

Abstract
Fatty acid (FA)-dependent oxidation is the predominant process for energy supply in normal heart. Impaired FA metabolism and metabolic insufficiency underlie the failing of the myocardium. So far, FA metabolism in normal cardiac physiology and heart failure remains undetermined. Here, we evaluate the mechanisms of FA and major metabolic substrates (termed NF) on the contraction, relaxation, and Ca2+ handling in rat left ventricular (LV) myocytes. Our results showed that NF significantly increased myocyte contraction and facilitated relaxation. Moreover, NF increased the amplitudes of diastolic and systolic Ca2+ transients ([Ca2+]i), abbreviated time constant of [Ca2+]i decay (tau), and prolonged the peak duration of [Ca2+]i. Whole-cell patch-clamp experiments revealed that NF increased Ca2+ influx via L-type Ca2+ channels (LTCC, ICa-integral) and prolonged the action potential duration (APD). Further analysis revealed that NF shifted the relaxation phase of sarcomere lengthening vs. [Ca2+]i trajectory to the right and increased [Ca2+]i for 50 % of sarcomere relengthening (EC50), suggesting myofilament Ca2+ desensitization. Butanedione monoxime (BDM), a myosin ATPase inhibitor that reduces myofilament Ca2+ sensitivity, abolished the NF-induced enhancement of [Ca2+]i amplitude and the tau of [Ca2+]i decay, indicating the association of myofilament Ca2+ desensitization with the changes in [Ca2+]i profile in NF. NF reduced intracellular pH ([pHi]). Increasing [pH]i buffer capacity with HCO3/CO2 attenuated Δ [pH]i and reversed myofilament Ca2+ desensitization and Ca2+ handling in NF. Collectively, greater Ca2+ influx through LTCCs and myofilament Ca2+ desensitization, via reducing [pH]i, are likely responsible for the positive inotropic and lusitropic effects of NF. Computer simulation recapitulated the effects of NF.
Electronic supplementary material
The online version of this article (doi:10.1007/s00424-016-1892-8) contains supplementary material, which is available to authorized users.
Keywords: Cardiac myocyte, Contraction, Intracellular Ca2+ transient, Metabolic substrates, Myofilament Ca2+ sensitivity, pH, Relaxation
The heart is an omnivorous organ that relies on both fatty acids and carbohydrates to produce myocardial ATP. Constant supply of ATP through cardiac metabolism is essential to maintain the normal functions of the heart [15, 20]. In healthy heart, fatty acid-dependent beta-oxidation accounts for 70–90 % of myocardial ATP and the remaining amount is produced through carbohydrate metabolism (such as glucose oxidation and glycolysis) [15]. However, during disease progression (hypertrophy and early stage heart failure), glucose oxidation and glycolysis become the predominant sources of cellular ATP, indicating a metabolic shift from fatty acids to glucose utilization [15]. At the end stage of heart failure, both glucose metabolism and beta-oxidation are dysregulated, resulting in energy deficiency and impaired myocardial contraction [15]. Until recently, glucose has been used as the sole metabolic substrate in most of cardiac physiological studies (despite the importance of fatty acids in cardiac metabolism). Accordingly, we aim to evaluate the impact of metabolic substrates’ supplementation on the mechanisms mediating cardiac contractility in normal rat hearts.
Excitation–contraction (E–C) coupling is the main scheme for the interpretation of myocyte contraction, where Ca2+ handling plays a central role to activate the event [2, 8]. Initially, Ca2+ influx through voltage-dependent L-type Ca2+ channels (LTCC, ICa-L) triggers more Ca2+ to release from the sarcoplasmic reticulum (SR) via ryanodine receptors (RyR). Intracellular Ca2+ ([Ca2+]i) binds to troponin C (TnC) in the thin filament (troponin complex), prompts actin association with myosin, and initiates contraction. Reuptake of Ca2+ into the SR via Ca2+-dependent ATPase (SERCA) and Ca2+ extrusion via Na+-Ca2+ exchanger (NCX) decrease intracellular Ca2+, lead to the dissociation of Ca2+ from TnC and myosin from actin, myofibril relaxes. As such, factors that increase or decrease the amount of intracellular Ca2+ plays important roles in myocardial contractile function. In addition, myofilament Ca2+ sensitivity is pivotal in determining myocyte contraction and relaxation in the functioning heart. On the other hand, recent consensus is that changes in the myofilament Ca2+ sensitivity, per se, regulates intracellular Ca2+ homeostasis and therefore myocyte contractility in normal and diseased hearts [4, 11, 19, 21]. Until recently, properties of intracellular Ca2+ handling and myofilament Ca2+ sensitivity in mediating metabolic substrates’ regulation of cardiac contractile function has not been reported yet.
Here, we aim to analyze the effects of metabolic substrate supplementation (fatty acids: palmitic acid, linoleic acid, and oleic acid; glucose; pyruvate; and lactate, termed nutrition with fatty acids, NF) on myocyte contraction, relaxation, intracellular Ca2+ handling, and myofilament Ca2+ sensitivity. Our results have shown that myocyte contraction and intracellular Ca2+ transients are enhanced and relaxation is facilitated by NF. Increased Ca2+ influx via LTCC and, counterintuitively, myofilament Ca2+ desensitization by reduced intracellular pH are involved in the effects of NF in rat cardiac myocytes.
