

Abstract
We have previously shown that in response to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-elicited NAFLD progression, central carbon, glutaminolysis, and serine/folate metabolism are reprogrammed to support NADPH production and ROS defenses. To further investigate underlying dose-dependent responses associated with TCDD-induced fibrosis, female C57BL/6 mice were gavaged with TCDD every 4 days (d) for 28 d or 92 d. RNA-Seq, ChIP-Seq (2 h), and 28 d metabolomic (urine, serum, and hepatic extract) analyses were conducted with complementary serum marker assessments at 92 d. Additional vehicle and 30 µg/kg treatment groups were allowed to recover for 36 d following the 92-d treatment regimen to examine recovery from TCDD-elicited fibrosis. Histopathology revealed dose-dependent increases in hepatic fat accumulation, inflammation, and periportal collagen deposition at 92 days, with increased fibrotic severity in the recovery group. Serum proinflammatory and profibrotic interleukins-1β, -2, -4, -6, and -10, as well as TNF-α and IFN-γ, exhibited dose-dependent induction. An increase in glucose tolerance was observed with a concomitant 3.0-fold decrease in hepatic glycogen linked to increased ascorbic acid biosynthesis and proline metabolism, consistent with increased fibrosis. RNA-Seq identified differential expression of numerous matrisome genes including an 8.8-fold increase in Tgfb2 indicating myofibroblast activation. Further analysis suggests reprogramming of glycogen, ascorbic acid, and amino acid metabolism in support of collagen deposition and the use of proline as a substrate for ATP production via the proline cycle. In summary, we demonstrate that glycogen, ascorbic acid, and amino acid metabolism are also reorganized to support remodeling of the extracellular matrix, progressing to hepatic fibrosis in response to chronic injury from TCDD.
Keywords: NAFLD, metabolomic, RNA-Seq, matrisome, AhR.
Exposure to the environmental contaminant 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) has been associated with the development of reversible hepatic steatosis, and the progression of steatosis to steatohepatitis with hepatic fibrosis (Fader et al., 2015; Pierre et al., 2014). These and the majority of responses to TCDD and related compounds, if not all, are mediated through the aryl hydrocarbon receptor (AhR) (Fernandez-Salguero et al., 1996). AhR activation leads to the dissociation of chaperone proteins and subsequent dimerization with the AhR nuclear translocator. The liganded heterodimer then binds to dioxin response elements (DREs; also known as AHRE or XRE) initiating changes in gene expression, although DRE-independent mechanisms have also been reported (Dere et al., 2011; Huang and Elferink, 2012). Not surprisingly, AhR activation plays an important role in TCDD-elicited fibrosis (Pierre et al., 2014) although the mechanisms involved in AhR-mediated fibrogenesis remain poorly understood.
Fibrosis is a wound-healing, protective response to repeated, and chronic injury leading to the accumulation of extracellular matrix (ECM) due to the unbalancing of deposition and turnover of the matrisome (Naba et al., 2016). Although the etiology of fibrosis in different organs is poorly defined, the process is believed to involve shared common mechanisms at the onset and during fibrotic progression. Following injury, cytokines and growth factors released by damaged tissue initiate a provisional ECM response allowing resident and infiltrating immune cells to adhere and initiate repair (Glaser et al., 2009). Further development is mediated by prolonged or persistent inflammation that triggers profibrotic signaling such as the overproduction/activation of cytokines and TGFβ, immune cell migration to damaged areas, and epithelial to mesenchymal transition (EMT) of hepatic stellate cells (HSC) and fibroblasts to myofibroblasts that synthesize excessive amounts of ECM proteins (Glaser et al., 2009; Kalluri and Weinberg, 2009; Omenetti et al., 2009). The accumulation of hepatocyte and cholangiocyte damage, increased ROS production, secretion of proinflammatory mediators, and the exhaustion of antioxidant defenses releases endogenous damage-associated molecular patterns (DAMPS) that further promote fibrogenesis. The deposition of ECM is continuously remodeled altering tissue tensile and compression strength and elasticity which ultimately impairs function and increases cell leakiness and death, promoting further inflammation.
We have recently shown that TCDD dosing over 28 days elicits a Warburg-like response well before hepatic ECM remodeling and myofibroblast activation (Nault et al., 2016). Interestingly, myofibroblast activation has also been linked to a Warburg-like response wherein cells increase glycolytic flux and lactate production (Chen et al., 2012). However, few studies have examined the metabolic reprogramming underlying the development of hepatic fibrosis. To further investigate the relationships between metabolic reprogramming and fibrosis, we evaluated systemic metabolic changes, serum cytokine and adipo/hepatokine levels, and differential gene expression in mice treated with TCDD every 4 days for 92 days. Furthermore, published metabolomic and transcriptomic data from mice exposed for 28 days (Nault et al., 2016) were used to investigate the temporal progression of these pathologies, supplemented with urinary metabolomic data. We show that, in addition to supporting antioxidant responses, hepatic metabolic reprogramming also supports collagen deposition through the redirection of glycogen, ascorbic acid, and proline metabolism.
MATERIALS AND METHODS
Animal Handling and Tissue Processing
Female C57BL/6 mice (Charles River Laboratories, Portage, MI) on postnatal day 25 (PND 25) were housed in polycarbonate cages with cellulose fiber chips (Aspen Chip Laboratory Bedding, Warrensberg, NY) at 30%–40% humidity and a 12 h light/dark cycle. Animals were fed ad libitum (Harlan Teklad 22/5 Rodent Diet 8940, Madison, WI) and had free access to deionized water. Following 4 days acclimation (PND 28), animals (n = 10) were orally gavaged with 0.1 ml sesame oil or 0.01, 0.03, 0.1, 0.3, 1, 3, 10, or 30 µg/kg TCDD (Dow Chemical Company, Midland, MI) every fourth day for 92 days for a total of 23 exposures. Two additional groups (vehicle and 30 µg/kg) were allowed to recover for 36 days following the 92 days treatment regimen. Body weight and food intake were monitored every 4 days. Urine was collected from individual mice over 2 h on day 26 in the absence of food or water and stored immediately at -80 °C. Oral glucose tolerance tests (OGTTs) were performed at 31, 63, and 96 days in sesame oil and 30 µg/kg TCDD groups only. Briefly, at time 0 min animals were orally gavaged with 2 g/kg glucose in a 25% solution and tail blood glucose was measured after 0, 15, 30, 60, and 120 min using a FreeStyle Lite handheld glucose meter (Abbott Laboratories, IL).
