

Abstract
Purpose of review
Cerebrovascular Disease (CeVD) remains a major cause of death and a leading cause of disability worldwide. CeVD is a complex and multifactorial disease caused by the interaction of vascular risk factors, environment and genetic factors. In the present article we discussed genetic susceptibility to CeVD, with particular emphasis on genetic studies of the associations between lipid traits and CeVD.
Recent findings
Several animal and clinical studies clearly defined genetic predisposition to atherosclerosis and CeVD, and particularly to ischemic stroke. Recent evidence has shown that traditional vascular risk factors explain only a small proportion of variance in atherosclerosis, suggesting that additional non-traditional factors and novel genetic determinants impact CeVD. With the help of genome-wide technology, novel genetic variants have been implicated in CeVD and lipid metabolism such as those in protein convertase subtilisin/kexin type 9 (PCSK9) gene in stroke and familial hypercholesterolemia. These studies are important since they contribute to our understanding of the genetic mechanisms underlying CeVD and to developing more effective CeVD prevention strategies.
Summary
CeVD is a complex and multifactorial disease and genetics likely plays an important role in its pathogenesis. The gene–gene and gene–environment interactions of genes involved in biology of vascular disease including the lipid metabolism are important factors for individual susceptibility to CeVD. Accounting for individual variation in genes, environment and lifestyle will bring us closer to precision medicine, which is an emerging and recently introduced new approach for disease treatment and prevention in clinical practice.
Keywords: Genetics, Cerebrovascular Disease, Monogenic Diseases, Lipids, Familial Hypercholesterolemia, Apolipoproteins
INTRODUCTION
Cardiovascular disease (CVD) is the leading cause of disability and mortality in the Western world [1]. Cerebrovascula disease (CeVD) is a subset of CVD and consists of a spectrum of subclinical and clinical disorders such as white matter hyperintensity (WMH), subclinical brain infarcts (SBI) and brain microbleeds detected by MRI; and ischemic stroke (IS) and cerebral hemorrhage [2]. CeVD is a complex and multifactorial disease caused by a combination of vascular, environment and genetic factors. Besides the multigenic disorders, several Mendelian disorders contribute to CeVD. Although these mutations are rare, they have a large relative risk. Well-described examples include familial forms of hypercholesterolemia, often caused by mutations in the low-density lipoprotein receptor (LDLR) gene or the apolipoprotein (apo) B gene (APOB), which encodes the major protein in the LDL particle [3]. However, the most common forms of predisposition to CeVD are multifactorial and result from interaction of many genes, each with a relatively small effect, working alone or in combination with modifier genes and/or environmental factors [4]. This genetic susceptibility to CeVD has been largely documented in animal models by using different experimental models and approaches, such as spontaneously hypertensive stroke-prone rats and genetic engineering to generate transgenic mice [5]. Moreover, various epidemiological studies in families and twins have revealed a genetic multifactorial component to CeVD, especially to IS [6, 7].
Both linkage and candidate gene association studies have identified a large number of genes involved in inflammation, the renin–angiotensin system, atherosclerosis and lipid metabolism associated with susceptibility to CeVD [4]. However, several of these associations were not been consistently replicated in population studies [8, 9], suggesting that the effect of the investigated single nucleotide polymorphisms (SNPs) may be weak or restricted to specific ethnic groups and/or CeVD subtype [10]. A genome-wide association study (GWAS) has recently helped replication of candidate gene studies in large consortia such as Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) [11] and the Stroke Genetics Network (SiGN) [12].
Since CeVD is a complex disease related to multiple genetic loci and interaction with environment, the American Heart Association (AHA) Council on Epidemiology and Prevention, the Stroke Council, and the Functional Genomics and Translational Biology Interdisciplinary Working Group, suggested that the study of the precursors of this complex phenotype may prove more rewarding [13]. Carotid plaque (CP) and carotid intima media thickness (cIMT) are markers of subclinical atherosclerosis and intermediate, preclinical phenotypes of vascular disease [14■■]. cIMT and CP are biologically and genetically different phenotypes of atherosclerosis [4]. In the multi-ethnic cohort from the Northern Manhattan Study (NOMAS), a large population study of stroke and cognitive decline incidence and risk factors, we have demonstrated high heritability for both cIMT and CP [15, 16]. We have also shown that traditional vascular risk factors, such as diabetes, lipids, hypertension, smoking, and obesity explain only a small proportion of variance (less than 20%) in atherosclerosis measured by cIMT [17] or CP [18]. These results suggest that genetic and novel environmental factors underlying unexplained subclinical atherosclerosis impact CeVD and deserve further investigation. We also used these subclinical markers of atherosclerosis as outcomes to investigate a novel and more efficient genetic approach, which analyze gene–gene or gene–environment interactions to emphasize the importance of the heterogeneity of genetic effects introduced with modification by environment risk factors. Using the genome wide interaction study (GWIS), we have demonstrated that a SNP (rs3751383) in exon 9 of RCBTB1 (encoding for RCC1 and BTB domain-containing protein 1) modulates the effect of smoking on cIMT and SNP (rs10205487) withinMXD1 (encoding for MAD protein), and SNP (rs7001413) within LY96 (encoding Lymphocyte antigen 96 protein) and JPH1 (encoding Junctophilin-1 protein) on CP burden in a sample of Caribbean Hispanics from NOMAS [19■■, 20■■]. We have also shown that genetic variants in LEKR1 (encoding for leucine, glutamate and lysine rich 1 protein) and GALNT10 (encoding for N-acetylgalactosaminyltransferase 10 protein), genes that have been associated with control of adiposity and weight that modulates sex-specific difference in developing cIMT [21■■]. We believe that GWIS may be particularly useful in studies that investigate genetic association with clinical CeVD, and can help identify genes that may be missed in GWAS by including gene-environment interactions.
In this article we review common single-gene disorders that lead to susceptibility to CeVD and summarize candidate gene and genome-wide association studies linked to risk factors for CeVD, with particular emphasis on genetic studies of lipid traits.
GENETIC of ISCHEMIC STROKE
Genetics of ischemic stroke (IS) has been widely investigated. The ultimate objective of IS genetic studies are to several fold. Most important among them is to prevent the devastating consequences of IS and to better understand the pathogenesis of IS and IS subtypes namely, large-artery disease, small-vessel disease, cardioembolic and cryptogenic stroke. This understanding may provide opportunities to develop new mechanistic and more effective therapeutic approaches. Many genes have been proposed in association with IS but few have been replicated [22]. Identifying the underlying multifactorial genetic variants predisposing individuals for IS has been a major challenge. The candidate gene approach identified several SNPs associated with risk to IS, including genes in pathways involved in endothelial function and nitric oxide release, the renin–angiotensin–aldosterone system, coagulation and hemostasis and inflammation [23]. However, most of these candidate genes studies have not replicated across different race-ethnic groups. A meta-analysis of IS candidate gene association studies conducted for 32 genes involving approximately 18,000 IS cases and 58,000 controls, identified factor V (F5) Leiden Arg506Gln, methylenetetrahydrofolate reductase (MTHFR) C677T, prothrombin (F2) G20210A, the angiotensin-converting enzyme (ACE) insertion deletion (I/D) polymorphism [24]. In non-European descent, another meta-analysis conducted in 12,883 IS cases and 19,548 controls, comprised mainly of Chinese, Japanese, and Korean individuals, confirmed the pivotal role of the ACE I/D polymorphism and MTHFR genetic variants, and suggested a strong association for apolipoprotein E (APOE) gene and risk for IS [25].