Methods and materials
Animals and isolation of LV myocytes
Sprague–Dawley rats (12 weeks old, male) were anesthetized with pentobarbital sodium (30 mg/kg, i.p.), and the hearts were extracted and rapidly mounted onto the Langendorff perfusion system which were then perfused with a nominal Ca2+-free solution for 10 min (in mM; NaCl 135, KCl 5.4, MgCl2 3.5, glucose 5, HEPES 5, Na2HPO4 0.4, taurine 20 at pH of 7.4, NaOH), followed by a further 8-min perfusion with the same solution containing collagenase (1 mg/ml, Worthington Biochemical Co.; protease, 0.133 mg/ml, BSA 1.65 mg/ml; Ca2+ 0.05 mM). Afterward, the LV free wall was dissected and incubated in fresh collagenase-only solution. Myocytes were harvested following a further 10-min digestion period, washed and resuspended in storage solution (in mM; NaCl 120, KCl 5.4, MgSO4 5, CaCl2 0.2, Na-pyruvate 5, glucose 5.5, taurine 20, HEPES 10, mannitol 29, pH 7.4, NaOH). The myocyte suspension was stored at room temperature and cells were used within 8 h of isolation. The study protocol was in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996). It is approved by the Institutional Animal Care and Use Committee (IACUC) in Seoul National University (IACUC approval no.: SNU-101213-1).
Scheme 1.

Schematic diagram of cardiac excitation–contraction coupling in the presence of major metabolic substrates including fatty acids and glucose at physiological concentration. Contraction is enhanced with metabolic substrates’ supplementation. Mechanistically, Ca2+ influx through LTCC is increased, leads to prolonged APD and greater Ca2+ release from the SR, and increases intracellular free Ca2+. Reuptake of Ca2+ via SERCA is facilitated. Myofilament Ca2+ sensitivity is reduced, possibly via reduced pHi, which in part is responsible for greater intracellular Ca2+ level and decline in rat LV myocytes
Chemical compositions in NT and “nutrition with fatty acids-NF” solution
Metabolic substrates (oleic acid 200 μM, palmitic acid 100 μM, linoleic acid 100 μM, lactate 1 mM, pyruvate 100 μM, and carnitine 50 μM) were supplemented to the normal Tyrode solution (NT, in mM: NaCl 141.4, KCl 4, NaH2PO4 0.33, MgCl2 1, HEPES 10, glucose 5.5, CaCl2 1.8). Fatty acids (oleic acid, palmitic acid, linoleic acid) were dissolved in 1.35 mM (0.09 g/ml) BSA in NT solution at 70 °C for 3 h to make respective stock solutions (10 mM). The composition and the concentrations of metabolic substrates are modified from blood sample results of normal rats [14].
Measurement of contraction and Ca2+ transients
Changes in sarcomere length and Ca2+ transients were measured in LV myocytes by using a video-sarcomere detection system (IonOptix Corp). For Ca2+ measurements, LV myocytes were preincubated with a Ca2+ indicator, acetoxymethyl ester of Fura-2 AM (2 μM) in perfusion solution containing 250 μM Ca2+ for 15 min at room temperature in the dark. LV myocytes were then washed in perfusion solution containing 500 μM Ca2+ for 10 min. The Fura-2 loaded LV myocytes were kept in perfusion solution (500 μM Ca2+) before being used. Measurements from at least 10 steady-state contractions were averaged for each myocyte and for each stage of the experimental protocols. All experiments were carried out at 36 ± 1 °C and field stimulated at 2 Hz.
Measurement of myofilament Ca2+ sensitivity
Sarcomere length and Fura-2 ratio (F360/F380) were recorded simultaneously (field stimulation, 2 Hz). The phase-plane loop of the Fura-2 ratio vs. sarcomere shortening of the same myocyte was plotted and the data were averaged for each group. In each plot, define Fura-2 ratio at 50 % relaxation (EC50). Compare both the loop and EC50 of each intervention.
Measurement of L-type Ca2+ current using whole-cell voltage clamp technique
L-type Ca2+ current (ICa) was measured with whole-cell voltage clamp technique (Axon instrument, 200 A) in LV myocytes in NT and in NF. ICa was elicited by 200-ms step depolarization protocol from a holding potential of −40 mV to test potentials from −60 mV to +40 mV. Current–voltage relationship, peak current density (pA/pF at 0 mV), and ICa integral at −20, 0, and +20 mV for 50 ms from the ICa peak were analyzed. Pipette solution for ICa measurement contained (in mM) 120 K aspartate, 20 KCl, 10 HEPES, 5 MgATP, 10 NaCl, pH adjusted to 7.2 using KOH.
Measurement of action potential using current clamp technique
The action potential profile of the myocyte was recorded with current clamp technique in NT and in NF (sampling rate: 10 kHz) at 35–36 °C (Axon Instruments, 200 A). The pipette (resistance 1–2 MΩ) solution contains (in mM) 110 K-aspartate, 30 KCl, 5 NaCl, 10 HEPES, 0.1 EGTA, 5 MgATP, 5 creatine phosphate, and 0.05 cAMP adjusted to pH 7.2.
Measurement of intracellular pH (pHi)
Temporal intracellular H+ was measured using a membrane-permeant acetoxymethyl ester form of the fluorescent H+-sensitive indicator, SNARF-1 AM (10 μM, 5–10 min, Molecular Probes, Eugene, OR). All experiments were performed at 37 °C. In some experiments, pH buffer capacity was increased with NaHCO3 (25 mM) + CO2 (5 %) instead of HEPES in the perfusion solution to examine the effect of pH on myocyte responses to NF.
Mathematical modeling
The endocardial myocyte model by Pandit et al. [17] was employed to simulate the effects of fatty acids on electrophysiology and Ca2+ handling of rat ventricular myocytes.