Prior to termination animals were fasted for 6 h. Blood was collected by submandibular vein puncture prior to cervical dislocation, centrifuged, and stored at -80 °C. Liver and gonadal white adipose tissue (gWAT) were excised, weighed, and frozen at -80 °C. Liver sections were stored in 10% neutral buffered formalin (Sigma-Aldrich, MO) or frozen in Tissue-Tek O.C.T compound (Sakura, CA) for Haematoxylin & Eosin (H&E), Oil Red O staining (ORO), and PicroSirius Red (PSR) staining as previously described (Nault et al., 2015a). Hepatic TCDD levels were determined as previously described (Fader et al., 2015). All animal procedures were approved by the Michigan State University Institutional Animal Care and Use Committee.
Clinical Chemistry, Glycogen Assay, and Protein Measurements
Serum cholesterol, triglycerides, and glucose were determined using commercially available reagents (Pointe Scientific, Canton, MI). Adiponectin (R&D systems), FGF21 (R&D systems), insulin (Crystal Chem Inc), and leptin (Crystal Chem Inc) were determined using commercially available ELISAs. Hepatic glucose and glycogen levels were determined as previously described by Keppler and Decker (1974). Briefly, liver samples (∼50 mg) or glycogen standards (Sigma-Aldrich) were homogenized in 6% PCA (250 µl) using a Polytron PT2100 homogenizer (Kinematica AG, Luzern, CH). A total of 25 µl of 1M NaHCO3 was added to 50 µl of the homogenate, and 125 µl of 2 mg/ml amyloglucosidase (Sigma-Aldrich) was added to each sample to hydrolyze glycogen. Samples were incubated and shaken for 2 h at 37 °C and then centrifuged to remove debris. Glycogen and glucose were assayed using the glucose assay kit (Pointe Scientific, Canton, MI) a M200 plate reader (Tecan, Durham, NC). Total hepatic levels were corrected using hepatic glucose levels, and expressed as glycosyl units. Serum cytokine levels were assayed using the Meso Scale V-PLEX Proinflammatory panel kit and Sector S 600 (Meso Scale Discovery, Rockville, MD). Protein determinations are described in Supplementary methods.
RNA Extraction and RNA Sequencing
Total RNA extraction and RNA-Seq analysis were performed as previously described (Nault et al., 2015b) for 3 animals (n = 3). Reads were mapped to the GRCm38 release 74 mouse reference genome. RNA-Seq data have been deposited in the Gene Expression Omnibus (GSE81990) and published 28 days RNA-Seq data (Nault et al., 2015b) used to complement the 92 days gene expression profiles are available on GEO (GSE62902).
Targeted Urinary Metabolomics
Urine samples, collected at 26 days (PND 54, 7 TCDD doses), were sent onto a trapping column (C18, 4 mm × 2 mm, Phenomenex) and desalted for 30 s with High Performance Liquid Chromatography (HPLC) grade water containing 10 mM tributylamine and 15 mM acetic acid (pH 4.95). Samples were examined by liquid chromatography (LC) and tandem mass spectrometry (MS/MS) using a Paradigm MS4 HPLC (Michrom Bioresources, Auburn, CA), and a Synergi Hydro column (4 µm particle size, 80 Å, 150 mm × 2 mm, from Phenomenex) (Lunt et al., 2015). HPLC was coupled with negative-mode electrospray ionization (ESI) to a TSQ Vantage Triple Stage Quadrupole Mass Spectrometer (Thermo Scientific) operating in multiple reaction monitoring (MRM) mode. The LC parameters were as follows: Autosampler temperature, 10ºC; injection volume, 10 µl; column temperature, room temperature; and flow rate, 200 µl/min. The LC solvents were Solvent A: 10 mM tributylamine and 15 mM acetic acid in 97:3 water:methanol (pH 4.95); and Solvent B: Methanol. Elution from the column was performed over 50 min with the following gradient: t = 0, 0% B; t = 5, 0% B; t = 10, 20% B; t = 20, 20% B; t = 35, 65% B; t = 38, 95% B; t = 42, 95% B, t = 43, 0% B; t = 50, 0% B. Electrospray ionization spray voltage was 3000 V. Nitrogen was used as the sheath gas at 30 psi and as the auxiliary gas at 10 psi, and argon as the collision gas at 1.5 mTorr, with the capillary temperature at 325ºC. Scan time for each MRM transition was 0.1 s with a scan width of 1 m/z. The LC runs were divided into time segments, with the MRM scans within each time segment containing compounds eluting during that time interval. For compounds eluting near boundaries between time segments, the MRM scan corresponding to the compound was conducted in both time segments. Instrument control, chromatographic control, and data acquisition were performed by the Xcalibar software (Thermo Scientific). Raw data and MRM parameters are deposited in www.ebi.ac.uk/metabolights (MTBLS225). Data analysis, including peak integration was performed using MAVEN (Clasquin et al., 2012). One-way ANOVA was performed in SAS 9.3. Urinary metabolomic data were supplemented with previously published serum and hepatic extract metabolomic assessments (Nault et al., 2016).
Pathway Enrichment Analysis and Integration of Datasets
Over-representation of metabolites within a pathway, as well as impact scores reflecting the connectivity of altered metabolites was determined using the Metabolic Pathway Enrichment Analysis tool (MetPA) (Xia and Wishart, 2010). CytoKegg 0.0.5 plugin (Cytoscape 3.2.0) was used to generate pathway templates that were also manually curated. Reactions catalyzed by several enzymes were streamlined by combining in a single node. Previously identified putative DREs (pDREs) and AhR enrichment peaks (Nault et al., 2016), as well as the largest change in gene expression at 92 days, and/or altered metabolite level for liver, serum (Nault et al., 2016), and urine, are represented for each node (eg, largest AhR enrichment fold-change, highest matrix similarity score of pDREs, or largest |fold-change| of gene expression) regardless of the dose.
RESULTS
TCDD Tissue Levels and Gross Pathology
In this study, animals were gavaged every 4 days for a total of 92 days in order to examine the progression of fibrosis. The higher doses used compensate for TCDD half-life differences in humans (1–11 years) compared with mice [8–12 days (Kopec et al., 2013)], the short duration of our study considering the bioaccumulative nature of halogenated AhR ligands, and the potential cumulative lifetime exposure from diverse AhR ligands, acknowledging the potential for nonadditive interactions for some responses. The hepatic TCDD levels achieved in mice using this dose range (0–30 µg/kg) and treatment regimen (oral gavage every 4 days for 28 days or 92 days) were comparable to the levels reported at 28 days, suggesting steady state levels of TCDD were achieved (Nault et al., 2015a). Furthermore, although the doses are not environmentally relevant, the TCDD levels achieved in mouse hepatic tissue approximate levels of AhR agonists reported in serum humans suggesting physiologically relevant perceived levels (Supplementary Figure 1). It remains challenging; however, to define the relationship between serum adjusted Toxic Equivalents (TEQs) to liver concentrations.
