nuclei from mouse brain |
EMD Millipore, mouse monoclonal A60, Cat# MAB3777, RRID: AB_2314890 |
1:200 (IHC) |
| Glial fibrillary acidic protein (GFAP) |
Purified GFAP from porcine spinal cord. |
EMD Millpore, mouse monoclonal GA5, Cat# MAB360, RRID: AB_212974 |
1:200 (IHC, IF); 1:1,000 (IB) |
| Slit homolog 2 protein precursor (Slit2) |
Synthetic peptide corresponding to residues in Human Slit2 |
Abcam, rabbit monoclonal, Cat# ab134166, RRID: YG100906C |
1:100 (IF); 1:1,000 (IB) |
| Myelin basic protein (MBP) |
Purified human myelin basic protein. |
QED Bioscience, mouse, monoclonal, Cat# 24201, RRID: AB_129975 |
1:200 (IHC); 1:500 (IB) |
| 2',3'-Cyclic-nucleotide 3'-phosphodiesterase (CNPase) |
Human cerebellum tissue |
Biolegend, mouse monoclonal SMI 91, Cat# 836401, RRID: AB_2565361 |
1:200 (IHC); 1:1,000 (IB) |
| Synaptophysin | Rat retina synaptosomes | Sigma-Aldrich, mouse monoclonal SVP 38, Cat# S5768, RRID: AB_477523 |
1:200 (IHC); 1:1,000 (IB) |
| βIII-tubulin | Microtubules derived from rat brain |
Biolegend, mouse, monoclonal, Clone TUJ1, Cat# 801201, RID: AB_2564639 |
1:2,000 (IB) |
| Glyceraldehyde 3 phosphate dehydrogenase (GAPDH) |
Rabbit GAPDH | Sigma-Aldrich, mouse, monoclonal, Clone GAPDH-71.1, Cat# G8795, RRID: AB_1078991 |
1:5,000 (IB) |
| Myelin-associated glycoprotein (MAG) |
Human MAG | Kind gift of Dr. Norman Latov, Cornell University, Ithaca, NY, Clone GEN-S3 |
1:500 (IB) |
| Nogo-A | Amino acids 701–1000 of Nogo A of human origin |
Santa Cruz Biotechnology, rabbit, polyclonal Clone H-300, Cat# sc25660, RRID: AB_2285559 |
1:500 (IB) |
| Oligodendrocyte myelin glycoprotein (OMgp) |
Amino acids 195–416 of human OMgp |
Santa Cruz Biotechnology, mouse, monoclonal, Clone E-8, Cat# sc271704, RRID: AB_10707816 |
1:500 (IB) |
Commercial supplier locations: EMD Millipore, Billerica, MA; Abcam, Cambridge, MA; QED Bioscience, San Diego, CA; BioLegend, San Diego, CA; Santa Cruz Biotechnology, Dallas, TX; and Sigma-Aldrich, St. Louis, MO.
IHC, immunohistochemistry; IF, immunofluorescence; IB, immunoblotting
Figure 3.

Immunoblots of neuronal, synaptic and myelin markers in extracts of midline structures from wild type and NgR123-null mice. Homogenates of midline structures dissected from 3 mice of each group were resolved by SDS-PAGE, transferred, and blotted with specific antibodies for neurons (anti-βIII-tubulin), synapses (anti-synaptophysin), myelin (anti-CNPase and anti-MBP) and a loading control protein (anti-GAPDH). Densitometry of each band was normalized to GAPDH in the same sample. Normalized data (relative to GAPDH) were compared by Student’s t-test (*, p < 0.05; **, p < 0.01). Data points are presented relative to the wild type average, with each data point for wild type (open symbols) and NgR123-null (grey symbols) presented. Averages for each genotype are denoted with a plus sign (+).
Figure 6.

Glial markers in midline extracts from wild type and NgR123-null mice. (A) Homogenates of cerebral midline tissue from wild type and NgR123-null mice (three mice each) were resolved by SDS-PAGE, transferred, and blotted with specific antibodies for astrocytes (anti-GFAP), Slit2 and ligands for NgRs including Nogo-A, OMgp and MAG. (B,C) Densitometry of each band was normalized to the loading control protein GAPDH and is presented by fold induction based on wild type. Data are presented as mean ± SE. Pairwise differences between mutant and wild type (Student t-test): *, p<0.05. Data points are presented relative to the wild type average, with each data point for wild type (open symbols) and NgR123-null (grey symbols) presented. Averages for each genotype are denoted with a plus sign (+).