The effects of intracellular acidosis induced by fatty acids [12] on cardiac ion channels and Ca2+ handling proteins were adopted from the results by Saegusa et al. [22]. In their report, the simulated effects of intracellular acidosis (~0.5 U) on ICaL were 24 % reduction in the maximum conductance, 9 mV leftward shift of activation, 3.5 mV leftward shift of inactivation, and 20 % slowing of inactivation. The simulated effects of intracellular acidosis on other cardiac ion channels and transporters were 50 % reduction in maximum conductance of Ito, 20 % reduction in maximum conductance of INaCa, 40 % reduction in maximum Ca2+ flux through RyR channel, and 40 % reduction in maximum pumping rate of sarcoplasmic reticulum Ca2+-ATPase (SERCA), respectively. As we assumed the change in intracellular pH to be ~0.1 U, all the changes were scaled down to 1/5 of those from Saegusa et al. [22] and are summarized in Table 1. The effects of fatty acids on myofilament Ca2+-sensitivity were simulated by reducing Ca2+ on rates for troponin high- and low-affinity sites (see Table 1).
Table 1.
Model parameters for simulation of the effects of NF on membrane currents and sarcoplasmic reticulum function
| Parameter | Unit | Description | NT | NF |
|---|---|---|---|---|
| gCaL | nS | Maximum conductance for ICaL | 31.000 | 29.512 |
| Vh,act,CaL | mV | Half-activation voltage for ICaL | −15.3 | −17.1 |
| kact,CaL | mV | Slope factor of activation for ICaL | 5.0 | 4.8 |
| Vh,inact,CaL | mV | Half-inactivation voltage for ICaL | −26.7 | −27.4 |
| SFtau,inact,CaL | – | Scaling factor for inactivation τ of ICaL | 1.00 | 1.04 |
| gto | nS | Maximum conductance of Ito | 569.2575 | 512.3318 |
| kNaCa | mM−4 | Scaling factor for INaCa | 9.984·10−3 | 9.584·10−3 |
| ν1 | ms−1 | Maximum RyR (ryanodine) channel Ca2+ flux | 1.800 | 1.656 |
| KSR | – | Scaling factor for Ca2+-ATPase | 1.00 | 0.92 |
| mM−1 s−1 | Ca2+ on rate for troponin high-affinity sites | 200 | 20 | |
| mM−1 s−1 | Ca2+ on rate for troponin low-affinity sites | 40 | 4 |
See glossary of Pandit’s model [17] for detailed explanation. All other model parameters are the same as those of endocardial myocyte in the Pandit’s model.
Statistics
Data were expressed as means ± S.E. or as relative to control (100 %) and n indicates the number of cells used. For all comparisons, cells were obtained from a minimum of three hearts per treatment group per protocol. Data were analyzed using Student’s t test and one-way ANOVA. A value of P < 0.05 was considered to be statistically significant.
Results
LV myocyte contraction and relaxation with metabolic substrates’ supplementation (NF)
As shown in Fig. 1a, b, NF increased LV myocyte contraction (P < 0.001 between NT and NF, n = 29 & 29) and facilitated relaxation (time to 50 % relaxation, P < 0.02) without changing diastolic sarcomere length (P = 0.5). Supplementation of three types of fatty acids (linoic acid, oleic acid and palmitic acid) increased sarcomere shortening (P < 0.01 between NT and 3FA, n = 15 and n = 15, Fig. 1c), similar to that observed with NF. These results suggest that metabolic substrates and fatty acids increase myocyte contraction in healthy rat heart.
Fig. 1.

Effect of NF on LV myocyte contraction. a Representative raw traces of sarcomere shortening and relengthening in the presence of NF. b Mean values of the diastolic sarcomere length and the amplitude of sarcomere shortening (peak height) and 50 % relaxation time (TR50). Diastolic sarcomere length was not different between NT and NF. Sarcomere shortening was significantly increased in NF and TR50 was shorter by NF. c Representatives raw traces and mean values of three fatty acids (3FAs) only on myocyte contraction. 3FAs increased myocyte contraction. d Representative raw traces and mean values of a potent antioxidant NAC on myocyte contraction in NF. NAC did not affect the increment of myocyte contraction by NF
Since fatty acids are shown to increase intracellular reactive oxygen species (ROS) in cardiac myocytes [20, 27], possibly due to increased beta-oxidation in mitochondria and ROS potentiates myocyte contraction in murine heart [27], we tested whether ROS is responsible for the inotropic effect of metabolic substrates. Incubation of LV myocytes with a potent antioxidant, N-acetyl-cysteine (NAC, 100 μM), did not prevent NF augmentation of myocyte contraction (P < 0.001 between NT + NAC and NF + NAC, n = 16, Fig. 1d). These results exclude the role of ROS in mediating NF-induced myocyte contraction in rat hearts.
Intracellular Ca2+ transient ([Ca2+]i) with NF
Next, we tested whether NF increased [Ca2+]i. As shown in Fig. 2a, b, NF significantly increased the diastolic and systolic [Ca2+]i (F360/380: P < 0.001 and P < 0.001, n = 18, n = 18). Analysis of [Ca2+]i profile showed that the time constant of [Ca2+]i decay (tau, ms) was abbreviated in NF (P < 0.001, Fig. 2b). Nevertheless, the peak [Ca2+]i duration (90 % peak time + 10 % time to relaxation, PT90 + TR10) and TR50 of [Ca2+]i were prolonged (P = 0.009 and P = 0.002, n = 18 and n = 18, respectively). These results suggest that NF increased [Ca2+]i transients and decay kinetics of [Ca2+]i but prolonged peak time of [Ca2+]i during systole.
Fig. 2.