Although our previous 28-day studies did not elicit overt toxicity (Nault et al., 2015a), there were losses in this 92 days study (Supplementary Figure 2). Individual losses within a dose group prior to 28 days were sudden, did not exhibit weight loss or decreased food intake, and did not appear to be treatment related. In contrast, later deaths were treatment related, and predicted based on sudden body weight decreases (∼15%), and therefore terminated. Survivors did not exhibit a difference in terminal body weight (Table 1). At 0.3 and 3 µg/kg TCDD, body weights increased as of days 43 and 27, respectively, with a corresponding increase in gWAT at 0.3 µg/kg (Supplementary Table 1 and Table 1). We have previously observed body weight gain at 3 µg/kg (Fader et al., 2015) suggesting TCDD may increase body weight gain within certain dose ranges using this treatment regimen, although additional studies are required to further evaluate this response. Food intake measurements do not indicate increased consumption at 0.3, or 3 µg/kg TCDD, although consumption was greater at 30 µg/kg TCDD (Supplementary Table 2). There was a dose-dependent increase in relative liver weight with a dose-dependent decrease in gWAT that was not statistically significant (Table 1). Visual examination found pale colored livers with a mottled surface and evidence of dimpling, consistent with Pierre et al. (2014).
TABLE 1.
Terminal Body and Tissue Weights
| Dose (µg/kg) | Terminal Body Weight (g) | Food Intake | RLWb | gWATc (g) | Serum Cholesterol (mg/dl) | Serum Glucose (mg/dl) | Serum Triglycerides (mg/dl) |
|---|---|---|---|---|---|---|---|
| Treated for 92 days | |||||||
| 0 | 21.86 ± 0.31 | 5.76 ± 0.35 | 0.27 ± 0.02 | 108.3 ± 15.1 | 112.3 ± 32.3 | 66.8 ± 15.2 | |
| 0.01 | 21.82 ± 0.58 | 5.42 ± 0.21 | 0.23 ± 0.03 | 102.4 ± 12.9 | 120.0 ± 45.4 | 62.8 ± 6.0 | |
| 0.03 | 21.91 ± 0.32 | 5.39 ± 0.11 | 0.24 ± 0.02 | 95.3 ± 10.8 | 159.0 ± 18.7 | 62.6 ± 20.8 | |
| 0.1 | 22.1 ± 0.31 | 5.66 ± 0.11 | 0.25 ± 0.02 | 121.1 ± 8.0 | 149.2 ± 26.7 | 66.9 ± 9.8 | |
| 0.3 | 23.24 ± 0.31a | 6.73 ± 0.54a | 0.41 ± 0.05a | 108.6 ± 22.3 | 149.1 ± 21.0 | 55.2 ± 13.1 | |
| 1 | 22.25 ± 0.28 | 6.44 ± 0.28a | 0.24 ± 0.03 | 94.1 ± 17.6 | 141.5 ± 15.9 | 54.0 ± 8.4 | |
| 3 | 23.62 ± 0.3a | 6.79 ± 0.61a | 0.33 ± 0.03 | 68.7 ± 13.1a | 139.0 ± 18.2 | 58.0 ± 5.4 | |
| 10 | 23.14 ± 0.41 | 6.89 ± 0.25a | 0.28 ± 0.02 | 59.2 ± 11.3a | 142.2 ± 28.2 | 57.9 ± 4.2 | |
| 30 | 21.27 ± 1.33 | 8.71 ± 0.39a | 0.18 ± 0.05 | 59.0 ± 21.3a | 125.5 ± 26.5 | 69.7 ± 11.4 | |
| Allowed to recover for 36 days | |||||||
| 0 | 23.19 ± 0.48 | 4.61 ± 0.12 | 0.28 ± 0.02 | NM | NM | NM | |
| 30 | 24.08 ± 0.89 | 7.46 ± 0.62a | 0.2 ± 0.01 | NM | NM | NM | |
NM indicates that the endpoint was not measured.
Significant difference compared with vehicle control determined by one-way ANOVA followed by Dunnett’s post hoc test.
Relative liver weight (%).
Gonadal white adipose tissue.
Histology of Hepatic NAFLD Features
Similarly to dosing every 4 days for 28 days (Nault et al., 2016), 92 days of treatment elicited dose-dependent increases in centrilobular hepatocyte vacuolation, inflammation, and fibrosis (Figure 1A; Supplementary Table 3). However, quantitative assessment revealed hepatic lipid accumulation at lower doses (0.3 vs 1 µg/kg; Figure 1B). Similarly, inflammatory foci were present at 1 µg/kg, with portal fibrosis at 10 µg/kg TCDD. In contrast, inflammation and fibrosis at 28 days were only observed at 10 and 30 µg/kg, respectively. Collagen deposition along portal tracts is in agreement with Pierre et al. (2014). Quantitative assessment of collagen deposition by PSR staining showed a ∼1.7-fold increase at 28 and 92 days (Figure 1C). Fibrotic portal regions extended outward with the deposited ECM, appearing to pull the visceral surface internally creating a regular dimpled pattern (Figure 2). There were also instances of minimal bile duct proliferation and extramedullary hematopoiesis at 30 µg/kg TCDD. Although reduced hepatocyte vacuolation was observed in the 36 days recovery group, inflammation, collagen deposition, and bile duct proliferation occurred at a similar or increased frequency and severity (Figure 1; Supplementary Table 3). It remains unclear whether increased severity upon removal of TCDD is a consequence of reduced TCDD levels or induction of repair mechanisms following reversal of steatosis.
FIG. 1.

Histological evaluation of livers from mice gavaged with sesame oil vehicle or 30 µg/kg TCDD every 4 days for 28 and 92 days, as well as following a 36 days recovery period (ie, no treatment) following the final dose at 88 days. A, Representative micrographs of sections stained with hematoxylin and eosin staining (H&E) was used to survey hepatic lesions. Oil Red O (ORO) was used to determine neutral lipid accumulation. PicroSirius Red (PSR) was used to verify collagen deposition. Scale bar represents 50 µm. Portal veins are labeled with the letter v and bile ducts with the letter b, whereas inflammation is denoted by a solid arrow and collagen deposition by a dashed arrow. Quantitation of (B) Oil Red O (ORO) and (C) PicroSirius Red (PSR) staining performed using the Quantitative Histological Analysis Tool (Nault et al., 2015a). Bars represent mean + SEM for at least 3 animals (n = 3–5). Asterisks (*) indicate a significant difference (P ≤ 0.05) between TCDD and vehicle treatment groups determined by two-way ANOVA and Dunnett’s post hoc test for ORO and Student’s t test for PSR.