Histology
Mice were anesthetized with isoflurane and subjected to intracardiac perfusion with Dulbecco’s phosphate buffered saline (PBS) and then with neutralized 4% paraformaldehyde in PBS. Brains were dissected, embedded in paraffin and sectioned to 5 µm thickness. Coronal sections of wild type and NgR123-null mice, spaced 1 mm apart, were stained with cresyl violet (Sigma-Aldrich, St. Louis, MO). White matter was stained with Luxol Fast Blue or Eriochrome/Eosin. Sections were deparaffinized, incubated in 0.1% of Luxol Fast Blue solution in 95% ethanol (Electron Microscopy Sciences, Hatfield, PA) at 56°C overnight, differentiated by aqueous 0.01% lithium carbonate. For Eriochrome/Eosin staining, the sections were deparaffinized, incubated in the staining solution containing 0.16% Eriochrome cyanine (Sigma-Aldrich) and 0.4% ferric chloride for 10 min and then differentiated by 3% ammonium hydroxide. Sections were then counterstained for 2 min with 0.5% Eosin Y in 90% ethanol (Sigma-Aldrich).
Immunohistochemistry and immunoblotting
Mice were intracardially perfused with PBS and 4% paraformaldehyde as above. Brains were embedded in paraffin and 5 µm sections were mounted on slides. Slides were briefly heated to boiling in 10 mM sodium citrate (pH 6.0) for antigen retrieval and endogenous peroxidase was inactivated by incubation in 0.3% aqueous hydrogen peroxide. Slides were blocked in 10% normal serum (appropriate to the antibody used) in PBS and then probed with primary antibodies (Table 2) to: neuronal antigen NeuN, glial fibrillary acidic protein (GFAP), myelin basic protein (MBP), 2’3’-cyclic nucleotide 3’-phosphodiesterase (CNPase), or synaptophysin. The sections were then incubated with biotin-conjugated secondary antibodies (1:500, Vector Laboratories, Burlingame, CA) and avidin-biotin complex using Vector ABC kits following the manufacturer’s protocols. Images were acquired and analyzed using a Nikon Eclipse 90i microscope with NIS-Elements image analysis software (Nikon, Melville, NY). GFAP-stained areas were quantified in coronal sections capturing the cortical midline, neocortex, striatum and hippocampus. Triplicate sections spaced 0.8 mm apart (AP, +0.6 to −1.0 mm based on Bregma for the midline, neocortex, and striatum; AP −1.0 to −2.6 for hippocampus) were immunostained and quantified as described above and normalized with GFAP staining normalized to wild type sections at the same levels.
For immunofluorescence, sections were prepared and mounted on slides as above, the slides were blocked with 10% normal serum appropriate to the primary antibody, and then were subjected primary antibodies (Table 2) to GFAP and Slit2 for 12–16 h at 4°C. Slides were washed and incubated with fluorescent secondary antibodies and 4’,6-diamidino-2-phenylindole (DAPI, Thermo Fisher, 62247) at 100 ng/ml for nuclear staining. Fluorescent images of washed and mounted slides were acquired and images analyzed as described above.
For protein immunoblotting, a block of brain tissue capturing the midline structures and CC (AP, +1.0 ~ −1.0 mm based on Bregma; ML, +2.0 to −2.0 mm; DV, 0 to −3.0 mm) was collected from wild type and NgR123-null mice and homogenized in CellLytic MT mammalian tissue lysis reagent (Sigma-Aldrich). Soluble proteins (30 µg or 3 µg) were electrophoretically resolved on 4–12% Bis-Tris NuPAGE gels, and then transferred to PVDF membranes using an iBlot system (ThermoFisher, Waltham, MA). Membranes were blocked with PBS containing 5% nonfat dry milk and 0.1% Tween 20 and then subjected to immunoblot analysis using primary antibodies (Table 2) against β-tubulin-III, synaptophysin, CNPase, MBP, GFAP, Slit2, glyceraldehyde 3-phosphate dehydrogenase (GAPDH), myelin-associated glycoprotein (MAG), Nogo-A, or OMgp. The proteins were probed with horseradish peroxidase-conjugated secondary antibodies (Cell Signaling, MA) and visualized using enhanced chemiluminescence (GE Healthcare, PA). Images were captured and band densities quantified using Carestream molecular imaging software (Carestream Health, NY) and normalized to GAPDH.
Behavior
Motor coordination was tested using a Rotarod apparatus. Mice were placed on a rotating cylinder (Rotamex, Columbus Instruments, Columbus, OH) that was initiated at 4 rpm then accelerated an additional 4 rpm every 30 sec for 5 min. Latency to fall was recorded for each mouse in 3 consecutive trials with a 5-min resting period between trials. Spontaneous locomotion was tested by placing each mouse in the center of a square acrylic box incorporating an automated activity monitor (Photobeam Activity System, San Diego Instruments, San Diego, CA) with dual horizontal grids of 16 × 16 infrared beams. The total number of beam breaks in both horizontal and vertical planes over a period of 30 min was recorded and analyzed.