NF regulation of intracellular Ca2+ transients. a Representative [Ca2+]i transients in NT and NF. b Mean values of [Ca2+]i transient parameters. NF significantly increased diastolic [Ca2+]i and peak amplitude of [Ca2+]i. Time to 50 % relaxation and peak time duration (PT90 + TR10) were prolonged; however, time constant of [Ca2+]i decay (tau) was facilitated in NF
L-type Ca2+ current (ICa) and action potential profile with NF
We aimed to test the key elements of excitation-contraction coupling responsible for increased myocyte [Ca2+]i and contraction in NF. Figure 3a, b showed raw traces and averaged current–voltage (I–V) relationships of ICa in LV myocytes before and after NF. And the inset in Fig. 3a showed raw traces of ICa at 0 mV for Ca2+ influx analysis. NF reduced the amplitude of ICa at 0 mV (current density of ICa at 0 mV, pA/pF: P = 0.03 between NT and NF, n = 34, n = 11) but slowed the inactivation of ICa at 0 mV (in ms, tauslow: P = 0.005; taufast: P = 0.3, Fig. 3c). As a result, total Ca2+ influx via LTCC (integral of ICa) was greater in NF (P = 0.05 between NT and NF at 0 mV, Fig. 3d). Similarly, integral of ICa was bigger at −20 mV (P = 0.05) but not at +20 mV.
Fig. 3.
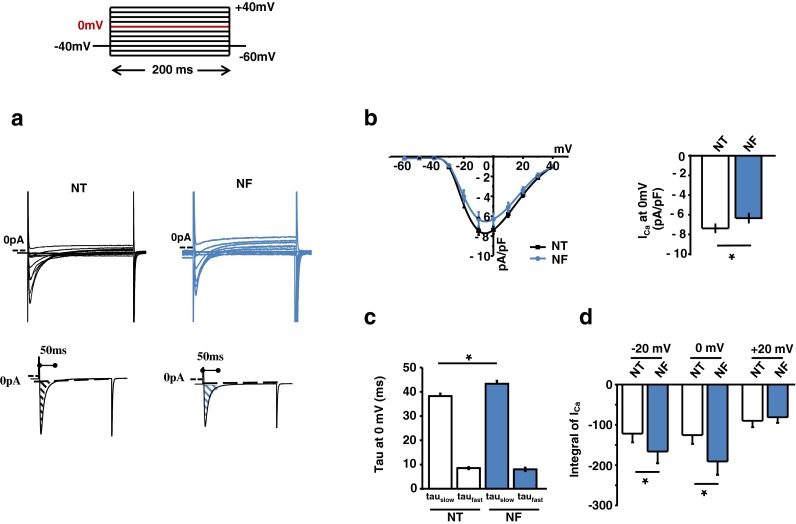
Patch-clamp recordings of LTCC activities in NF. a, b Pulse protocol, representative ICa and the corresponding I-V relationship, peak ICa at 0 mV, inactivation parameters, and the integral of Ca2+ influx in NF. NF reduced peak ICa density, prolonged slow inactivation of ICa, and increased integral of ICa at 0 mV and −20 mV
Greater ICa influx in NF may be responsible for the prolongations of action potential (AP) plateau (and therefore APD) and [Ca2+]i duration. Accordingly, we examined the APD duration in NF. As shown in Fig. 4a, b, APD20, APD50, and APD90 were significantly prolonged in NF (P < 0.05, P < 0.05, and P < 0.05, n = 12). Collectively, these results suggest that Ca2+ influx via LTCC is greater and APD is prolonged, which may be responsible for increased [Ca2+]i amplitudes and prolonged peak time of [Ca2+]i in NF.
Fig. 4.

Effect of NF on action potential profile. a Representative action potential profile in NT and NF. b Mean value of action potential duration parameters. NF significantly prolonged the repolarization duration of APD (APD20, APD50, APD90)
Myofilament Ca2+ sensitivity with NF
To test whether myofilament Ca2+ sensitivity is regulated by NF and contributes to greater contraction and faster relaxation, we recorded sarcomere shortening and [Ca2+]i simultaneously in Fura-2-loaded LV myocytes and plotted a phase-plane loop of the changes in sarcomere length vs. [Ca2+]i transient with and without NF. As shown in Fig. 5a, b, relaxation phase of the sarcomere length-[Ca2+]i transient relationship shifted to the right in NF; [Ca2+]i at 50 % sarcomere relengthening (EC50) was significantly increased (P < 0.001, n = 18). These results suggest that myofilament Ca2+ sensitivity was significantly reduced by NF.
Fig. 5.

NF regulation of myofilament Ca2+ sensitivity. a Simultaneous recordings of LV myocyte sarcomere shortening/relengthening and intracellular Ca2+ transients in NT and in NF. b Phase-plane loop of sarcomere shortening/relengthening vs. [Ca2+]i transient. The relaxation phase of the loop shifted to the right in NF compared to that in NT. Ca2+ concentration at 50 % sarcomere relengthening (EC50) was significantly larger in NF
Myofilament Ca2+ desensitization regulates [Ca2+]i in NF
Myofilament buffers Ca2+ and changing myofilament Ca2+ sensitivity affects [Ca2+]i homeostasis [4, 11]. Accordingly, we tested whether myofilament Ca2+ desensitization with a potent myosin ATPase inhibitor, BDM, prevents increased [Ca2+]i and facilitated tau of [Ca2+]i in NF. As shown in Fig. 6a, b, incubation of LV myocytes with BDM (5 mM) abolished NF-induced increase in diastolic and systolic [Ca2+]i and faster tau (P = 0.8 in diastole; P = 0.22 in systole; P = 0.96 for tau between NT and NF with BDM; n = 12). These results suggest that reduced myofilament Ca2+sensitivity in NF may, at least in part, increase [Ca2+]i and facilitate [Ca2+]i decline in rat LV myocytes. The duration of [Ca2+]i peak (90 % time to peak +10 % [Ca2+]i decline, PT90 + TR10), however, remain prolonged in NF in the presence of BDM (P < 0.001, between NT + BDM and NF + BDM, n = 12, Fig. 6b).