FIG. 2.

Representative histological micrographs of liver sections from mice gavaged with (A, C) sesame oil vehicle or (B, D) 30 µg/kg TCDD every 4 days for 92 days demonstrating dimpled regions of the visceral surface. Hematoxylin and eosin staining (H&E; A, B) illustrates inflammatory infiltration while PicroSirius Red (PSR; C, D) demonstrates collagen deposition. Scale bar represents 50 µm. Portal veins are labeled with the letter v while inflammation is denoted by a solid arrow and collagen deposition by a dashed arrow.
Differential Gene Expression in the Liver
In our published 28 days dataset (GSE62902), 17 464 unique Ensembl annotated genes were detected, 3406 of which were differentially expressed while at 92 days 19 935 unique genes were expressed (min count ≥ 5) with 4575 differentially expressed using |fold-change| ≥ 1.5 and P1(t) ≥ 0.8 criteria. The larger number of detected genes may be due to differing sequencing depths which confounds direct read count comparisons, but still allows for fold-change comparisons. A total of 2233 DEGs were conserved in both datasets with those unique to a single time-point largely representing lower fold changes. Overall, the same genes exhibited the greatest induction or repression at both time-points. Comparative analysis found >90% of the 2233 common DEGs were positively correlated (similar responses) for both fold-change and P1(t) values (Supplementary Figure 3; quadrant I). A 98% of the expression patterns were positively correlated illustrating the similarity between these datasets (Supplementary Figure 3; quadrants I and IV). In many cases, 92 days DEGs exhibited differential expression at lower doses (∼36% or 80% at lower or equal dose, respectively) and larger fold changes (∼60%) compared with 28 days DEGs. For example, Cd36, a lipid transporter integral to AhR-mediated hepatic steatosis (Lee et al., 2010; Yao et al., 2016) was induced at 0.03 µg/kg at 92 days compared with 1 µg/kg at 28 days with 9.2- and 3.2-fold increases, respectively. Not surprisingly, Cyp1a1 and Cyp1b1 exhibited the highest induction at 28 days (1250- and 807-fold, respectively) and 92 days (1228- and 578-fold, respectively).
Increased Ascorbic Acid Biosynthesis Supports Fibrosis
LC–MS/MS analysis detected 132 urinary metabolites, of which 39 were altered by treatment (Supplementary Table 4). MetPA analysis identified phenylalanine and tryptophan metabolism as the most enriched pathways (over-represented altered metabolites within a pathway) while most impacted pathways (high connectivity among altered metabolites) included the TCA cycle, alanine, aspartate and glutamate metabolism, glycolysis and gluconeogenesis, and ascorbate and aldarate metabolism (Figure 3).
FIG. 3.
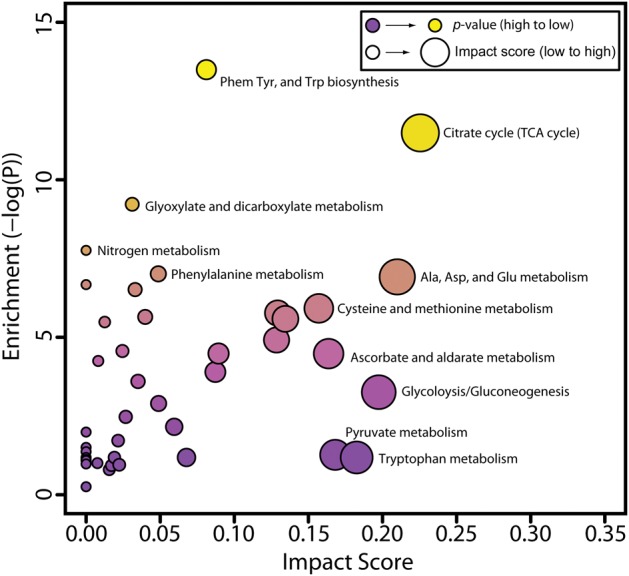
Metabolic pathway enrichment analysis (MetPA) of urinary metabolites altered by TCDD (P ≤ 0.05). Pathway alterations were considered significant when enrichment values (pathway over-representation) ≥ 4.0. Impact scores (connectedness of altered metabolites) were used to rank pathways most affected by TCDD treatment (Supplementary Tables 4 and 6).
Urinary metabolomic analysis was marked with a 124-fold increase in ascorbic acid, the largest measured metabolite change in TCDD treated animals (Supplementary Table 4). Similarly, hepatic levels of ascorbic acid were also increased 1.3-fold suggesting rapid elimination of increased hepatic biosynthesis via the urine. Interestingly, glycogen is used as substrate for ascorbic acid biosynthesis which serves important roles in anti-oxidant response, iron homeostasis, and collagen synthesis (De Tullio, 2012). Unlike humans and other vertebrates, mice synthesize ascorbic acid due to the expression of Gulo (21). Downstream from glycogenolysis, Rgn, a gene involved in ascorbic acid synthesis was repressed 2.8-fold with AhR enrichment (Figure 4). However, Gulo, the rate-limiting step in ascorbic acid synthesis was induced 2.0-fold, consistent with increased urinary levels. Ascorbic acid is a required cofactor for prolyl 4-hydroxylase (P4h), a 2-oxoglutarate dioxygenase that catalyzes the hydroxylation of free proline and the post-translational hydroxylation of proline in collagen. Consequently, changes in ascorbic acid metabolism are consistent with TCDD-elicited fibrosis.
FIG. 4.

Integrative analysis of (A) glucose and glycogen; B, ascorbic acid; and C, proline metabolism. The color scale represents the log2(fold-change) for genes and metabolites. Gray indicates metabolites not measured or detected. Genes are identified as rectangles, metabolites as circles, and transporters as hexagons. Values in the upper left corner of genes provides the maximum AhR enrichment fold-change, whereas upper right corner indicates the highest pDRE MSS. An expandable and interactive version that provides additional information can be viewed at http://dbzach.fst.msu.edu/index.php/supplementarydata.html, last accessed 31 August 2016.