Statistics
Results for each parameter were analyzed for genotype (wild type vs NgR123-null) using Student’s t-test with a significance level set to p < 0.05. All values are presented as the mean ± standard error of the mean (SE).
RESULTS
Agenesis of the corpus callosum in NgR123-null mice
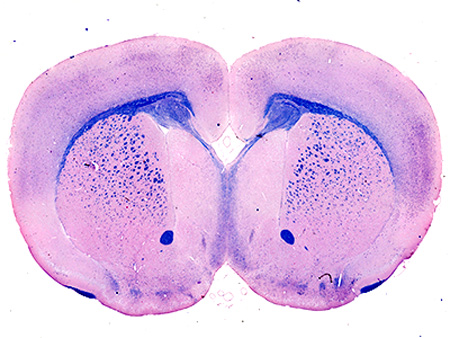
Nissl-stained adult brains of NgR123-null mice displayed normal brain size, gross brain anatomy and normal cortical lamination (data not shown). However, medial brain structures were markedly disrupted (Fig. 1A–D). At comparable anatomic levels, the fasciola cinereum was missing and the CC was interrupted (Fig. 1E,F). In contrast, the size of the striatum and septum, thickness of cortex and size of ventricles were not significantly different between wild type and NgR123-null mice. These data suggest selective impairment of medial fiber development in NgR123-null mice. This observation was evaluated using Eriochrome/Eosin and Luxol Fast Blue staining of coronal sections of 12-week old mice (Fig. 1G–N). Compared to wild type mice, NgR123-null mice routinely displayed medial agenesis with formation of bilateral Probst bundles characteristic of failure of callosally-projecting neurons to extend axons across the midline (Paul et al., 2007). The medial agenesis of the CC was observed from the rostral level (striatum and septal area) to the caudal level (hippocampal level) in NgR123-null mice and was observed in all of the NgR123-null mice examined (n=16) but in none of the wild type control mice (n=10). White matter axon bundles in the striatum, anterior commissure, internal capsule and external capsule of NgR123-null mice appeared normal. At comparable anatomic levels, the cingulum bundle was clearly delineated in NgR123-null mice (n=5) extending past the Probst bundle, then projecting ventrally (Fig. 1H). The sizes of the cingulum bundles were comparable between wild type (n=4) and NgR123-null (n=5) mice (Fig. 1G,H and data not shown).
Figure 1.

Neuroanatomy of NgR123-null mice. A–D: Nissl-stained coronal brain sections from 12-week old wild type (A,C) and NgR123-null (B,D) mice at two different levels. Callosal agenesis was found in 16/16 NgR123-null and 0/10 wild type (WT) mice. Scale bar = 1 mm. E–F: NeuN immunostained caudal midline structures in a 12-week old wild type (E) and NgR123-null (F) mouse. CC, Corpus callosum; FC, Fasciola cinereum. Arrows highlight CC agenesis. Scale bar = 200 µm. G–N: CC agenesis and Probst bundle formation in NgR123-null mice. Coronal brain sections from 12-week old wild type (G,I,K,M) and NgR123-null (H,J,L,N) mice were stained with Eriochrome/Eosin (G–J) and Luxol Fast Blue (K–N). Insets are magnified images at the midline. Arrows indicate loss of the CC and arrowheads indicate Probst bundles. Scale bar is 1 mm for the larger image and 250 µm for the insets.
To verify aberrant genesis of commissural fibers, adjacent coronal brain sections from wild type and NgR123-null mice were immunostained with anti-MBP and anti-CNPase (myelin-specific) antibodies (Fig. 2). In addition to confirming Probst bundle formation, immunostaining revealed ectopic white matter bundles in the cingulate cortex adjacent to the midline of NgR123-null mice that were stained equally with Eriochrome/Eosin, anti-MBP antibody, and anti-CNPase antibody. Ectopic white matter bundles ranged from 20 to 500 µm in diameter, extended anterior to posterior throughout a relatively long longitudinal axis (bregma −1.0 mm to 0.6 mm) and were present in 14 of the 16 mice examined (88%). Despite altered fiber morphology and ectopic myelination, medial brain myelin was quantitatively equivalent in wild type and NgR123-null mice as measured by extraction and immunoblotting of the major myelin proteins MBP and CNPase (Fig. 3). In contrast, and consistent with axon misdirection, synapses were reduced in the same area in NgR123-null mice as measured by synaptophysin immunohistochemistry (Fig. 2G,H) and immunoblotting (Fig. 3). Although anti-synaptophysin immunohistological staining was not significantly different across the whole brains of wild type compared to NgR123-null mice, the midline region of the cingulate cortex, notably in the same area as ectopic white matter bundles, was visibly less stained (Fig. 2G,H) and synaptophysin in extracts of this region was decreased 40% in NgR123-null mice (Fig. 3). Together, these data indicate developmental defects in axon navigation at the midline of NgR123-null mice.