Fig. 6.

Effect of NF on [Ca2+]i in BDM-pretreated LV myocytes. Representative [Ca2+]i (a) and mean vales of [Ca2+]i transient parameters (b). NF-induced increase in diastolic and systolic [Ca2+]i were abolished by BDM (5 mM). Time constant of Ca2+ decay (tau) was not facilitated in NF in the presence of BDM. Peak time duration remained unaltered by BDM pretreatment
Regulation of intracellular pHi by NF
To investigate whether intracellular pH ([pHi]) is reduced by NF and contributes to reduced myofilament Ca2+ sensitivity, we detected changes of pHi in SNARF-1/AM-loaded LV myocytes. As shown in Fig. 7a, the pHi was gradually decreased with NF (3–5 min). Averaged results showed that NF significantly reduced pHi (P = 0.03, n = 10, Fig. 7b).
Fig. 7.

NF regulation of intracellular pHi. a Representative recording of pHi in NT and NF using HEPES buffer. b Mean values of pHi in NT and NF, pHi was significantly reduced by NF
Increasing pH buffer capacity in NF using NaHCO3 + CO2 (5 %) weaken the changes in pHi by NF (Supplementary Fig. 1a, b). Importantly, such a modification in pHi abolished NF-induced myofilament Ca2+ desensitization, increase in the amplitude of [Ca2+]i and the prolongation of the peak time of [Ca2+]i (supplementary Fig. 2a, b). These results suggest that reduced pHi may play an important role in mediating the effects of NF on the myofilament Ca2+ sensitivity and its regulation of [Ca2+]i handling.
Computer simulation of ICa, [Ca2+]i and APD with NF
Next, we employed the computer simulation to further confirm whether reduced pHi and concomitant myofilament Ca2+-desensitization affect [Ca2+]i, ICa, and AP in the same way as NF. Indeed, the peak amplitude of [Ca2+]i was increased from 1.34 to 1.63 μM, qualitatively similar to the experimental results in Fig. 2 (Fig. 8a). In addition, peak [Ca2+]i duration was also prolonged (Fig. 8a), in line with the results observed in Fig. 2b. I–V relationship of ICa and the peak ICa at 0 mV were slightly reduced in the simulation while the integral of ICa was increased (Fig. 7b). In addition, the AP duration was prolonged in the simulation (APD50 from 32.8 to 37.4 ms and APD90 from 82.6 to 96.4 ms), which is in line with the experimental results.
Fig. 8.
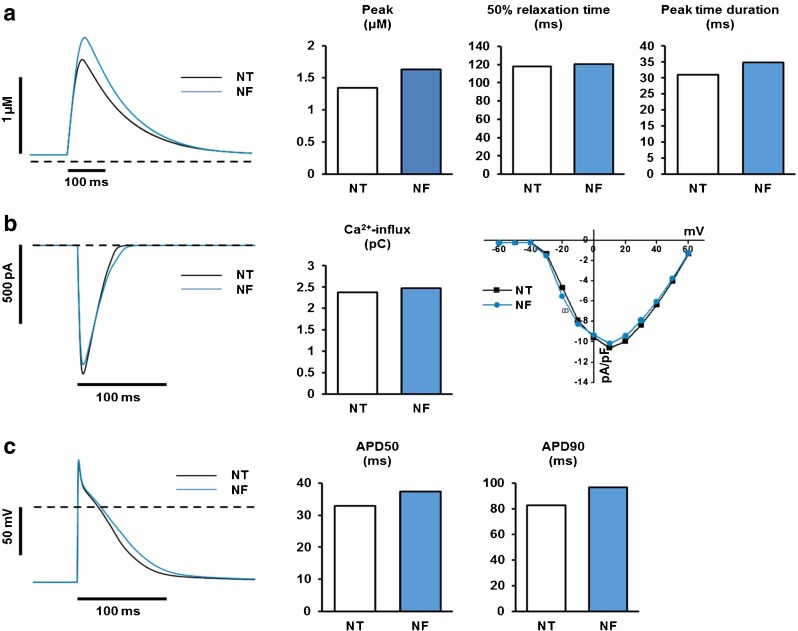
Simulated effects of NF on electrical properties and Ca2+ regulation of rat ventricular myocytes. Ca2+ transient morphology (a), ICaL (b), and APD (c), a Simulated effects of NF on morphology and relaxation time of Ca2+ transient. Measurements of 50 % relaxation time and peak time duration were conducted in the same way as those in Fig. 2b. The dashed line indicates 10 nM [Ca2+]i level. b Simulated effects of NF on ICaL and Ca2+-influx through ICaL. The left panel shows the effects of NF on ICaL during AP. The middle panel compares the total Ca2+ influx through ICaL between NT and NF. The right panel shows the effects of NF on ICaL density obtained by a voltage-clamp protocol used in animal experiments (see Fig. 3b). The dashed line indicates zero current level. c Simulated effects of NF on morphology and duration of AP. Pacing frequency is 1 Hz. APD50 was calculated as the difference between time at 50 % depolarization and time at 50 % repolarization. APD90 was calculated as the difference between time at 10 % depolarization and time at 90 % repolarization. The dashed line indicates zero voltage level
Discussion
The present study showed that supplementation of metabolic substrates (including fatty acids, physiological concentration of glucose) increased LV myocyte contraction and facilitated relaxation from rat heart by increasing Ca2+ influx via LTCC and increasing [Ca2+]i transients, despite that myofilament Ca2+ sensitivity was reduced. Importantly, both experimental and computer simulation results suggest that myofilament Ca2+ desensitization, at least in part, contributes to increased [Ca2+]i , greater contraction, abbreviated tau, and faster relaxation by NF. Intracellular pH, which is reduced by NF, is important in mediating the effects of NF on myocyte contraction, relaxation, and [Ca2+]i. These results suggest that supplementation of metabolic substrates (including fatty acids) potentiates myocyte inotropy and lusitropy by regulating key elements of Ca2+ handling processes and myofilament Ca2+ sensitivity, in addition to their effects on cardiac metabolism.