Altered Glucose and Glycogen Metabolism
Insulin, which plays an important role in glucose homeostasis, was within the normal range (∼0.5 ng/ml) in all groups (Supplementary Table 5). However, adiponectin that is secreted by adipocytes to regulate glucose and lipid metabolism, and improve insulin sensitivity (Lin et al., 2013), decreased from 2.2 µg/ml in controls to 1.2 µg/ml at 30 µg/kg TCDD (Figure 5A). Conversely, hepatic FGF21, which also regulates glucose and lipid metabolism by increasing sensitivity to insulin (Lin et al., 2013), was induced from undetectable levels to 8.2 ng/ml at 30 µg/kg TCDD (Figure 5B). Fgf21 mRNA was induced 17.2- and 6.2-fold at 28 and 92 days, respectively. No AhR enrichment as observed, unlike previous reports (Cheng et al., 2014; Girer et al., 2016) that could be accounted for by differences in treatment duration and model.
FIG. 5.

Serum levels: A, adiponectin; B, FGF21; C, IFN-γ; D, IL-4; E, IL-10; F, IL-6; G, IL-2; and H, TNF-α were determined in mice orally gavaged with TCDD every 4 days for 92 days. Cytokines were determined using Meso Scale Discovery V-Plex assay kits. Adiponectin and FGF21 were determined by ELISA. Bars represent mean + SEM (n = 4-5). Asterisks (*) indicate a significant difference (P ≤ 0.05) compared with vehicle control determined by one-way ANOVA and Dunnett’s post-hoc test. Insulin and leptin as well as the cytokines IL-12p70, IL-1B, IL-5, and KC/GRO were all unchanged (Supplementary Table 5).
Oral glucose tolerance tests were performed at days 31, 63, and 96 on vehicle and 30 µg/kg TCDD-treated groups to further examine hepatic glucose metabolism reprogramming (Figure 6A–C). Although basal fasted blood glucose levels did not differ (Table 1), consistent with unaltered insulin levels, TCDD-elicited a dose-dependent decrease in hepatic glucose and glycogen levels (Figure 6D–E), and improved glucose tolerance suggesting increased glucose uptake and metabolism. This is consistent with the 1.7- and 2.0-fold induction of rate-limiting hexokinases Hk2 and Hk3, although the glucose transporter Glut2 (Slc2a2) and glucokinase (Gck) were repressed 3.4- and 8.0-fold, respectively (Figure 4). TCDD-elicited metabolic reprogramming is marked by pyruvate kinase (PKM) and glutaminase (GLS1) isoform switching (Nault et al., 2016) that may partially account for the increase in hepatic glucose metabolism. At 92 days, total PKM protein levels increased 2.7-fold, matched by a 2.7-fold increase in PKM2 and no change PKM1 (Supplementary Figure 4). A concomitant decrease in pyruvate carboxykinase (PEPCK-C; Pck1) mRNA (2.0-fold at 28 and 92 days) and protein (1.4-fold; P-value = 0.06) with a 4.0-fold increase in AhR enrichment is in line with TCDD-elicited repression of gluconeogenesis (Supplementary Figure 4) (Ahmed et al., 2015).
FIG. 6.

Glucose and glycogen homeostasis. Oral glucose tolerance tests (OGTT): A, 31 days; B, 63 days; and C, 96 days following oral gavage with sesame oil (red) or 30 µg/kg TCDD (blue) every 4 days in female C57BL/6 mice. Values represent mean ± SEM for 5 animals (n = 5). Dose-dependent changes in hepatic (D) glucose or (E) glycogen levels at 92 days. Values represent mean + SEM for 5 animals (n = 5). Asterisks (*) indicate a significant difference (P ≤ 0.05) determined by repeated measures two-way ANOVA for the OGTT and one-way ANOVA followed by Dunnett’s post hoc test for hepatic glucose and glycogen levels.
Moreover, genes associated with glycogen catabolism including Pgm, Ugdh, and Ugt1a6a were induced 1.7-, 7.5-, and 2.1-fold, respectively. However, expression of glycogen phosphorylase (Pygl) which catalyzes the rate-limiting step in glycogen breakdown was repressed 2.1-fold. The repression was not reflected at the protein level (Supplementary Figure 4). ChIP-Seq analysis identified enrichment within Pygl, Pgd, Ugdh, Ugt1a6a genomic regions (Figure 4) implicating AhR regulation. Glycogen synthase (Gys2) which is involved in the biosynthesis of glycogen was downregulated 1.9-fold and showed a 2.7-fold increase in AhR genomic binding. Taken together, these results suggest that glucose is not being stored as glycogen and is likely being catabolized in TCDD treated animals.
Effects of TCDD on the Matrisome
Fibrosis is an initial form of tissue repair that progressively impairs function following chronic disease or persistent damage. It is defined by the excessive deposition of ECM, the noncellular component of all tissues and organs that provide not only scaffolding, but also initiate signaling required for morphogenesis, differentiation, homeostasis, and function. In addition, it acts as a growth factor reservoir and selective filter to control the flow of materials between cells. Qualitative and quantitative changes in ECM composition affect not only tissue function, elasticity, tension, and compression strength, but also cell integrity and viability. While characterized by collagen deposition, fibrosis involves interactions between the ∼1100 ECM structural and associated proteins that constitute the matrisome, including a core set ∼300 human and mouse collagens, proteoglycans, and glycoproteins (Naba et al., 2016). This includes: (1) fibrous proteins such as collagens, elastins, fibronectins, and lamins; (2) interstitial proteoglycans that provide unique tissue buffering, hydration, binding, lubrication, and force-resistance properties (Frantz et al., 2010). In addition to PSR staining along hepatic portal tracts (Figure 1), 344 matrisomal genes exhibited differential expression with 55 possessing AhR enrichment (Figure 7; Supplementary Figure 4).
FIG. 7.

Hepatic gene expression changes of the matrisome (http://matrisomeproject.mit.edu/, last accessed 31 August 2016) in mice gavaged every 4 days for 92 days categorized: A, structural genes; B, secreted factors; or C, inflammatory chemokines and interleukins; D anchoring genes of the cadherin, integrin, and selectin families; and E, epithelial and mesenchymal markers. The presence of pDREs (DRE; MSS ≥ 0.89) and AhR enrichment peaks (FDR ≤ 0.05) determined by ChIP-Seq (Nault et al., 2016) are shown. Relative transcript representation describing the raw number of aligned reads to each transcript is shown to indicate potential importance of the genes function in the liver. Larger heat maps with 28 days expression data included are available in Supplementary Figure 6.