Figure 2.

Ectopic white matter tracts in the cingulate cortex of NgR123-null mice. Myelinated fibers in coronal sections from brains of 12-week old wild type (A,C,E) and NgR123-null mice (B,D,F) were stained with Eriochrome/Eosin (A,B) or immunostained with anti-MBP (C,D) or anti-CNPase (E,F). Synaptic staining of wild type (G) and NgR123-null (H) coronal sections used anti-synaptophysin antibody. The dotted line indicates loss of synapses in the cingulate cortex of NgR123-null mice. Boxed areas at the cortical midline (top row) are presented at higher magnification below each complete coronal section (middle row), with selected areas corresponding to regions of ectopic fibers at higher power (bottom row). Scale bar = 50 µm (bottom row), 280 µm (middle row) and 1.9 mm (top row).
Commissural fiber defects in developing NgR123-null mice
Altered development of the CC was apparent by the first postnatal week in NgR123-null mice (Fig. 4). At the septal level the fibers of the CC crossed the midline in wild type mice, but collected in Probst bundle-like structures in NgR123-null mice and a gap between hemispheres was notable. During the following weeks, as myelination proceeded, misdirected axons that failed to cross the midline in NgR123-null mice were myelinated and Probst bundles were clearly apparent. The midline gap between hemispheres narrowed but persisted to adulthood (Fig. 4). At this level of resolution, other myelinated fiber tracks did not appear different between wild type and NgR123-null mice. Prenatal developmental analyses would be revealing, but were not feasible due to loss of colony fecundity (see Materials and Methods).
Figure 4.

Callosal agenesis during postnatal development in NgR123-null mice. Coronal sections from wild type (A,C,E,G,I) and NgR123-null (B,D,F,H,J) mice were stained with Eriochrome/Eosin. Brains from mice of the following post-natal ages are shown: 1 week (A,B), 2 weeks (C,D), 4 weeks (E,F), 12 weeks (G,H) and 1 year (I,J). Midline structures (right column) of NgR123-null mice at higher power are presented to the right of the corresponding complete coronal sections. Arrows indicate disconnected corpus callosum and arrowheads indicate Probst bundles. The scale bar is 1 mm for complete coronal sections and 200 µm for midline images.
Midline glia in NgR123-null mice
Midline glia, including the glial wedge, glia of the indusium griseum, and midline zipper glia are directive in the development of the corpus callosum, in part via appropriately placed repellent migratory guidance signals. Mutations that disrupt these cells result in CC agenesis. To evaluate the potential role of glia in CC agenesis in NgR123-null mice, coronal cross sections of the midline region were stained with the general glial marker anti-GFAP (Fig. 5A–D). At 12 weeks of age, the area where the CC had already developed in wild type mice displayed bilateral gliosis with a 2.5-fold increase in GFAP immunostaining in the areas of the glial wedge and indusium griseum in NgR123-null mice (Fig. 5B,E). That difference resolved to wild type levels by one year of age despite the complete absence of the CC (Fig. 5D). Except at the neocortical midline, the hippocampus, striatum and neocortex showed no significant differences in staining for GFAP (Fig. 5E and data not shown). Concurrent with CC agenesis, interhemispheric fusion was incomplete resulting in a gap bordered by the indusium griseum and unfused cingulate cortices. This gap persisted through adulthood (Fig. 5A–D,F).
Figure 5.

Glial marker and Slit2 expression in midline structures of wild type and NgR123-null mice. A–D: Coronal sections of brains from 12-week (A,B) and 1-year old (C,D) wild type (A,C) and NgR123-null (B,D) mice were immunostained for GFAP. The cingulate cortex (Cg, one of two bilateral labeled), CC, glial wedge (gw, one of two bilateral labeled) and indusium griseum (Ig) are labeled. E: Average GFAP-immunostained midline and hippocampal binary pixel areas from 5 animals from each group at each age. Values were normalized to the 12-week old wild type for presentation. Pairwise comparisons (Student t-test) revealed a significant increase in GFAP immunostaining only in the midline of 12-week old NgR123-null mice (**, p < 0.01). F: Failure of midline fusion in NgR123-null mice. Relative areas bounded by the bilateral cingulate cortices and the indusium griseum of wild type and NgR123-null mice (5 animals each) are presented (**, p < 0.01). G–N: DAPI, GFAP and Slit2 immunostaining of midline cortical regions in coronal brain sections from 12-week old wild type (G,I,K,M) and NgR123-null (H,J,L,N) mice. Staining by DAPI (blue nuclei, G,H), anti-GFAP (green, I,J), anti-Slit2 (magenta, K,L), and anti-GFAP/anti-Slit2 overlay (M,N) are shown.