The advent of GWAS allowed identification of a number of novel genetic variants associated with risk of IS. Initially, PITX2 and ZFHX3, as genes linked with atrial fibrillation were found associated with risk of IS in Caucasians [26, 27]. In CHARGE that consists of four prospective epidemiological cohorts of nearly 19,600 subjects with 1,544 incident strokes [28], two SNPs were identified on chromosome 12, in the region of 12p13, in association with IS while replication was obtained only for one SNP (rs12425791) [28]. Recently, a meta-analysis of GWAS in 14,746 African Americans (1,365 ischemic and 1,592 total stroke cases) from Consortium of Minority Population Genome-Wide Association Studies of Stroke (COMPASS) identified 18 variants in or near genes implicated in cell cycle/mRNA presplicing (PTPRG, CDC5L), platelet function (HPS4), blood-brain barrier permeability (CLDN17), immune response (ELTD1, WDFY4, and IL1F10-IL1RN), and histone modification (HDAC9) [29]. The NHLBI Exome Sequence Project recently analyzed approximately 6,000 participants from numerous cohorts of European and African ancestry and identified 2 novel genes associated with an increased risk of IS: a protein-coding variant in PDE4DIP (rs1778155) (encoding for phosphodiesterase 4D interacting protein) with an intracellular signal transduction mechanism and in ACOT4 (rs35724886) (encoding for Acyl-coenzyme A thioesterase 4 protein) with a role in fatty acid metabolism. Confirmation of PDE4DIP was observed in affected sib-pair families with large-vessel stroke subtype and in African Americans [30]. Currently, collaborative efforts have been established to form the largest datasets for genetic investigations of IS as a part of several consortia such as METASTROKE [31], International Stroke Genetics Consortium [32], NINDS SiGN study and Wellcome Trust [31, 33]. The recent results from 17,970 cases of IS and 70,764 controls from these consortia report a novel association on chromosome 12q24 (rs10744777) with IS [34] and a role of for SNPs near the opioid receptor μ1 (OPRM1) gene in prediction for large artery atherosclerosis and small vessel disease subtypes of IS [32]. Taking together these findings, we may conclude that GWAS has had a major influence on our understanding of IS physiopathology and may help to develop better and more individually tailored IS therapy. Collaborative efforts are critical to achieve this goal.
MONOGENIC DISEASESAND SUSCEPTIBILITY FOR CeVD
Monogenic disorders related with CeVD are uncommon and in most cases these disorders occur in the absence of traditional CeVD risk factors. These disorders and associated CeVD phenotypes are listed in Table 1. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) is an autosomal dominant small-vessel disease caused by mutations in the NOTCH3 gene [35, 36]. The frequency and the prevalence ofNotch3 gene mutations has been estimated from 2 to 4 per 100,000 adults [35]. The onset of symptoms occurs around the age of 20 to 30 years, with subsequent cerebral vascular accidents, mainly recurrent IS, progressive cognitive impairment, as well as depression and migraine [7, 35]. Notch3 is mainly expressed in vascular smooth muscle cells and pericytes and plays an important role in the control of different processes of vascular development and vascular smooth muscle cell differentiation [37]. Pathogenic mutations, consisting of loss or gain of cysteine residues of extracellular domains lead to the deposition of Notch3 fragments in the vascular basal membrane [37].
Table 1.
Monogenic Disorders Associated with Cerebrovascular Disease
| Syndrome | Inheritance | Gene | Chromosome region | Symptoms | Vascular pathology |
|---|---|---|---|---|---|
| CADASIL | AD | NOTCH3 | 19p13.1 | Migraine, cognitive problems, depression, seizures, stroke | Small-vessel vasculopathy |
| CARASIL | AR | HTRA1 | 10q | Spasticity, stroke, cognitive problems, scalp hair loss, back pain | |
| MELAS | Maternal | MTTL1 | mtDNA | Muscle weakness, headache episodes, seizures, strokelike episodes | |
| Fabry disease | XL-R | GLA | Xq22 | Episodes of pain in hands and feet, angiokeratomas, corneal opacity, renal affection, heart affection, stroke | Small and large artery vasculopathy |
|
MYMY1
MYMY2 MYMY3 MYMY4 MYMY5 |
AD? AD? AD? XL-R AD |
n.d.
RNF213 n.d. BRCA1/2-containing complex, subunit 3 MTCP1/MTCP1NB ACTA2 |
3p24.2-p26 17q25.3 8q23 Xq28 Xq28 10q23.31 |
Progressive, occlusive, cerebrovascular arteriopathy, bilateral progressive stenosis of the distal internal carotid arteries, with particular involvement of the circle of Willis | Large-artery vasculopathy |
| Marfan syndrome type 1 and 2 | AD | FBN1, TGFBR2 | 15q21.1-3p24.1 | Tall build, long arms, legs, scoliosis, flat feet, fatigue, shortness of breath, heart palpitations, chest pain, partial lens dislocation, spontaneous pneumothorax | Arterial dissection |
| Hereditary cerebral hemorrhage with amyloidosis of the Dutch type | AD | APP | 21q21.3 | Lobar intracerebral hemorrhage, cerebral microbleeds, cognitive problems | Inherited cerebral amyloid angiopathies |
| Cystatin C-related familial cerebral amyloid angiopathy | AD | CST3 | 20p11.21 | Intracerebral haemorrhage, stroke and dementia | |
| Transthyretin-related CAA | AD | TTR | 18q12.1 | Pain, paresthesia, muscular weakness, autonomic dysfunction, sensory and motor polyneuropathy | |
| Autosomal dominant polycystic kidney disease adult type I and II | AD | PDK1, PDK2 | 16p13.3-4q22.1 | Pain in the abdomen, side or lower backhaematuria, hypertension, kidney stones recurrent urinary tract infections, loss of kidney function | Cerebrovascular malformations |
| Cerebral cavernous malformations | AD | KRIT1, C7orf22, PDCD10 | 7q21-q22, 7p13-p15, 3q25.2-q27 | Some are silent, others cause seizures, hemorrhage, or focal neurologic deficit | |
| Hereditary hemorrhagic telangiectasia (Osler-Weber-Rendu disease) | AD | ENG, ACVRL1 | 9q34.1, 12q11-q14 | Telangiactasia, arteriovenous malformations in lungs, brain, liver, intestines, intracerebral hemorrhage, ischemic stroke |
AD: Autosomal dominant; AR: Autosomal recessive; CAA: Cerebral amyloid angiopathy; CADASIL: Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy; CARASIL: Cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy; MELAS: Mitochondrial encephalomyopathy, lactic acidosis and stroke episodes; MYMY: Moyamoya disease; n.d.: Not determined; XL-R: X-linked recessive. NOTCH3: Neurogenic locus notch homolog protein 3; HTRA1: Serine protease HTRA1; MTTL1: Mitochondrially encoded tRNA leucine 1; GLA: Alpha-galactosidase; RNF213: Ring finger protein 213; BRCA1/2: Breast cancer 1/2; MTCP1/MTCP1NB: Mature T-cell proliferation 1 and mature T-cell proliferation 1 neighbor; ACTA2: Alpha-actin-2; FBN1: Fibrillin-1; TGFBR2: Transforming growth factor, beta receptor II; APP: Amyloid precursor protein; CST3: Cystatin C or cystatin 3; TTR: Transthyretin; PDK1 and PDK2: Pyruvate dehydrogenase lipoamide kinase isozyme 1 and 2; KRIT1: Krev interaction trapped protein 1; C7orf22: Malcavernin; PDCD10: Programmed cell death protein 10; ENG: Endoglin; ACVRL1: Activin A Receptor Type II-Like 1.
Cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL) is a non-hypertensive cerebral small vessel arteriopathy transmitted in an autosomal recessive manner [38]. The mean age of symptom onset is 32 years, and common clinical features are atherosclerotic leukoencephalopathy, alopecia, lumbago, spondylosis deformans and psychiatric disorders [38]. A study conducted in CARASIL families suggests an association of this disease with mutations in the HTRA1 gene [39, 40] which is localized on ch10q and is expressed in blood vessels, skin and bone. It encodes a serine protease that represses signaling by TGF-b family members.
Fabry disease is an X-linked recessive lysosomal storage disorder, which occurs usually during childhood [41]. Clinical features of this pathology include multiple neurological infarcts and neuropathic pain [41]. The multisystemic involvement derives from mutations affecting a-Gal A gene at Xq22 [42]. The enzyme deficiency results in uncleaved glycosphingolipids, which accumulates in lysosomes within the intima and media of blood vessels in different organs and tissues including vascular smooth muscle and endothelial cells, carotid, heart, brain, peripheral nerves and kidney leading to cell dysfunction, organ failure and development of tissue ischemia and infarction [43].
Mitochondrial encephalomyopathy, lactic acidosis and stroke episodes (MELAS) is a maternally inherited multisystem disorder caused by mutations in the mtDNA [44]. It typically develops during childhood with symptoms characterized by tonic–clonic seizures, migraine headaches, anorexia and vomiting. In some cases monosymptomatic stroke episodes have been described [44]. The stroke-like episodes in MELAS differ from typical ischemic infarcts involving not only the vascular territories but also the brain parenchyma leading to tissue ischemia, since the underlying cause is an energy imbalance instead of vascular occlusion [45]. The most frequent mutation of MELAS, reported in approximately 80% of patients with typical clinical findings, is an A to G transition at nucleotide 3243 on the MTTL1 mitochondrial gene, which encodes tRNALeu (UUR). mtDNA mutations lead to dysfunctional mitochondrial oxidative phosphorylation with impairment of cellular respiratory capacity and ATP synthesis [46].
Moyamoya disease (MYMY) is a progressive, occlusive, cerebrovascular arteriopathy, characterized by bilateral progressive stenosis of the distal internal carotid arteries, with particular involvement of the circle of Willis [47]. MYMY is rare in western countries, with an incidence of 0.086 per 100,000 persons per year, while it is more common in African, American or Asian descendants (up to ten-times more common in Japan) [47]. The disease has two peaks of age at onset: a juvenile type that occurs at 5 years and an adult type that occurs at 30–50 years [47]. Children suffer from transient ischemic accidents and epileptic seizures due to vascular stenosis (juvenile type) [48]. Adults more often suffer from intracerebral hemorrhages (subarachnoid, intraparenchymal or intraventricular) caused by the rupture of the collateral vessels that have developed during childhood [43] and headache from dilated transdural collaterals [43]. To date, five genetic loci have been linked to different familial forms of the disease [49]. However, the pattern of inheritance for each of these forms is still not clear. A polymorphism in c.14576G>A in RNF213 was identified in 95% of familial patients with MYMY disease and 79% of sporadic cases, and patients having this SNP were found to have significantly earlier disease onset and a more severe form of MYMY, such as the presentation of cerebral infarction and posterior cerebral artery stenosis [50].
In some cases polygenic inheritance and an autosomal dominant mode of transmission with incomplete penetrance have been suggested [49]. Moreover, environmental factors could be involved in the pathogenesis of MYMY [49]. Loci for the disorder have been mapped to ch3p (MYMY1) and ch8q23 (MYMY3). MYMY-5 (MYMY5) is caused by mutation in the ACTA2 gene on ch10q23.3 [51]. Finally, an X-linked recessive syndromic disorder characterized by MYMY, short stature, hypergonadotropic hypogonadism and facial dysmorphism has been identified in members of three unrelated families (MYMY4) [52]. The deleted region was mapped on chXq28 and included exon 1 of the MTCP1/MTCP1NB gene and the first three exons of the BRCC3 gene [52, 53].
GENETICS OF LIPID TRAITS AND SUSCEPTYBILITY FOR CeVD
There is mounting evidence that the genetics of lipid disorders may play a great role in the assessment of risk for atherosclerosis and CeVD, and particularly for IS [54].
Familial hypercholesterolemia (FH) is an autosomal dominant disorder (prevalence 1:200-500) [55]. The clinical phenotype of FH is characterized by elevated cholesterol levels, tendinous xanthomata, and premature vascular disease, especially coronary heart disease (CHD). The symptoms are more severe for homozygotes than heterozygotes [55]. The genetic mutations underlaying the FH phenotype are primarily within the low-density lipoprotein receptor gene (LDLR), with fewer mutations in the APOB gene, encoding apoliproprotein B and PCSK9 gene, encoding proprotein convertase subtilisin kexin 9 [56]. However, despite high cholesterol levels being a well-defined risk factor for IS [54], the association between FH and CeVD is not as definitive as with CHD [57]. In fact, a study conducted in a cohort of 1,405 men and 1,466 women with heterozygous treated FH showed, after a median duration of 7.9 years of follow-up, a mortality rate from stroke of 0.39 per 1000 person-years (95% CI, 0.18 to 0.74), which was non-significantly lower than in the general population, suggesting that these patients were not at increased risk of fatal stroke [58]. However, the possibility of increased risk for vascular accidents cannot be excluded for untreated individuals with FH. A Finnish study prospectively followed 54 subjects aged 21–50 years with clinical FH for an average of 10 years [59]. The incidence of stroke was 7.4/1,000 years, which was 20 times higher compared to general population [59]. However, in this study many of enrolled patients had preexisting CHD, and therefore might have represented individuals with more severe forms of FH, likely biasing the results. So far, the direct association between FH and CeVD is unclear. Further studies should directly analyze the impact of specific variants of LDLR, APOB, and PCSK9 genes in association with FH.