Several collagen genes exhibited differential expression including Col4a3, Col4a4, and 9a2 that were upregulated 5.7-, 2.1-, and 206-fold at 92 days, respectively (Figure 7A), with AhR enrichment, although only Col4a3 contained a pDRE. Biosynthesis of collagen triple helixes is a complex multistep process beginning with post-translational proline hydroxylation and crosslinking by prolyl 4-hydroxylases (P4h) a2 (induced 1.8-fold) and b subunits requiring oxygen, Fe2+, ascorbic acid, and α-ketoglutarate (Figs. 4 and 7B). Hydroxylated proline is essential for the stabilization of the collagen triple helix. Similarly, lysyl oxidase (Lox) and lysyl oxidase-like (Loxl) 1, 2, and 3 that were induced 3.0-, 1.4-, 1.5-, and 2.1-fold, respectively, and Loxl4 repressed 7.7-fold also participate in the crosslinking of collagen. In the endoplasmic reticulum, the procollagen triple helix peptide is then folded with the assistance of chaperone proteins such as Hsp47 (Serpinh1) that was induced 1.7-fold. Procollagen is transported through the Golgi stacks and secretory vesicles followed by cleavage of N- and C- terminal propeptides producing tropocollagen which spontaneously self-assemble into fibrils, and undergoes further LOX and LOXL crosslinking to produce mature collagen fibrils. Elastin which was induced 2.1-fold also associates with collagen, whereas transglutaminase (Tgm) 1, 2, and 3 which add more cross-links that stiffen and decrease the solubility of the ECM were induced 1.9-, 1.8-, and 11.5-fold, respectively.
Despite the prolonged induction of ECM structural genes, there is a lack of additional fibrotic accumulation between 28 and 92 days (Figure 1). Extracellular matrix is continuously turned over by enzymes associated with synthesis and degradation that allow the fine tuning of ECM remodeling. This includes the induction of various MMP, Adam, Adamt, heparanase proteases, and their inhibitors (eg, Timp, Serpin, A2m) (Figure 7B). For example, MMPs degrade ECM to open up avenues for migration, and activate profactors stored within the ECM such as TGFβ. Among the 115 differentially expressed ECM regulators, only 19 showed AhR enrichment including Adamts14 which was induced 2.4-fold with a 2.6-fold increase in AhR genome binding. Although typically associated with converting plasminogen to plasmin, urokinase/tissue type plasminogen activators (Plau/Plat) which were induced 1.9- and 2.3-fold, respectively, also activate latent forms of TGFβ and pro-MMPs, and aid in the migration of collagen producing cells to damaged tissue (Loskutoff and Quigley, 2000). Serpine1 (commonly known as PAI-1), the predominant inhibitor of PLAU/PLAT, was induced 54.3-fold by TCDD. Therefore, SERPINE1 interaction with PLAU/PLAT could attenuate these responses. Conversely, Lrp1 which clears proteases, protease inhibitors, and other fibrogenic factors following endocytosis, was repressed 1.6-fold (Van Gool et al., 2015). In contrast to Serpine1, Serpin a1, a3, b, c1, f2, g and i1 clades and isoforms, which inhibit trypsin, chymotrypsin, urokinase/tissue plasminogen activator, and fibrinolysis activity, were largely repressed with some exceptions (Figure 7B).
Effects of TCDD on Proline and Hydroxyproline Metabolism
Proline/hydroxyproline represents ∼23% of the amino acids in collagen with glycine (∼30%) and alanine (∼20%) the other major residues (Barbul, 2008). Although hepatic extract levels of glycine increased 20.3-fold, alanine and proline were reduced 2.0- and 2.4-fold, respectively (Figure 4). Proline serves multiple roles in metabolism including protein synthesis, cell signaling, and as a precursor for other amino acids. Moreover, during periods of cell stress, proline released during ECM remodeling can be used as substrate for ATP production (Phang et al., 2015). Extracellular matrix fragments from remodeling can serve as a substrate for prolidase (Pepd), a peptidase that hydrolyzes di- and tri-peptides to release C-terminal proline or hydroxyproline residues. Subsequently, proline dehydrogenase (Prodh) oxidizes proline to pyrroline-5-carboxylate (P5C) producing mitochondrial ATP and ROS, and the cytosolic P5C reductase 2 (Pycr2) then reduces P5C to proline at the expense of NADPH. TCDD induced Pepd, Prodh and Pycr2 1.9-, 2.1-, and 1.3-fold, respectively (Figure 4). Although hepatic proline levels are lower in treated animals, a 4.6-fold increase was observed in the serum. However, there was no evidence of altered expression of the proline transporters Slc6a7 or Sit1 suggesting intracellular hepatic proline is sequestered in collagen and that proline released by extrahepatic protein catabolism and Pepd activity may be elevating serum levels. Proline can also be synthesized from glutamine and Slc1a5, a glutamine transporter which is induced 10.7-fold by TCDD with a 2.9-fold increase in AhR enrichment. Glutamine is then deaminated to glutamate by glutaminase (Gls) which was induced 1.9-fold. Glutamate is a substrate for P5C synthetase (Aldh18a1) producing glutamic-γ-semialdehyde that spontaneously converts to P5C which can cycle back to proline producing ATP. Consequently, glutaminolysis may support proline cycling in addition to GSH biosynthesis in response to TCDD. However, the relevance of proline cycling to cellular bioenergetics in combating TCDD-elicited energy depletion and hepatotoxicity remains to be determined.
Hydroxyproline released during collagen turnover is not re-incorporated into protein (Phang et al., 2015) and is directed to glyoxylate synthesis to produce oxalate, glycolate, or glycine (Jiang et al., 2012). Paradoxically, Prodh2, Got1, Got2, and Hoga1 involved in the metabolism of hydroxyproline to glyoxylate are down-regulated 5.0-, 4.3-. 1.8-, and 1.7-fold, respectively (Figure 4), suggesting pathway repression. Although glyoxylate, a highly reactive aldehyde, can be metabolized to glycine that was increased 20.3-fold in liver extracts, Agxt was repressed 2.6-fold. Similarly, glyoxylate reductase/hydroxypyruvate reductase (Grhpr) that uses NADPH to reduce glyoxylate to glycolate was also repressed 1.6-fold despite 2.3- and 4.0-fold increases in hepatic extracts and urine. Notably, Hao1 which uses NADPH to convert glycolate to glyoxylate while generating ROS and was the most repressed gene with a 137.0- and 71.4-fold, at 28 and 92 days, respectively. Therefore, HAO1 repression likely lowers oxidative stress, NADPH consumption, and the production of a reactive aldehyde. Lastly, glyoxylate metabolism by lactate dehydrogenase (Ldh) produces oxalate, a poorly metabolized dicarboxylic acid that can form tissue-damaging calcium oxalate crystals in the kidney. However, Slc26a1 which exports oxalate from the liver into the blood was repressed 1.9-fold. Hydroxyproline tracer studies are required to determine the fate of hydroxyproline derived from collagen remodeling.