Glial-derived migratory guidance signals, most notably the released soluble inhibitory molecule Slit2, have been implicated in directing axon guidance across the midline (Shu et al., 2003b; Fothergill et al., 2014). Therefore, coronal midline sections were dual immunostained for GFAP and Slit2 (Fig. 5G–N), revealing an increase in Slit2 immunostaining in the midline region of 12-week old NgR123-null mice that matches the increase in gliosis (GFAP immunostaining). Slit2 and GFAP overexpression at the NgR123-null mouse midline was further established by extraction of midline structures, resolution by gel electrophoresis and immunoblotting (Fig. 6A). Slit2 and GFAP were both increased 3-fold in NgR123-null mice compared to wild type mice (Fig. 6B).
NgR has multiple complementary binding ligands including Nogo-A, oligodendrocyte-myelin glycoprotein (OMgp) and myelin-associated glycoprotein (MAG). Immunoblots of midline structures for these proteins indicated that while Nogo-A and OMgp were equivalent in wild type and NgR123-null mice, MAG was sharply decreased (Fig. 6C), perhaps because it is the only one of the three that is nearly exclusively expressed on myelin and not neurons (Table 1). The decrease in MAG, per se, is not likely to be causative in callosal agenesis, since Mag-null mice do not display this phenotype (Li et al., 1994).
The observed increases in the number of glia and expression of repellant guidance cues in NgR123-null mice may inhibit axon crossing at the midline. To test whether apoptotic loss of neurons in the cingulate cortex, some of which extend axons to the contralateral hemisphere, also contribute to loss of the CC, the cingulate cortices were stained with TUNEL (data not shown). The data revealed no significant differences between wild type and NgR123-null mice. Furthermore, the cingulum bundle in NgR123-null mice and is generally equivalent in area to that of wild type mice. This leaves gliosis and sustained secretion of Slit2 at the midline as reasonable mechanisms for the agenesis of CC in NgR123-null mice.
Behavior
Bilateral sensorimotor coordination was impaired in NgR123-null mice compared to wild type mice as measured using an accelerating rotating rod (Rotarod) test. Significant deficits were detected equally in young adult (12 week old) and mature (1 year old) mice (Fig. 7A). Despite impaired sensorimotor coordination, NgR123-null mice displayed more spontaneous locomotion in the open field (Fig. 7B). Locomotion was increased equally in the center and periphery of the activity box (data not shown) indicating generalized increased activity rather than investigative behavior.
Figure 7.

Behaviors of NgR123-null mice. (A) Motor coordination was evaluated by Rotarod at 12 weeks and 1 year of age. Duration time on the drum was measured starting at 4 rpm and accelerating an additional 4 rpm every 30 sec. (B) Open field activity of 1 year-old mice was evaluated using infrared beam breaks in an open field frame. Total beam breaks in 120 sec were averaged for 5 trials for each mouse. Data are presented as mean ± SE. Pairwise differences were determined by Student t-test (n=8; *, p<0.05; **, p<0.01).
DISCUSSION
Mice lacking all three Nogo receptors displayed completely penetrant CC agenesis (16/16 mice) whereas wild type mice of the parent strain (129S2/SvPasCrl) showed no CC agenesis or dysgenesis (0/10 mice). Absent undetected secondary genetic alterations, these data indicate that, like some other axon directive molecules (Richards et al., 2004; Paul et al., 2007), NgRs play an essential role in corpus callosum development. The CC agenesis in these mice was characterized by anatomic pathology (Probst bundles) indicating differentiation of the neurons and outgrowth of axons that normally contribute to the CC, but failure of those axons to cross the midline. The presence of ectopic nerve tracts in NgR123-null mice further support the conclusion that cortical axon outgrowth was robust, but that normal axon migration patterns were disrupted. One straightforward hypothesis consistent with these data is that NgRs on extending axons bind to cognate ligands on midline structures to funnel the axons across the midline. If the NgRs are missing, the axons are misdirected. In considering related mutant mouse models that result in CC dysgenesis, protein tyrosine phosphatase sigma (PTPσ) is of potential relevance (Meathrel et al., 2002). NgR1 and NgR3 are receptors for axon inhibitory chondroitin sulfate proteoglycans (Dickendesher et al., 2012), as is PTPσ (Shen et al., 2009). Although this relationship is intriguing, genetic loss of PTPσ in mice results in a much less severe CC dysgenesis than we report in NgR123-null mice.