Several candidate gene association studies, linkage studies and GWAS have identified variants associated with lipid traits and risk of CeVD. Moreover, studies demonstrated that lipid traits have a significant genetic component, with the estimates of heritability of 40–70% for total cholesterol (TC), 30–70% for LDL-C, 30–60% for high-density lipoprotein cholesterol (HDL-C), 20–55% for triglycerides (TG), and 30–50% for LDL-C/HDL-C [60-62]. In NOMAS, first we have demonstrated the strong association between lipids and apolipoproteins with CP as phenotype of atherosclerosis [63], and then we identified genetic loci affecting blood lipid levels among Caribbean Hispanics [64]. We found suggestive evidence of linkage of LOD score>2.0 on 15q23 for TG, 16q23 for LDL-C, 19q12 for TC and LDL-C, and 20p12 for LDL-C. In the association analysis of the linkage peaks, we also found that seven SNPs near FLJ45974 were associated with LDL-C/HDL-C with a nominal P<3.5×10−5, in addition to associations (P<0.0001) for other lipid traits with SNPs in or near CDH13, SUMF2, TLE3, FAH, ARNT2, TSHZ3, ZNF343, RPL7AL2, and TMC3 genes [64]. Replication of these genes is still required in other populations before specifically targeting them for intervention.
The Ser447Ter gain of function in exon 9 (S447X), the c.1127A4G in exon 6, resulting in p.Asn291Ser, and the p.Asp9Asn, all in the LPL gene encoding Lipoprotein Lipase, the central enzyme in hydrolysis of triglycerides from chylomicrons and VLDL, have been associated with susceptibility to IS [65]. Most investigated association has been between apolipoprotein genes and CeVD. Figure 1 shows schematic representation of APOE polymorphisms and associated degenerative disorders and CeVD. Apolipoproteins, mostly the E4 allele have been demonstrated to play a role in in Alzheimer's disease [66]. However, APOoE4 may also be a predisposing genetic marker for CeVD [67]. In NOMAS, we have shown that none of the plasma lipid profile components are associated with WMH. However, the association between lipids and WMH was modified by ApoE status suggesting that this association is dependent on APOE4 genotype and worsening of the lipid profile over time [68]. Recently, a systematic meta-analysis pooled together 41 studies (with a total of 9,027 cases and 61,730 controls) and reported on association between APOE genotype and IS. The results suggested a strong association of APOE E2/E2 genotype with IS, particularly in individuals of European ancestry [69]. Another meta-analysis conducted in a total of 29,965 patients, showed that the E4 allele carrier status and APOE E4/4 genotype were associated with increasing WMH and presence of cerebral microbleeds, especially lobar. APOE E2 carrier status was associated with increasing WMH load and risk of subclinical brain infarct [70]. Variants in LPA, encoding apolipoprotein (a) which is the key protein in Lp(a) also have been associated with risk for CeVD, however these associations are still debated and not validated [71].
Figure 1. ApoE polymorphisms and their association with neurological degenerative and cerebrovascular disease.
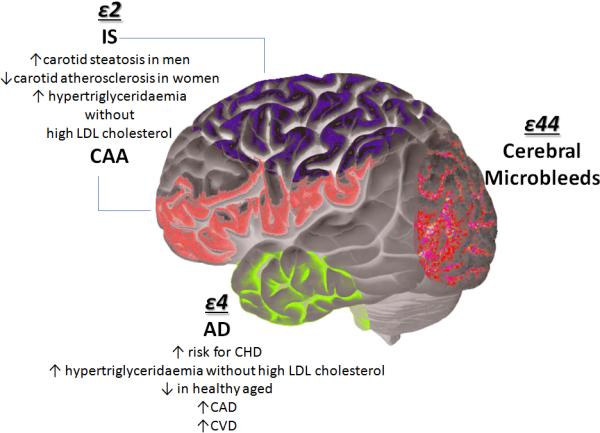
Four main polymorphisms of ApoE have been associated with risk for both neurodegenerative and cerebrovascular disease.IS: ischemic stroke; CAA: Cerebral amyloid angiopathy; CHD: coronary heart disease; CVD: cardiovascular disease; CAD: coronary artery disease; AD: Alzheimer disease; LDL: low density lipoprotein.
Comparative genome-wide linkage analysis had indicated that the chromosome 11p14.1–q12.1 contains a sequence variant(s) for cholesterol and triglyceride traits [72]. Specific genetic variants in PON1 encoding Paraxonase, , an antioxidant protein able to prevent lipid peroxidation and consequently exerts antiatherosclerotic effects, have been linked with increased risk for IS [73].
Further studies are necessary to understand the actual impact of genetics in control of lipid profile as an important risk factor for CeVD. This knowledge would result in better prevention since it would allow us to identify subjects with particular susceptibility to CeVD and to plan a personal preventive therapeutic strategy. However, considering the emerging concept of precision medicine, an approach for disease prevention and treatment that takes into account people's individual variations in genes, environment, and lifestyle [74]; treatment of dyslipidemia by considering genetic determinants of lipid traits, pharmacogenetic response to lipid lowering medications, diet, and physical activity in a unique algorithm may help reduce CeVD risk.
CONCLUSION
Recent evidence shows that genetic factors may have unique effects for CeVD predisposition and that these effects may vary by age and sex [75]. In several studies conducted in the NOMAS, the relevance of genetic susceptibility to developing atherosclerosis and CeVD was demonstrated among different ethnic groups [4]. Therefore, we need to take into consideration multiple factors (e.g., age, sex, ethnic groups, environment) that play a pivotal role in the risk for CeVD, even more so because most studies os stroke using GWAS did not reach consistent results. Several factors may account for inconsistent findings, including a lack of power due to limited sample sizes of study populations; inappropriate hypotheses for candidate gene studies; and differences in measurement methods and reproducibility of phenotypes across the studies. To avoid these biases, improved approaches would need to consider gene–gene or gene–environment interactions. GWIS may be useful methodological approach for future analyses. Including genetics of lipid traits could ensure novel discoveries for treatment and prevention of cerebrovascular disease and bring us a step closer to precision medicine.
KEY POINTS.
Cerebrovascular disease (CeVD) is a complex and multifactorial disease caused by the combination of vascular risk factors, environment and genetic factors.
Monogenic disorders even if uncommon are highly associated with risk for vascular accidents and neurological degeneration in most cases independently of common risk factors.
Several studies identified a large number of genes involved in inflammation, renin–angiotensin system, atherosclerosis and lipid metabolism, among many others, associated with susceptibility to CeVD. However, these associations have not been consistently replicated in different populations, suggesting the effect of unaccounted environmental factors and/or that the effect of the investigated SNPs may be restricted to specific race-ethnicities and/or subtypes of CeVD.
Genetic of lipid disorders, such as familial hypercholesterolemia, play a great role in the assessment of risk for atherosclerosis as well as for CeVD, particularly for ischemic stroke. Among genes regulating lipid traits those encoding apoliproproteins have been among the most investigated genes.
We propose that investigating the gene–gene or gene–environment interactions in the susceptibility to cerebrovascular disease may help to better understand this complex process.
Acknowledgements
Financial support and sponsorship
This work was supported by NIH/NINDS K24 NS 062737 Genetic Determinants of Extreme Phenotypes of Subclinical Atherosclerosis (TR), and University and Research PON03PE_00146_1/10 BIBIOFAR (DDM).