Proinflammatory Signaling and Fibrosis
As fibrosis progresses, the ECM is enriched with hyaluronan and versican (Vcan) which bind and trap inflammatory cells infiltrating the damaged tissue. Hyaluronan is a non-sulfated glycosaminoglycan polymer that increases in length with the addition of glucuronic acid and N-acetylglucosamine. It forms a pericellular coat through interaction with CD44 (induced 3.2-fold) that maintains the myofibroblast phenotype. TGFβ also increases hyaluronan retention in the ECM, thus creating an autostimulatory cycle because myofibroblasts secrete TGFβ (Wight and Potter-Perigo, 2011). Glucuronic acid, which is increased 2.7- and 2.6-fold in hepatic extracts and urine, respectively, is a metabolite in the TCDD-induced ascorbic acid biosynthesis pathway. N-acetylglucosamine is a product of the hexosamine biosynthesis pathway. Gfpt1 and 2, the rate limiting steps in the hexosamine biosynthesis, were induced 1.4- and 3.1-fold, respectively. In addition, Uap1l1, a paralog of Uap1, which catalyzes the last step in N-acetylglucosamine synthesis, was also induced 4.4-fold. Furthermore, Vcan, an ECM proteoglycan, was induced 3.9-fold. High molecular weight hyaluronan also promotes the induction of IL-10, which was induced 3.0-fold (Figure 5E).
Injury to cholangiocytes, activated myofibroblasts, and hepatic macrophages also recruit immune cells such as circulating monocytes, T cells, and neutrophils due to proinflammatory mediator secretion. In accordance with hepatic inflammation (Figure 1), serum levels of IL-1β, IL-2, IL-4, IL-6, IL-10, TNF-α, and IFN-γ exhibited dose-dependent induction (Figure 5C–H; Supplementary Table 5). The 2.4- and 28.7-fold increases of proinflammatory factors IL-1β and IL-6, respectively, and myofibroblast activation are consistent with an inflammatory-profibrogenic cycle resulting in ECM deposition along portal tracts. Myofibroblasts also release soluble hedgehog ligands which stimulate cholangiocytes to produce Cxcl16 (induced 2.1-fold) and recruit NKT cells (Omenetti et al., 2009) that produce IL-2, IL-4, TNF-α, and IFNy which were induced 13.3-, 20.3-, 3.3-, and 12.5-fold, respectively. Osteopontin (Spp1), a target of hedgehog transcription factors commonly induced in fibrotic liver disease, was induced 4.3-fold at 92 days in the absence of AhR enrichment (Figure 7A). Other TCDD-elicited proinflammatory and immunogenic activities include the hepatic induction of Il1a, Ccl2, Ccl3, and Ccl4 mRNA levels 1.9-, 1.7-, 1.7-, and 1.7-fold, respectively (Figure 7C). Countering these proinflammatory signals was the 28.7- and 58.1-fold induction of glycoprotein non-metastatic melanoma B (Gpnmb), a macrophage expressed transmembrane glycoprotein that negatively regulates inflammation (Katayama et al., 2015).
TCDD-Elicited EMT and Altered cell Adhesion
Epithelial to mesenchymal transition (EMT) is commonly associated with fibrosis (Brenner et al., 2012). Collagen producing myofibroblasts are not detected in normal liver, but appear during the course of chronic liver disease, leading to fibrosis. Hepatic stellate cells, portal fibroblasts, bone marrow cells, epithelial cells, and endothelial cell have been identified as possible ECM sources (Brenner et al., 2012). These cells transition from an epithelial to a mesenchymal phenotype allowing them to polarize, migrate, invade, resist apoptosis, and produce ECM (Kalluri and Weinberg, 2009). The EMT is induced in response to DAMPS, immune cell derived cytokines such as TGFβ, and the mechanical tension produced by infiltrating cells. Although Tgfβ1 was only induced ∼1.5-fold, Tgfβ2 was induced 7.7- and 8.8-fold at 28 and 92 days, respectively. These isoforms are reported to possess interchangeable activities (Roberts et al., 1991). TGFβ-induced EMT is mediated by the activation of SMADs (SMAD2, 3, 4, and 7; unchanged), Zeb1 (repressed 2.0-fold), Zeb2 (induced 1.8-fold), and Hmga2 (induced 2.2-fold) (Lamouille et al., 2014). TCDD induced both epithelial [eg, E-cadherin (Cdh1), cadherin3, plakophilin 1 (Pkp1)] and mesenchymal [eg, N-cadherin (Cdh2), vimentin (Vim), snail 1/2/3] markers. More specifically, Cdh1 was induced 3.8-fold at 92 days while Cdh2 and Vim were unchanged (Figure 7E). Conversely, the 2.0-fold repression of Zeb1 is consistent with a mesenchymal phenotype as well as the 43.9- and 13.4-fold increase of Pkp1 at 28 and 92 days, respectively. Conversely, OVol15402, a transcription factor that induces mesenchymal to epithelial transition, was induced 33.2- and 6.4-fold, respectively, at 28 and 92 days possibly indicating EMT reversion (Roca et al., 2013).
Although cadherins play an integral role in defining epithelial and mesenchymal phenotypes, integrins, selectins, and other matrisome proteins (eg, laminins, proteoglycans, nidogens, fibulins, and fibronectin) are implicated in cell adhesion and interactions with the ECM. The majority of integrins were induced with Itg4 and 9 demonstrating a 1.9- and 1.5-fold induction, respectively, with concomitant AhR enrichment. Selectins Selp and Sell were also induced 1.8- and 1.7-fold, respectively. Tenascin C (Tnc) that inhibits interactions between syndecans, and fibronectin was induced 7.8-fold 92 days, whereas syndecans (Sdc1 and 2) and fibronectin (Fn1) were repressed 2.4-, 2.6-, and 2.2-fold, respectively (Figure 7B).
DISCUSSION
TCDD and related compounds have been linked to the development of metabolic disorders. In rodents, TCDD induces dose-dependent increases in hepatic steatosis, and steatohepatitis with underlying AhR-mediated differential gene expression driving metabolic reprogramming (Nault et al., 2015a, 2016; Pierre et al., 2014). In this study, we examined the metabolic changes associated with TCDD-elicited fibrogenesis. Hepatic fibrosis is an important risk factor in hepatocellular carcinoma and other liver disease-related deaths (Baffy et al., 2012). Our studies revealed further AhR-mediated metabolic reprogramming that affected glycogen and proline metabolism, as well as the expression of matrisomal genes associated with TCDD-elicited hepatotoxicity and fibrosis.