The main theme of the study was that supplementation of metabolic substrates essential for cardiac metabolism (e.g. FAs for beta-oxidation in addition to glucose, NF) influenced myocyte function from normal rat hearts. That is, NF enhanced LV myocyte contraction and promoted myocyte relaxation in almost all the myocytes studied. Furthermore, NF increased diastolic and systolic [Ca2+]i, facilitated Ca2+ reuptake into the SR via SERCA, and prolonged peak time duration of [Ca2+]i. Since LTCC and Ca2+ release from the SR are important in shaping AP and [Ca2+]i profiles and myofilament Ca2+ sensitivity and myofilament mechanics dominate myocyte contraction, we analyzed these parameters in the presence of NF. As shown in both experimental and computer simulation results, peak ICa density was reduced by NF; however, the integral of ICa (total Ca2+ influx) was increased due to slower inactivation of LTCC (especially at the voltage range of AP plateau, −20 and 0 mV), suggesting more Ca2+ is fluxed into the myocyte under these conditions. Accordingly, APD was prolonged remarkably with NF. These results suggest that [Ca2+]i elevation with NF may be due to increased Ca2+ influx via LTCC and the changes in LTCC are likely responsible for the prolongations of APD and the peak duration of [Ca2+]i.
NF regulation of myofilament Ca2+ sensitivity was analyzed in contracting LV myocytes. Our results showed a significant reduction in myofilament Ca2+ sensitivity with NF but myocyte contraction was increased. The seemingly counterintuitive results stress the importance of Ca2+ handling in greater myocyte contraction with NF. It should be noted that alterations in the myofilament Ca2+ sensitivity, in general, is associated with changes in Ca2+ binding affinity to TnC and [Ca2+]i buffering capacity [11, 19]. For example, Robinson et al. showed that the Ca2+ binding affinity of the myofilament complex was reduced in dilated cardiomyopathy due to the mutations in thin filament, where myofilament Ca2+ sensitivity was reduced [19]. Conversely, hypertrophic cardiomyopathy with myofilament Ca2+-sensitizing mutant increased Ca2+-binding affinity and Ca2+ buffering [19, 21]. In fact, previous results from our laboratory have shown that myofilament Ca2+ desensitization in LV myocytes from angiotensin II-induced hypertensive rat hearts was associated with greater [Ca2+]i [13] and myofilament Ca2+ desensitization with BDM increased [Ca2+]i in sham but not in hypertension (where myofilament Ca2+ sensitivity was reduced). Furthermore, myofilament Ca2+ desensitization in hypertension, with BDM or with high stimulation frequency, prompted greater ICa inhibition due to [Ca2+]i increment [25]. Here, the current findings that myofilament Ca2+ desensitization with BDM prevented NF-induced increase in the amplitude of [Ca2+]i are consistent to our previous results. Furthermore, computer simulation with reduced Ca2+ binding and myofilament Ca2+ desensitization recapitulated greater [Ca2+]i and the prolongation of the peak duration of [Ca2+]i, supporting the experimental results with NF. These results strongly suggest that both LTCC and myofilament contribute to increased [Ca2+]i and myocyte contraction/relaxation with NF in rat heart.
Multiple mechanisms may underlie the regulation of myofilament Ca2+ sensitivity and LTCC by NF. It is possible that enhanced mitochondrial activity secondary to the supplementation of metabolic substrates may produce greater ROS via oxidative phosphorylation [15] and ROS can potentiate myocyte contraction via the activation of second messengers including protein kinase A (PKA) [6, 27]. As such, we tested whether intracellular ROS is involved in greater myocyte contraction with NF. Although we did not detect the ROS production from LV myocytes with NF, preincubation with a potent antioxidant, NAC failed to prevent the effect of NF on myocyte shortening. Furthermore, inhibition of PKA (with membrane permeable PKA inhibitor, PKA-amide 14-22) failed to affect NF regulation of myocyte contraction (data not shown). These results excluded the involvement of ROS in mediating the effect of NF.
Our results showed that intracellular pH was reduced by NF. Since low pH is a potent modulator of myofilament Ca2+ sensitivity in cardiac and skeletal muscle [1, 9, 16, 24], reduced pH may mediate the reduced myofilament Ca2+ sensitivity and the subsequent regulation of [Ca2+]i. To confirm, when intracellular pH buffering was increased with NaHCO3 + CO2, the effects of NF on myofilament Ca2+ sensitivity and [Ca2+]i were diminished. Alternatively, reduced pH may increase [Ca2+]i via enhancing the activity of Na+-H+ exchanger and the reverse mode of cardiac Na+-Ca2+ exchanger [5]. It is possible that increased cellular metabolic status in NF reduced intracellular pH. Alternatively, it is well established that pyruvate or lactate enters the myocyte with one proton through the sarcolemmal monocarboxylate-proton symporter [7] and consequently reduces intracellular pH [18]. Mechanism leading to the changes in intracellular pH needs further investigation.