Less direct hypotheses are also consistent with these data. CC agenesis in NgR123-null mice was accompanied by other developmental alterations in the cortical midline, such as diminished midline fusion. Since midline glia contribute significantly both to midline structures and axon migration, we suspected that they might be diminished in NgR123-null mice as they are in other mouse models of callosal agenesis and dysgenesis (Shu et al., 2003a). Quite the opposite, a glial marker (GFAP) was greatly increased in both glial wedge and indusium griseum midline structures. These data indicate dysregulation of glial proliferation or glial migration to the cortical midline in NgR123-null mice and suggest an alternate contributing mechanism for CC agenesis. A careful balance of attractive factors such as Netrin1 on the floor plate and Slits from glia ensure that axons migrate ventrally to the appropriate level and then cross the midline (Fothergill et al., 2014). Whereas experimental reduction of glia or Slits at the midline results in callosal agenesis and dysgenesis (Shu et al., 2003a; Shu et al., 2003b; Unni et al., 2012), it is equally valid to propose that excessive repulsive signals would do the same by generating unbalanced repulsive signaling. Immunohistochemistry and immunoblotting of the diffusible axon repellant Slit2 supported this hypothesis, with equivalent increases in glia and Slit2 at the midline. Interestingly, gliosis that was readily apparent in young adults was not maintained into maturity (1 year-old mice) suggesting moderating factors in gliosis later in adulthood. An alternative hypothesis is that gliosis may be secondary to CC agenesis rather than a contributing factor.
There are several genetic mutations that result in human CC agenesis or dysgenesis (Paul et al., 2007). The behavioral and neurological consequences of CC agenesis in humans are variable, from little impairment to neuropsychological deficits. Initial behavioral studies reported here indicate reduced motor coordination and hyperactivity in NgR123-null mice.
The cellular basis for the finding that members of the NgR family are involved in CC development remains unresolved. Data are strong that NgRs are involved in axon regeneration (Sandvig et al., 2004; Yiu and He, 2006), and directly or indirectly control synaptic density (Wills et al., 2012). However, roles of Nogo-A beyond axon outgrowth inhibition have been reported, including regulation of neuronal stem cells, microglia and angiogenesis (Seiler et al., 2016). The earlier focus on the roles of these molecules in axon regeneration during development or after regeneration established that NgR1 is expressed by neurons and found on their axons in the developing and adult brain (Wang et al., 2002; Josephson et al., 2002; Hasegawa et al., 2005). More recent transcriptome analyses of isolated cell populations from the cerebral cortex of young mice (Table 1) revealed that NgR1 and NgR2 transcripts are more abundant in neurons, but also are well represented in oligodendrocytes and less so in astrocytes and microglia (Zhang et al., 2014). NgR3 transcripts are expressed more abundantly in microglia and less so in neurons, oligodendrocytes and astrocytes. The question of which cell(s) and which NgR(s) are required for CC genesis, which mechanisms – direct axon guidance, gliosis, or both – are responsible for agenesis, and which NgR ligands (Nogo-A, MAG, OMgp, CSPGs) are related to CC development remain unresolved.
Acknowledgments
Grant sponsor: National Institutes of Health; Grants numbers: NS037096 and NS057338.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
Study concept and design: S-WY. Acquisition of data S-WY and MGM. Data analysis and interpretation: S-WY and RLS. Manuscript preparation: S-WY and RLS.