Footnotes
Conflicts of interest
There are no conflicts of interest.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
■of special interest
■ ■of outstanding interest
- 1.Mozaffarian D, Benjamin EJ, Go AS, et al. Heart disease and stroke statistics--2015 update: a report from the American Heart Association. Circulation. 2015;131:e29–322. doi: 10.1161/CIR.0000000000000152. [DOI] [PubMed] [Google Scholar]
- 2.Fisher M. The challenge of mixed cerebrovascular disease. Annals of the New York Academy of Sciences. 2010;1207:18–22. doi: 10.1111/j.1749-6632.2010.05758.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hutter CM, Austin MA, Humphries SE. Familial hypercholesterolemia, peripheral arterial disease, and stroke: a HuGE minireview. American journal of epidemiology. 2004;160:430–435. doi: 10.1093/aje/kwh238. [DOI] [PubMed] [Google Scholar]
- 4.Della-Morte D, Guadagni F, Palmirotta R, et al. Genetics of ischemic stroke, stroke-related risk factors, stroke precursors and treatments. Pharmacogenomics. 2012;13:595–613. doi: 10.2217/pgs.12.14. [DOI] [PubMed] [Google Scholar]
- 5.Herrera VL, Ruiz-Opazo N. Genetic studies in rat models: insights into cardiovascular disease. Current opinion in lipidology. 2005;16:179–191. doi: 10.1097/01.mol.0000162323.77666.5e. [DOI] [PubMed] [Google Scholar]
- 6.Flossmann E, Schulz UG, Rothwell PM. Systematic review of methods and results of studies of the genetic epidemiology of ischemic stroke. Stroke; a journal of cerebral circulation. 2004;35:212–227. doi: 10.1161/01.STR.0000107187.84390.AA. [This is a very good and comprehensive review on genetic epidemiology of ischemic stroke.] [DOI] [PubMed] [Google Scholar]
- 7.Francis J, Raghunathan S, Khanna P. The role of genetics in stroke. Postgraduate medical journal. 2007;83:590–595. doi: 10.1136/pgmj.2007.060319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rosand J, Bayley N, Rost N, de Bakker PI. Many hypotheses but no replication for the association between PDE4D and stroke. Nature genetics. 2006;38:1091–1092. doi: 10.1038/ng1006-1091. author reply 1092-1093. [DOI] [PubMed] [Google Scholar]
- 9.Kostulas K, Gretarsdottir S, Kostulas V, et al. PDE4D and ALOX5AP genetic variants and risk for Ischemic Cerebrovascular Disease in Sweden. Journal of the neurological sciences. 2007;263:113–117. doi: 10.1016/j.jns.2007.06.042. [DOI] [PubMed] [Google Scholar]
- 10.Matarin M, Brown WM, Dena H, et al. Candidate gene polymorphisms for ischemic stroke. Stroke; a journal of cerebral circulation. 2009;40:3436–3442. doi: 10.1161/STROKEAHA.109.558015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ikram MA, Seshadri S, Bis JC, et al. Genomewide association studies of stroke. The New England journal of medicine. 2009;360:1718–1728. doi: 10.1056/NEJMoa0900094. [The first large genomewide association studies demonstratting the association of genetic locus on chromosome 12p13 with an increased risk of stroke.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meschia JF, Arnett DK, Ay H, et al. Stroke Genetics Network (SiGN) study: design and rationale for a genome-wide association study of ischemic stroke subtypes. Stroke; a journal of cerebral circulation. 2013;44:2694–2702. doi: 10.1161/STROKEAHA.113.001857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arnett DK, Baird AE, Barkley RA, et al. Relevance of genetics and genomics for prevention and treatment of cardiovascular disease: a scientific statement from the American Heart Association Council on Epidemiology and Prevention, the Stroke Council, and the Functional Genomics and Translational Biology Interdisciplinary Working Group. Circulation. 2007;115:2878–2901. doi: 10.1161/CIRCULATIONAHA.107.183679. [The most comprehensive review on the effecto of genetics and genomics on cardiovascuolar disease.] [DOI] [PubMed] [Google Scholar]
- 14■■.Rundek T, Gardener H, Della-Morte D, et al. The relationship between carotid intima-media thickness and carotid plaque in the Northern Manhattan Study. Atherosclerosis. 2015;241:364–370. doi: 10.1016/j.atherosclerosis.2015.05.027. [The first study demonstrating in a large multi-ethnic population the relationship between carotid plaque and intima media thickness as differne phenotypes pf atherosclerosis.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sacco RL, Blanton SH, Slifer S, et al. Heritability and linkage analysis for carotid intima-media thickness: the family study of stroke risk and carotid atherosclerosis. Stroke; a journal of cerebral circulation. 2009;40:2307–2312. doi: 10.1161/STROKEAHA.109.554121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dong C, Beecham A, Slifer S, et al. Genomewide linkage and peakwide association analyses of carotid plaque in Caribbean Hispanics. Stroke; a journal of cerebral circulation. 2010;41:2750–2756. doi: 10.1161/STROKEAHA.110.596981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rundek T, Blanton SH, Bartels S, et al. Traditional risk factors are not major contributors to the variance in carotid intima-media thickness. Stroke; a journal of cerebral circulation. 2013;44:2101–2108. doi: 10.1161/STROKEAHA.111.000745. [This study showed as traditional vascular risk factors explained only for a small proportion of variance in atherosclerosis.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuo F1, Gardener H, Dong C, Cabral D, Della-Morte D, Blanton SH, Elkind MS, Sacco RL, Rundek T. Traditional cardiovascular risk factors explain the minority of the variability in carotid plaque. Stroke. 2012;43:1755–60. doi: 10.1161/STROKEAHA.112.651059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19 ■■.Wang L, Rundek T, Beecham A, Hudson B, Blanton SH, Zhao H, Sacco RL, Dong C. Genome-wide interaction study identifies RCBTB1 as a modifier for smoking effect on carotid intima-media thickness. Arterioscler Thromb Vasc Biol. 2014;34:219–25. doi: 10.1161/ATVBAHA.113.302706. [The first study that using a novel genetic approach identified the interaction between specific genetic variant on RCBTB1 gene as modifier for smoking effect on subclinical marker of atherosclerosis.