Another possible mechanism for myofilament Ca2+ desensitization is intracellular ATP. This is because ATP desensitizes myofilament response to Ca2+ in muscle and myocardium [3, 10]. Preliminary results showed that cellular ATP level tended be elevated in normal rat LV myocyte homogenates preincubated with palmitic acid (30 min) (data not shown). The level of ATP was not detected with NF due to the lack of possibility for real-time temporal measurement in the current experimental setting. The details of myofilament Ca2+ desensitization and myofilament buffering of Ca2+ with metabolic substrates need further investigation.
Simulated effects of NF on Ca2+ transients, ICaL, and AP
Most of experimentally observed effects of NF on AP, Ca2+ transients, and ICa were successfully reproduced by computer simulation which assumed that the effects of NF are all mediated by lowered intracellular pH. Saegusa et al. [22] proposed that the increased H+ concentration at low intracellular pH affects ICa by modulating charge screening/binding of surface charge and permeability of LTCC. As NF was found to reduce intracellular pH ~0.1 U (Fig. 8a) in our experimental condition, modifications proposed by Saegusa et al. [22] were all scaled down to 1/5 and were applied to our simulation. The increased intracellular H+ is also known to raise intracellular Ca2+ by unloading Ca2+ from Ca2+-buffering proteins (coupled Ca2+/H+ transport by cytoplasmic buffers regulates local Ca2+ and H+ ion signaling [23]). In our simulation, the ability of increased intracellular H+ to unload Ca2+ from Ca2+-buffering proteins was embodied by assuming it reduces Ca2+ on rates for TnC. Therefore, the effects of NF were reproduced in our simulation by assuming that L-type Ca2+ channels and Ca2+-buffering are both affected in response to increased intracellular H+. The altered LTCC was found to be responsible for both increased amplitude of Ca2+ transients and the prolongation of AP in our simulation, whereas the altered Ca2+-buffering was found to be mainly responsible for increased amplitude of Ca2+ transients rather than prolongation of AP (data not shown).
In conclusion, our results clearly show that supplementation of metabolic substrates, including fatty acids, is important in strengthening cardiac metabolism and contractile function. Under these conditions, Ca2+ handling processes and myofilament play in concert to potentiate intracellular Ca2+ level and induce greater contraction. Our results provide useful evidence for better understanding of cardiac excitation–contraction coupling with comprehensive metabolic supplies.
Electronic supplementary material
(PDF 17.5 kb)
(PDF 161 kb)
Compliance with ethical standards
Conflicts of interests
The authors declare that they have no conflicts of interest.
Funding
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science, and Technology (2013068067); by the Brain Korea 21 Graduate Program of the Korean Ministry of Education, Science, and Technology, Seoul National University Hospital; the Korean Society of Hypertension (2013); SK Telecom Research Fund (no. 3420130290); and from the National Natural Science Foundation of China (NSFC, 31460265).
Footnotes
Zai Hao Zhao and Jae Boum Youm equal contribution.
Contributor Information
Lan Cui, Email: lancui@ybu.edu.cn.
Yin Hua Zhang, Phone: +82 2 740 8223, Email: yinzhang87@gmail.com.
References
- 1.Ashley CC, Moisescu DG. Effect of changing the composition of the bathing solutions upon the isometric tension-pCa relationship in bundles of crustacean myofibrils. J Physiol. 1977;270(3):627–652. doi: 10.1113/jphysiol.1977.sp011972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 3.Best PM, Donaldson SK, Kerrick WG. Tension in mechanically disrupted mammalian cardiac cells: effects of magnesium adenosine triphosphate. J Physiol. 1977;265(1):1–17. doi: 10.1113/jphysiol.1977.sp011702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Briston SJ, Dibb KM, Solaro RJ, Eisner DA, Trafford AW. Balanced changed in Ca buffering by SERCA and troponin contribute to Ca handling during β-adrenergic stimulation in cardiac myocyte. Cardiovasc Res. 2014;104(2):347–354. doi: 10.1093/cvr/cvu201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cingolani HE, Ennis IL, Aiello EA, Pérez NG. Role of autocrine/paracrine mechanisms in response to myocardial strain. Pflugers Arch. 2011;462(1):29–38. doi: 10.1007/s00424-011-0930-9. [DOI] [PubMed] [Google Scholar]
- 6.de Hemptinne A, Marrannes R, Vanheel B. Influence of organic acids on intracellular pH. Am J Phys. 1983;245(3):C178–C183. doi: 10.1152/ajpcell.1983.245.3.C178. [DOI] [PubMed] [Google Scholar]
- 7.Dong F, Zhang X, Ren J. Leptin regulates cardiomyocyte contractile function through endothelin-1 receptor-NADPH oxidase pathway. Hypertension. 2006;47(2):222–229. doi: 10.1161/01.HYP.0000198555.51645.f1. [DOI] [PubMed] [Google Scholar]
- 8.Eiser DA, Isenberg G, Sipido KR (2003) Normal and pathological excitation-coupling in the heart – an overview 546(Pt 1):3–4 [DOI] [PMC free article] [PubMed]