LITERATURE CITED
- Bagri A, Marin O, Plump AS, Mak J, Pleasure SJ, Rubenstein JL, Tessier-Lavigne M. Slit proteins prevent midline crossing and determine the dorsoventral position of major axonal pathways in the mammalian forebrain. Neuron. 2002;33:233–248. doi: 10.1016/s0896-6273(02)00561-5. [DOI] [PubMed] [Google Scholar]
- Belichenko PV, Kleschevnikov AM, Salehi A, Epstein CJ, Mobley WC. Synaptic and cognitive abnormalities in mouse models of Down syndrome: exploring genotype-phenotype relationships. J Comp Neurol. 2007;504:329–345. doi: 10.1002/cne.21433. [DOI] [PubMed] [Google Scholar]
- Blaise S, Kneib M, Rousseau A, Gambino F, Chenard MP, Messadeq N, Muckenstrum M, Alpy F, Tomasetto C, Humeau Y, Rio MC. In vivo evidence that TRAF4 is required for central nervous system myelin homeostasis. PLoS ONE. 2012;7:e30917. doi: 10.1371/journal.pone.0030917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrie SC, Baeumer BE, Bandtlow CE. The Nogo-66 receptor family in the intact and diseased CNS. Cell Tissue Res. 2012;349:105–117. doi: 10.1007/s00441-012-1332-9. [DOI] [PubMed] [Google Scholar]
- Carulli D, Laabs T, Geller HM, Fawcett JW. Chondroitin sulfate proteoglycans in neural development and regeneration. Curr Opin Neurobiol. 2005;15:116–120. doi: 10.1016/j.conb.2005.01.014. [DOI] [PubMed] [Google Scholar]
- Delloye-Bourgeois C, Jacquier A, Charoy C, Reynaud F, Nawabi H, Thoinet K, Kindbeiter K, Yoshida Y, Zagar Y, Kong Y, Jones YE, Falk J, Chedotal A, Castellani V. PlexinA1 is a new Slit receptor and mediates axon guidance function of Slit C-terminal fragments. Nat Neurosci. 2015;18:36–45. doi: 10.1038/nn.3893. [DOI] [PubMed] [Google Scholar]
- Dickendesher TL, Baldwin KT, Mironova YA, Koriyama Y, Raiker SJ, Askew KL, Wood A, Geoffroy CG, Zheng B, Liepmann CD, Katagiri Y, Benowitz LI, Geller HM, Giger RJ. NgR1 and NgR3 are receptors for chondroitin sulfate proteoglycans. Nat Neurosci. 2012;15:703–712. doi: 10.1038/nn.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donahoo AL, Richards LJ. Understanding the mechanisms of callosal development through the use of transgenic mouse models. Semin Pediatr Neurol. 2009;16:127–142. doi: 10.1016/j.spen.2009.07.003. [DOI] [PubMed] [Google Scholar]
- Fothergill T, Donahoo AL, Douglass A, Zalucki O, Yuan J, Shu T, Goodhill GJ, Richards LJ. Netrin-DCC signaling regulates corpus callosum formation through attraction of pioneering axons and by modulating Slit2-mediated repulsion. Cereb Cortex. 2014;24:1138–1151. doi: 10.1093/cercor/bhs395. [DOI] [PubMed] [Google Scholar]
- Gil V, Bichler Z, Lee JK, Seira O, Llorens F, Bribian A, Morales R, Claverol-Tinture E, Soriano E, Sumoy L, Zheng B, Del Rio JA. Developmental expression of the oligodendrocyte myelin glycoprotein in the mouse telencephalon. Cereb Cortex. 2010;20:1769–1779. doi: 10.1093/cercor/bhp246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa T, Ohno K, Sano M, Omura T, Omura K, Nagano A, Sato K. The differential expression patterns of messenger RNAs encoding Nogo-A and Nogo-receptor in the rat central nervous system. Brain Res Mol Brain Res. 2005;133:119–113. doi: 10.1016/j.molbrainres.2004.10.004. [DOI] [PubMed] [Google Scholar]
- Islam SM, Shinmyo Y, Okafuji T, Su Y, Naser IB, Ahmed G, Zhang S, Chen S, Ohta K, Kiyonari H, Abe T, Tanaka S, Nishinakamura R, Terashima T, Kitamura T, Tanaka H. Draxin, a repulsive guidance protein for spinal cord and forebrain commissures. Science. 2009;323:388–393. doi: 10.1126/science.1165187. [DOI] [PubMed] [Google Scholar]
- Josephson A, Trifunovski A, Widmer HR, Widenfalk J, Olson L, Spenger C. Nogo-receptor gene activity: cellular localization and developmental regulation of mRNA in mice and humans. J Comp Neurol. 2002;453:292–304. doi: 10.1002/cne.10408. [DOI] [PubMed] [Google Scholar]
- Jouhilahti EM, Peltonen S, Peltonen J. Class III beta-tubulin is a component of the mitotic spindle in multiple cell types. J Histochem Cytochem. 2008;56:1113–1119. doi: 10.1369/jhc.2008.952002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kottis V, Thibault P, Mikol D, Xiao ZC, Zhang R, Dergham P, Braun PE. Oligodendrocyte-myelin glycoprotein (OMgp) is an inhibitor of neurite outgrowth. J Neurochem. 2002;82:1566–1569. doi: 10.1046/j.1471-4159.2002.01146.x. [DOI] [PubMed] [Google Scholar]