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20■■.Della-Morte D, Wang L, Beecham A, et al. Novel genetic variants modify the effect of smoking on carotid plaque burden in Hispanics. J Neurol Sci. 2014;15344:27–31. doi: 10.1016/j.jns.2014.06.006. [The first study that using a novel genetic approach identified the interaction between specific genetic variant on MXD1 gene as modifier for smoking effect on subclinical marker of atherosclerosis.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21■■.Dong C, Della-Morte D, Beecham A, et al. Genetic variants in LEKR1 and GALNT10 modulate sex-difference in carotid intima-media thickness: a genome-wide interaction study. Atherosclerosis. 2015;240:462–467. doi: 10.1016/j.atherosclerosis.2015.04.019. [The first study that using a novel genetic approach identified the interaction between specific genetic variants on LEKR1 and GALNT10 genes as modifier for sex effect on subclinical marker of atherosclerosis.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim SJ1, Moon GJ, Bang OY. Biomarkers for stroke. J Stroke. 2013;15:27–37. doi: 10.5853/jos.2013.15.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Markus HS. Stroke genetics. Hum Mol Genet. 2011;20:R124–31. doi: 10.1093/hmg/ddr345. [DOI] [PubMed] [Google Scholar]
- 24.Casas JP, Hingorani AD, Bautista LE, Sharma P. Meta-analysis of genetic studies in ischemic stroke: thirty-two genes involving approximately 18,000 cases and 58,000 controls. Arch Neurol. 2004;61:1652–61. doi: 10.1001/archneur.61.11.1652. [A large meta-analisys conducted for the most important candidate genes found associated with the risk for ischemic stroke.] [DOI] [PubMed] [Google Scholar]
- 25.Ariyaratnam R, Casas JP, Whittaker J, Smeeth L, Hingorani AD, Sharma P. Genetics of ischaemic stroke among persons of non-European descent: a meta-analysis of eight genes involving approximately 32,500 individuals. PLoS Med. 2007;4:e131. doi: 10.1371/journal.pmed.0040131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gretarsdottir S, Thorleifsson G, Manolescu A, Styrkarsdottir U, Helgadottir A, Gschwendtner A, Kostulas K, Kuhlenbäumer G, Bevan S, Jonsdottir T, Bjarnason H, Saemundsdottir J, Palsson S, Arnar DO, Holm H, Thorgeirsson G, Valdimarsson EM, Sveinbjörnsdottir S, Gieger C, Berger K, Wichmann HE, Hillert J, Markus H, Gulcher JR, Ringelstein EB, Kong A, Dichgans M, Gudbjartsson DF, Thorsteinsdottir U, Stefansson K. Risk variants for atrial fibrillation on chromosome 4q25 associate with ischemic stroke. Ann Neurol. 2008;64:402–9. doi: 10.1002/ana.21480. [DOI] [PubMed] [Google Scholar]
- 27.Gudbjartsson DF, Holm H, Gretarsdottir S, Thorleifsson G, Walters GB, Thorgeirsson G, Gulcher J, Mathiesen EB, Njølstad I, Nyrnes A, Wilsgaard T, Hald EM, Hveem K, Stoltenberg C, Kucera G, Stubblefield T, Carter S, Roden D, Ng MC, Baum L, So WY, Wong KS, Chan JC, Gieger C, Wichmann HE, Gschwendtner A, Dichgans M, Kuhlenbäumer G, Berger K, Ringelstein EB, Bevan S, Markus HS, Kostulas K, Hillert J, Sveinbjörnsdóttir S, Valdimarsson EM, Løchen ML Ma RC, Darbar D, Kong A, Arnar DO, Thorsteinsdottir U, Stefansson K. A sequence variant in ZFHX3 on 16q22 associates with atrial fibrillation and ischemic stroke. Nat Genet. 2009;41:876–8. doi: 10.1038/ng.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ikram MA, Seshadri S, Bis JC, Fornage M, DeStefano AL, Aulchenko YS, Debette S, Lumley T, Folsom AR, van den Herik EG, Bos MJ, Beiser A, Cushman M, Launer LJ, Shahar E, Struchalin M, Du Y, Glazer NL, Rosamond WD, Rivadeneira F, Kelly-Hayes M, Lopez OL, Coresh J, Hofman A, DeCarli C, Heckbert SR, Koudstaal PJ, Yang Q, Smith NL, Kase CS, Rice K, Haritunians T, Roks G, de Kort PL, Taylor KD, de Lau LM, Oostra BA, Uitterlinden AG, Rotter JI, Boerwinkle E, Psaty BM, Mosley TH, van Duijn CM, Breteler MM, Longstreth WT, Jr, Wolf PA. Genomewide association studies of stroke. N Engl J Med. 2009;360:1718–28. doi: 10.1056/NEJMoa0900094. [The first large GWAS conducted from a consortium aimed to investigate genetic of ischemic stroke in a large population.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carty CL, Keene KL, Cheng YC, Meschia JF, Chen WM, Nalls M, Bis JC, Kittner SJ, Rich SS, Tajuddin S, Zonderman AB, Evans MK, Langefeld CD, Gottesman R, Mosley TH, Shahar E2, Woo D, Yaffe K, Liu Y, Sale MM, Dichgans M, Malik R, Longstreth WT, Jr, Mitchell BD, Psaty BM, Kooperberg C, Reiner A, Worrall BB, Fornage M, COMPASS and METASTROKE Consortia Meta-Analysis of Genome-Wide Association Studies Identifies Genetic Risk Factors for Stroke in African Americans. Stroke. 2015;46:2063–8. doi: 10.1161/STROKEAHA.115.009044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Auer PL, Nalls M, Meschia JF, Worrall BB, Longstreth WT, Jr, Seshadri S, Kooperberg C, Burger KM, Carlson CS, Carty CL, Chen WM, Cupples LA, DeStefano AL, Fornage M, Hardy J, Hsu L, Jackson RD, Jarvik GP, Kim DS, Lakshminarayan K, Lange LA, Manichaikul A, Quinlan AR, Singleton AB, Thornton TA, Nickerson DA, Peters U, Rich SS, National Heart, Lung, and Blood Institute Exome Sequencing Project Rare and Coding Region Genetic Variants Associated With Risk of Ischemic Stroke: The NHLBI Exome Sequence Project. JAMA Neurol. 2015;72:781–8. doi: 10.1001/jamaneurol.2015.0582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meschia JF, Arnett DK, Ay H, Brown RD, Jr, Benavente OR, Cole JW, de Bakker PI, Dichgans M, Doheny KF, Fornage M, Grewal RP, Gwinn K, Jern C, Conde JJ, Johnson JA, Jood K, Laurie CC, Lee JM, Lindgren A, Markus HS, McArdle PF, McClure LA, Mitchell BD, Schmidt R, Rexrode KM, Rich SS, Rosand J, Rothwell PM, Rundek T, Sacco RL, Sharma P, Shuldiner AR, Slowik A, Wassertheil-Smoller S, Sudlow C, Thijs VN, Woo D, Worrall BB, Wu O, Kittner SJ, NINDS SiGN Study Stroke Genetics Network (SiGN) study: design and rationale for a genome-wide association study of ischemic stroke subtypes. Stroke. 2013;44:2694–702. doi: 10.1161/STROKEAHA.113.001857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holliday EG, Traylor M, Malik R, Bevan S, Falcone G, Hopewell JC, Cheng YC, Cotlarciuc I, Bis JC, Boerwinkle E, Boncoraglio GB, Clarke R, Cole JW, Fornage M, Furie KL, Ikram MA1, Jannes J, Kittner SJ, Lincz LF, Maguire JM, Meschia JF, Mosley TH, Nalls MA, Oldmeadow C, Parati EA, Psaty BM, Rothwell PM, Seshadri S, Scott RJ, Sharma P, Sudlow C, Wiggins KL1, Worrall BB, Rosand J, Mitchell BD, Dichgans M, Markus HS, Levi C, Attia J, Wray NR, Australian Stroke Genetics Collaborative. Wellcome Trust Case Control Consortium. International Stroke Genetics Consortium Genetic overlap between diagnostic subtypes of ischemic stroke. Stroke. 