- 9.Fabiato A, Fabiato F. Effects of pH on the myofilaments and the sarcoplasmic reticulum of skinned cells from cardiac and skeletal muscles. J Phyisol. 1978;276:233–255. doi: 10.1113/jphysiol.1978.sp012231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Godt RE. Calcium-activated tension of skinned muscle fibers of the frog. Dependence on magnesium adenosine triphosphate concentration. J Gen Physiol. 1974;63(6):722–739. doi: 10.1085/jgp.63.6.722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huke S, Knollmann BC. Increased myofilament Ca2+ sensitivity and arrhythmias susceptibility. J Mol Cell Cardiol. 2010;48(5):824–833. doi: 10.1016/j.yjmcc.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hamilton JA, Civelek VN, Kamp F, Tornheim K, Corkey BE. Changes in internal pH caused by movement of fatty acids into and out of clonal pancreatic beta-cells (HIT) J Biol Chem. 1994;269(33):20852–20856. [PubMed] [Google Scholar]
- 13.Jin CZ, Jang JH, Kim HJ, Wang Y, Hwang IC, Sadayappan S, Park BM, Kim SH, Jin ZH, Seo EY, Kin KH, Kim YJ, Kim SJ, Zhang YH. Myofilament Ca2+ desensitization mediates positive lusitropic effect of neuronal nitric oxide synthase in left ventricular myocytes from murine hypertensive heart. J Mol Cell Cardiol. 2013;60:107–115. doi: 10.1016/j.yjmcc.2013.04.017. [DOI] [PubMed] [Google Scholar]
- 14.Khairallah M, Labarthe F, Bouchard B, Danialou G, Petrof BJ, Des Rosiers C. Profiling substrate fluxes in the isolated working mouse heart using 13C-labeled substrates: focusing on the origin and fate of pyruvate and citrate carbons. Am J Physiol Heart Circ Physiol. 2004;286(4):H1461–H1470. doi: 10.1152/ajpheart.00942.2003. [DOI] [PubMed] [Google Scholar]
- 15.Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Res. 2010;90(1):207–258. doi: 10.1152/physrev.00015.2009. [DOI] [PubMed] [Google Scholar]
- 16.Liou YM, Kuo SC, Hsieh SR. Differential effects of a green tea-derived polyphenol (−)-epigallocatechin-3-gallate on the acidosis-induced decrease in the Ca2+ sensitivity of cardiac and skeletal muscle. Pflugers Arch. 2008;456(5):787–800. doi: 10.1007/s00424-008-0456-y. [DOI] [PubMed] [Google Scholar]
- 17.Pandit SV, Clark RB, Giles WR, Demir SS. A mathematical model of action potential heterogeneity in adult rat left ventricular myocytes. Biophys J. 2001;81(6):3029–3051. doi: 10.1016/S0006-3495(01)75943-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Poole RC, Halestrap AP. Transport of lactate and other monocarboxylates across mammalian plasma membranes. Am J Phys. 1993;264:C761–C782. doi: 10.1152/ajpcell.1993.264.4.C761. [DOI] [PubMed] [Google Scholar]
- 19.Robinson P, Griffiths PJ, Watkins H, Redwood CS. Dilated and hypertrophic cardiomyopathy mutation in troponin and alpha-tropomyosin have opposing effects on the calcium affinity of cardiac thin filaments. Circ Res. 2007;101(12):1266–1273. doi: 10.1161/CIRCRESAHA.107.156380. [DOI] [PubMed] [Google Scholar]
- 20.Shipp JC, Opie LH, Challoner D. Fatty acid and glucose metabolism in the perfused heart. Nature. 1961;189:1018–1019. doi: 10.1038/1891018a0. [DOI] [Google Scholar]
- 21.Schober T, Huke S, Venkataraman R, Gryshchenko O, Kryshtal D, Hwang HS, Baudenbacher FJ, Knollmann BC. Myofilament Ca sensitization increase cytosolic Ca biding affinity, alters intracellular Ca homeostasis and causes pause-dependent Ca-triggered arrhythmia. Cir Res. 2012;111(2):179–179. doi: 10.1161/CIRCRESAHA.112.270041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saequsa N, Moorhouse E, Vaughan-Jones RD, Spitzer KW. Influence of pH on Ca2+ current and Ca2+ signaling in ventricular myocytes. J Gen Physiol. 2011;138(5):537–559. doi: 10.1085/jgp.201110658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Swietach P, Yunm JB, Saequsa N, Leam CH, Spitzer KW, Vaughan-Jones RD. Coupled Ca2+/H+ transport by cytoplasmic buffers regulates local Ca2+ and H+ ion signaling. Proc Natl Acad Sci U S A. 2013;110(22):E2064–E2073. doi: 10.1073/pnas.1222433110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takahashi R, Shimazaki Y, Endoh M. Decrease in Ca2+-sensitizing effect of UD-CG 212 Cl, a metabolite of pimobendan, under acidotic condition in canine ventricular myocardium. J Pharmacol Exp Ther. 2001;298(3):1060–1066. [PubMed] [Google Scholar]
- 25.Wang Y, Youm JB, Jin CZ, Shin DH, Zhao ZH, Seo EY, Jang JH, Kim SJ, Jin ZH, Zhang YH. Modulation of L-type Ca2+ channel activity by neuronal nitric oxide synthase and myofilament Ca2+ sensitivity in cardiac myocytes from hypertensive rat. Cell Calcium. 2015;58(3):264–274. doi: 10.1016/j.ceca.2015.06.004. [DOI] [PubMed] [Google Scholar]
- 26.Yao Y, Li R, Ma Y, Wang X, Li C, Zhang X, Ma R, Ding Z, Liu L. α-Lipid acid increases tolerance of cardiomyoblasts to glucose/glucose oxidase-induced injury via ROS- dependent ERK1/2 activation. Biochim Biophys Acta. 2012;1823(4):920–929. doi: 10.1016/j.bbamcr.2012.02.005. [DOI] [PubMed] [Google Scholar]
- 27.Zhang YH, Dingle L, Hall R, Casadei B. The role of nitric oxide and reactive oxygen species in the positive inotropic response to mechanical stretch in the mammalian myocardium. Biochim Biophys Acta. 2009;1797(7):811–817. doi: 10.1016/j.bbabio.2009.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 17.5 kb)
(PDF 161 kb)