- Li C, Tropak MB, Gerial R, Clapoff S, Abramow-Newerly W, Trapp B, Peterson A, Roder J. Myelination in the absence of myelin-associated glycoprotein. Nature. 1994;369:747–750. doi: 10.1038/369747a0. [DOI] [PubMed] [Google Scholar]
- Meathrel K, Adamek T, Batt J, Rotin D, Doering LC. Protein tyrosine phosphatase sigma-deficient mice show aberrant cytoarchitecture and structural abnormalities in the central nervous system. J Neurosci Res. 2002;70:24–35. doi: 10.1002/jnr.10382. [DOI] [PubMed] [Google Scholar]
- Mullen RJ, Buck CR, Smith AM. NeuN, a neuronal specific nuclear protein in vertebrates. Development. 1992;116:201–211. doi: 10.1242/dev.116.1.201. [DOI] [PubMed] [Google Scholar]
- Nobile-Orazio E, Hays AP, Latov N, Perman G, Golier J, Shy ME, Freddo L. Specificity of mouse and human monoclonal antibodies to myelin- associated glycoprotein. Neurology. 1984;34:1336–1342. doi: 10.1212/wnl.34.10.1336. [DOI] [PubMed] [Google Scholar]
- Paul LK, Brown WS, Adolphs R, Tyszka JM, Richards LJ, Mukherjee P, Sherr EH. Agenesis of the corpus callosum: genetic, developmental and functional aspects of connectivity. Nat Rev Neurosci. 2007;8:287–299. doi: 10.1038/nrn2107. [DOI] [PubMed] [Google Scholar]
- Richards LJ, Plachez C, Ren T. Mechanisms regulating the development of the corpus callosum and its agenesis in mouse and human. Clin Genet. 2004;66:276–289. doi: 10.1111/j.1399-0004.2004.00354.x. [DOI] [PubMed] [Google Scholar]
- Samanta J, Alden T, Gobeske K, Kan L, Kessler JA. Noggin protects against ischemic brain injury in rodents. Stroke. 2010;41:357–362. doi: 10.1161/STROKEAHA.109.565523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandvig A, Berry M, Barrett LB, Butt A, Logan A. Myelin-, reactive glia-, and scar-derived CNS axon growth inhibitors: Expression, receptor signaling, and correlation with axon regeneration. Glia. 2004;46:225–251. doi: 10.1002/glia.10315. [DOI] [PubMed] [Google Scholar]
- Seiler S, Di SS, Widmer HR. Non-canonical actions of Nogo-A and its receptors. Biochem Pharmacol. 2016;100:28–39. doi: 10.1016/j.bcp.2015.08.113. [DOI] [PubMed] [Google Scholar]
- Shen Y, Tenney AP, Busch SA, Horn KP, Cuascut FX, Liu K, He Z, Silver J, Flanagan JG. PTPsigma is a receptor for chondroitin sulfate proteoglycan, an inhibitor of neural regeneration. Science. 2009;326:592–596. doi: 10.1126/science.1178310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu T, Butz KG, Plachez C, Gronostajski RM, Richards LJ. Abnormal development of forebrain midline glia and commissural projections in Nfia knock-out mice. J Neurosci. 2003a;23:203–212. doi: 10.1523/JNEUROSCI.23-01-00203.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu T, Sundaresan V, McCarthy MM, Richards LJ. Slit2 guides both precrossing and postcrossing callosal axons at the midline in vivo. J Neurosci. 2003b;23:8176–8184. doi: 10.1523/JNEUROSCI.23-22-08176.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unni DK, Piper M, Moldrich RX, Gobius I, Liu S, Fothergill T, Donahoo AL, Baisden JM, Cooper HM, Richards LJ. Multiple Slits regulate the development of midline glial populations and the corpus callosum. Dev Biol. 2012;365:36–49. doi: 10.1016/j.ydbio.2012.02.004. [DOI] [PubMed] [Google Scholar]
- Wang X, Chun SJ, Treloar H, Vartanian T, Greer CA, Strittmatter SM. Localization of Nogo-A and Nogo-66 receptor proteins at sites of axon-myelin and synaptic contact. J Neurosci. 2002;22:5505–5515. doi: 10.1523/JNEUROSCI.22-13-05505.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wills ZP, Mandel-Brehm C, Mardinly AR, McCord AE, Giger RJ, Greenberg ME. The nogo receptor family restricts synapse number in the developing hippocampus. Neuron. 2012;73:466–481. doi: 10.1016/j.neuron.2011.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yiu G, He Z. Glial inhibition of CNS axon regeneration. Nat Rev Neurosci. 2006;7:617–627. doi: 10.1038/nrn1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, Deng S, Liddelow SA, Zhang C, Daneman R, Maniatis T, Barres BA, Wu JQ. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34:11929–11947. doi: 10.1523/JNEUROSCI.1860-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]