2015;46:615–9. doi: 10.1161/STROKEAHA.114.007930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Malik R, Bevan S, Nalls MA, Holliday EG, Devan WJ, Cheng YC, Ibrahim-Verbaas CA, Verhaaren BF, Bis JC, Joon AY, de Stefano AL, Fornage M, Psaty BM, Ikram MA, Launer LJ, van Duijn CM, Sharma P, Mitchell BD, Rosand J, Meschia JF, Levi C, Rothwell PM, Sudlow C, Markus HS, Seshadri S, Dichgans M, Wellcome Trust Case Control Consortium 2 Multilocus genetic risk score associates with ischemic stroke in case-control and prospective cohort studies. Stroke. 2014;45:394–402. doi: 10.1161/STROKEAHA.113.002938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kilarski LL, Achterberg S, Devan WJ, Traylor M, Malik R, Lindgren A, Pare G, Sharma P, Slowik A, Thijs V, Walters M, Worrall BB, Sale MM, Algra A, Kappelle LJ, Wijmenga C, Norrving B, Sandling JK, Rönnblom L, Goris A, Franke A, Sudlow C, Rothwell PM, Levi C, Holliday EG, Fornage M, Psaty B, Gretarsdottir S, Thorsteinsdottir U, Seshadri S, Mitchell BD, Kittner S, Clarke R, Hopewell JC, Bis JC, Boncoraglio GB, Meschia J, Ikram MA, Hansen BM, Montaner J, Thorleifsson G, Stefanson K, Rosand J, de Bakker PI, Farrall M, Dichgans M, Markus HS, Bevan S, GARNET Collaborative Research Group. Wellcome Trust Case Control Consortium, Australian Stroke Genetic Collaborative, the METASTROKE Consortium, and the International Stroke Genetics Consortium Meta-analysis in more than 17,900 cases of ischemic stroke reveals a novel association at 12q24.12. Neurology. 2014;83:678–85. doi: 10.1212/WNL.0000000000000707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Andre C. CADASIL: pathogenesis, clinical and radiological findings and treatment. Arquivos de neuro-psiquiatria. 2010;68:287–299. doi: 10.1590/s0004-282x2010000200026. [DOI] [PubMed] [Google Scholar]
- 36.Joutel A, Corpechot C, Ducros A, et al. Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature. 1996;383:707–710. doi: 10.1038/383707a0. [The first study which demonstrated the mutation of Notch3 gene in CADASIL development and risk for neurological accidents.] [DOI] [PubMed] [Google Scholar]
- 37.Mawet J, Vahedi K, Aout M, et al. Carotid atherosclerotic markers in CADASIL. Cerebrovascular diseases. 2011;31:246–252. doi: 10.1159/000321932. [DOI] [PubMed] [Google Scholar]
- 38.Nozaki H, Nishizawa M, Onodera O. Features of cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy. Stroke; a journal of cerebral circulation. 2014;45:3447–3453. doi: 10.1161/STROKEAHA.114.004236. [DOI] [PubMed] [Google Scholar]
- 39.Bianchi S, Di Palma C, Gallus GN, et al. Two novel HTRA1 mutations in a European CARASIL patient. Neurology. 2014;82:898–900. doi: 10.1212/WNL.0000000000000202. [DOI] [PubMed] [Google Scholar]
- 40.Hara K, Shiga A, Fukutake T, et al. Association of HTRA1 mutations and familial ischemic cerebral small-vessel disease. The New England journal of medicine. 2009;360:1729–1739. doi: 10.1056/NEJMoa0801560. [DOI] [PubMed] [Google Scholar]
- 41.Thomas AS, Hughes DA. Fabry disease. Pediatric endocrinology reviews : PER. 2014;12(Suppl 1):88–101. [PubMed] [Google Scholar]
- 42.Laney DA, Bennett RL, Clarke V, et al. Fabry disease practice guidelines: recommendations of the National Society of Genetic Counselors. Journal of genetic counseling. 2013;22:555–564. doi: 10.1007/s10897-013-9613-3. [DOI] [PubMed] [Google Scholar]
- 43.Ballabio E, Bersano A, Bresolin N, Candelise L. Monogenic vessel diseases related to ischemic stroke: a clinical approach. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2007;27:1649–1662. doi: 10.1038/sj.jcbfm.9600520. [DOI] [PubMed] [Google Scholar]
- 44.El-Hattab AW, Adesina AM, Jones J, Scaglia F. MELAS syndrome: Clinical manifestations, pathogenesis, and treatment options. Molecular genetics and metabolism. 2015;116:4–12. doi: 10.1016/j.ymgme.2015.06.004. [DOI] [PubMed] [Google Scholar]
- 45.Iizuka T, Sakai F. Pathogenesis of stroke-like episodes in MELAS: analysis of neurovascular cellular mechanisms. Current neurovascular research. 2005;2:29–45. doi: 10.2174/1567202052773544. [DOI] [PubMed] [Google Scholar]
- 46.Finsterer J. Genetic, pathogenetic, and phenotypic implications of the mitochondrial A3243G tRNALeu(UUR) mutation. Acta neurologica Scandinavica. 2007;116:1–14. doi: 10.1111/j.1600-0404.2007.00836.x. [DOI] [PubMed] [Google Scholar]
- 47.Smith ER, Scott RM. Moyamoya: epidemiology, presentation, and diagnosis. Neurosurgery clinics of North America. 2010;21:543–551. doi: 10.1016/j.nec.2010.03.007. [DOI] [PubMed] [Google Scholar]
- 48.Rafay MF, Armstrong D, Dirks P, et al. Patterns of cerebral ischemia in children with moyamoya. Pediatric neurology. 2015;52:65–72. doi: 10.1016/j.pediatrneurol.2014.10.007. [DOI] [PubMed] [Google Scholar]
- 49.Achrol AS, Guzman R, Lee M, Steinberg GK. Pathophysiology and genetic factors in moyamoya disease. Neurosurgical focus. 2009;26:E4. doi: 10.3171/2009.1.FOCUS08302. [DOI] [PubMed] [Google Scholar]
- 50.Fujimura M, Sonobe S, Nishijima Y, et al. Genetics and Biomarkers of Moyamoya Disease: Significance of RNF213 as a Susceptibility Gene. Journal of stroke. 2014;16:65–72. doi: 10.5853/jos.2014.16.2.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guo DC, Papke CL, Tran-Fadulu V, et al. Mutations in smooth muscle alpha-actin (ACTA2) cause coronary artery disease, stroke, and Moyamoya disease, along with thoracic aortic disease. American journal of human genetics. 2009;84:617–627. doi: 10.1016/j.ajhg.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Herve D, Touraine P, Verloes A, et al. A hereditary moyamoya syndrome with multisystemic manifestations. Neurology. 2010;75:259–264. doi: 10.1212/WNL.0b013e3181e8ee3f. [An interesting study that by describing a family affected by a hereditary multisystem disorder associated with moyamoya syndrome demonstrated a hereditary of this disease with X-linked recessive pattern of inheritance.] [DOI] [PubMed] [Google Scholar]
- 53.Miskinyte S, Butler MG, Herve D, et al. Loss of BRCC3 deubiquitinating enzyme leads to abnormal angiogenesis and is associated with syndromic moyamoya. American journal of human genetics. 2011;88:718–728. doi: 10.1016/j.ajhg.2011.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ansell BJ. Cholesterol, stroke risk, and stroke prevention. Current atherosclerosis reports. 2000;2:92–96. doi: 10.1007/s11883-000-0101-5. [DOI] [PubMed] [Google Scholar]
- 55.Nicholls P, Young IS, Graham CA. Genotype/phenotype correlations in familial hypercholesterolaemia. Current opinion in lipidology. 1998;9:313–317. doi: 10.1097/00041433-199808000-00005. [DOI] [PubMed] [Google Scholar]
- 56.Di Taranto MD, D'Agostino MN, Fortunato G. Functional characterization of mutant genes associated with autosomal dominant familial hypercholesterolemia: Integration and evolution of genetic diagnosis. Nutrition, metabolism, and cardiovascular diseases : NMCD. 2015 doi: 10.1016/j.numecd.2015.06.007. [DOI] [PubMed] [Google Scholar]
- 57.Braenne I, Kleinecke M, Reiz B, et al. Systematic analysis of variants related to familial hypercholesterolemia in families with premature myocardial infarction. European journal of human genetics : EJHG. 2015 doi: 10.1038/ejhg.2015.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Huxley RR, Hawkins MH, Humphries SE, et al. Risk of fatal stroke in patients with treated familial hypercholesterolemia: a prospective registry study. Stroke; a journal of cerebral circulation. 2003;34:22–25. doi: 10.1161/01.str.0000047123.14312.3e. [DOI] [PubMed] [Google Scholar]
- 59.Kaste M, Koivisto P. Risk of brain infarction in familial hypercholesterolemia. Stroke; a journal of cerebral circulation. 1988;19:1097–1100. doi: 10.1161/01.str.19.9.1097. [DOI] [PubMed] [Google Scholar]
- 60.Bielinski SJ, Tang W, Pankow JS, et al. Genome-wide linkage scans for loci affecting total cholesterol, HDL-C, and triglycerides: the Family Blood Pressure Program. Human genetics. 2006;120:371–380. doi: 10.1007/s00439-006-0223-0. [A large genome-wide linkage analisys that identified loci affecting lipid phenotypes.] [DOI] [PubMed] [Google Scholar]
- 61.Adeyemo AA, Johnson T, Acheampong J, et al. A genome wide quantitative trait linkage analysis for serum lipids in type 2 diabetes in an African population. Atherosclerosis. 2005;181:389–397. doi: 10.1016/j.atherosclerosis.2004.12.049. [DOI] [PubMed] [Google Scholar]
- 62.Yu Y, Wyszynski DF, Waterworth DM, et al. Multiple QTLs influencing triglyceride and HDL and total cholesterol levels identified in families with atherogenic dyslipidemia. Journal of lipid research. 2005;46:2202–2213. doi: 10.1194/jlr.M500137-JLR200. [DOI] [PubMed] [Google Scholar]
- 63.Gardener H, Della Morte D, Elkind MS, et al. Lipids and carotid plaque in the Northern Manhattan Study (NOMAS). BMC cardiovascular disorders. 2009;9:55. doi: 10.1186/1471-2261-9-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dong C, Beecham A, Wang L, et al. Genetic loci for blood lipid levels identified by linkage and association analyses in Caribbean Hispanics. Journal of lipid research. 2011;52:1411–1419. doi: 10.1194/jlr.P013672. [A study that identified in100 Dominican familiessignificant linkage evidence for all lipid traits on chromosome 7p12 and 16q23.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bersano A, Ballabio E, Bresolin N, Candelise L. Genetic polymorphisms for the study of multifactorial stroke. Human mutation. 2008;29:776–795. doi: 10.1002/humu.20666. [DOI] [PubMed] [Google Scholar]
- 66.Roses AD. Apolipoprotein E in neurology. Current opinion in neurology. 1996;9:265–270. doi: 10.1097/00019052-199608000-00004. [DOI] [PubMed] [Google Scholar]
- 67.Huang Y. Mechanisms linking apolipoprotein E isoforms with cardiovascular and neurological diseases. Current opinion in lipidology. 2010;21:337–345. doi: 10.1097/MOL.0b013e32833af368. [A very interesting review discussing as ApoE isoforms, with their multiple cellular origins and multiple structural and biophysical properties, contribute to cardiovascular and neurological diseases by interacting with different factors through various pathways.] [DOI] [PubMed] [Google Scholar]
- 68.Willey JZ, Gardener H, Moon YP, et al. Lipid profile components and subclinical cerebrovascular disease in the northern Manhattan study. Cerebrovascular diseases. 2014;37:423–430. doi: 10.1159/000362920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Khan TA, Shah T, Prieto D, et al. Apolipoprotein E genotype, cardiovascular biomarkers and risk of stroke: systematic review and meta-analysis of 14,015 stroke cases and pooled analysis of primary biomarker data from up to 60,883 individuals. International journal of epidemiology. 2013;42:475–492. doi: 10.1093/ije/dyt034. [A meta-analysis which clarify the role of ApoE on cardiovascular disease, especially with ischemic stroke.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schilling S, DeStefano AL, Sachdev PS, et al. APOE genotype and MRI markers of cerebrovascular disease: systematic review and meta-analysis. Neurology. 2013;81:292–300. doi: 10.1212/WNL.0b013e31829bfda4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Enas EA, Chacko V, Senthilkumar A, et al. Elevated lipoprotein(a)--a genetic risk factor for premature vascular disease in people with and without standard risk factors: a review. Disease-a-month : DM. 2006;52:5–50. doi: 10.1016/j.disamonth.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 72.Naoumova RP, Bonney SA, Eichenbaum-Voline S, et al. Confirmed locus on chromosome 11p and candidate loci on 6q and 8p for the triglyceride and cholesterol traits of combined hyperlipidemia. Arteriosclerosis, thrombosis, and vascular biology. 2003;23:2070–2077. doi: 10.1161/01.ATV.0000095975.35247.9F. [DOI] [PubMed] [Google Scholar]
- 73.Kim NS, Kang K, Cha MH, et al. Decreased paraoxonase-1 activity is a risk factor for ischemic stroke in Koreans. Biochemical and biophysical research communications. 2007;364:157–162. doi: 10.1016/j.bbrc.2007.09.119. [DOI] [PubMed] [Google Scholar]
- 74.Jameson JL, Longo DL. Precision medicine--personalized, problematic, and promising. N Engl J Med. 2015;372:2229–34. doi: 10.1056/NEJMsb1503104. [DOI] [PubMed] [Google Scholar]
- 75.Traylor M, Rutten-Jacobs LC, Holliday EG, et al. Differences in Common Genetic Predisposition to Ischemic Stroke by Age and Sex. Stroke; a journal of cerebral circulation. 2015 doi: 10.1161/STROKEAHA.115.009816. [DOI] [PMC free article] [PubMed] [Google Scholar]