The cumulative burden of TCDD on hepatic lipid accumulation and differential gene expression change was more evident at lower doses following 92 days of treatment compared with 28 days despite comparable steady state hepatic levels of TCDD, revealing the importance of duration of exposure in addition to levels. In humans TCDD has a longer half-life which, in addition to lifetime exposure, sustains AhR activation and may be a potential link between metabolic disease and persistent environmental contaminant exposure (Taylor et al., 2013). However, as exposure duration increased glucose tolerance improved, contrary to expectations (Taylor et al., 2013). No changes in insulin levels were observed, consistent with a previous report (Takuma et al., 2015), suggesting insulin signaling is not responsible for improved tolerance. Both adiponectin (ADIPOQ) and FGF21 regulate insulin sensitivity and glucose uptake, with increased ADIPOQ enhancing FGF21-mediated insulin sensitivity (Lin et al., 2013). Although ADIPOQ was reduced by ∼50% in treated mice, suggesting that improved glucose tolerance could be due to gluconeogenesis inhibition, it has also been reported that AhR-mediated induction of FGF21 could also plays a critical role in increasing insulin sensitivity and preventing development of type II diabetes (Lu et al., 2015).
Integrating 28 and 92 days transcriptomic datasets with published hepatic and serum metabolomic data (Nault et al., 2016), as well as complementary urinary metabolomic data, identified changes in glycogen, ascorbic acid, and proline metabolism, in addition to the reprogramming of glycolysis, gluconeogenesis and the TCA cycle. In murine hepatocytes, glycogen metabolism and ascorbic acid biosynthesis are associated with anti-oxidant responses as well as proline hydroxylation in ECM remodeling (Braun et al., 1996). Notably, glycogenolysis is required for ascorbic acid synthesis in mouse hepatocytes (Braun et al., 1996). Moreover, xenobiotics including AhR ligands, increase urinary excretion of ascorbic acid and induce UDP glucuronosyltransferase (UGTs) that participates in glycogenolysis (Hassan et al., 1987). Coincidentally, UGTs are also members of the AhR “gene battery” including Ugt1a6 suggesting a direct regulatory role of the AhR in glycogenolysis (Yeager et al., 2009). Moreover, glutathione and oxidative stress regulate ascorbate synthesis (Braun et al., 1996). These data suggest that the hepatic metabolism is reprogrammed to use glycogen in support of ECM remodeling and antioxidant responses. Nevertheless these, and other ROS defense mechanisms induced by AhR (He et al., 2013), are overwhelmed leading to NAFLD development.
The ECM is constantly remodeled by fine tuning the balance between the deposition of structural proteins, expression and/or activity of ECM degradation enzymes, and interactions with other matrisome associated proteins (Naba et al., 2016). TCDD affected the expression of >30% of the matrisome, resulting in the deposition of ECM along hepatic portal tracts. This pattern is similar to the fibrosis seen in chronic viral hepatitis and cholestatic disorders, suggesting deposition is largely mediated by myofibroblasts resulting from the EMT of portal fibroblasts and migrating HSCs (Brenner et al., 2012). Portal fibroblasts, cholangiocytes, and IL-6 are also interact to promote bile duct proliferation in biliary fibrosis (Dranoff and Wells, 2010) which may explain the bile duct proliferation in some TCDD treated mice. Furthermore, the forces exerted by ECM deposition along portal ducts may cause the dimpled appearance of the visceral surface of TCDD-treated livers. Despite conflicting epithelial and mesenchymal marker expression, it is clear that HSCs and portal fibroblasts undergo EMT to generate ECM producing myofibroblasts (Kalluri and Weinberg, 2009) consistent with previous reports of HSC activation by TCDD (Harvey et al., 2016; Pierre et al., 2014). However, OVol15402 induction suggests possible EMT reversal to replace dead and damaged cells (Syn et al., 2009). Reductions in adiponectin were contrary to improved glucose tolerance, but consistent with increased fibrosis because adiponectin inhibits myofibroblast proliferation and mobility (Ramezani-Moghadam et al., 2015). TIMP-1 which was induced by TCDD cooperates with adiponectin to reduce fibrogenic activity (Ramezani-Moghadam et al., 2015), although other studies suggest profibrogenic interactions (Yoshiji et al., 2000). IFN-γ blocks fibrogenesis by inhibiting TGFβ/Smad signaling (Weng et al., 2007). Additionally, AhR-mediated changes in the gut microbiome (Zhang et al., 2015) and intestinal permeability may increase endotoxin levels resulting in hepatic injury via activation of TLR4, promoting collagen deposition (Brenner et al., 2015). Collectively, the differential expression of the matrisome suggests TCDD elicits qualitative and quantitative changes in ECM composition and structure that could influence cell function, organization, migration, differentiation, proliferation, apoptosis, and adhesion.
In addition to providing tensile strength, regulating cell adhesion, supporting chemotaxis and migration, and directing tissue development, collagen can serve as reservoir for proline/hydroxyproline, an important anaplerotic substrate for the TCA cycle and ATP source from proline cycling at the expense of NADPH (Phang et al., 2015). Consequently, collagen can be considered as proline storage, similar to glycogen serving as glucose storage or triglycerides as storage for fatty acids (Phang et al., 2015). Continuous ECM remodeling by TCDD-induced metalloproteinases (eg, MMPs, Adams, and Adamts) can mobilize collagen stored proline/hydroxyproline, making it available for the ATP production in response to AhR-mediated PKM isoform switching (Nault et al., 2016). Excess hydroxyproline can also be diverted towards glycolate, oxalate, and glycine synthesis (Jiang et al., 2012) although this pathway is repressed by TCDD with the fate of released hydroxyproline being unclear. In addition, proline can also be converted to arginine and subsequently used for creatinine, urea, and polyamine synthesis (Morris, 2009). Limited proline availability is likely associated with reduced polyamine production (Thomas et al., 1990), but little is known about effects on the urea cycle or proline-glutamine-arginine interconversion. The relevance of proline cycling on ATP production and overall cellular bioenergetics in combating TCDD-elicited energy depletion and hepatotoxicity remains to be determined.
In summary, TCDD-elicited metabolic reprogramming extends to ascorbic acid synthesis, glycogen metabolism, and ECM remodeling/proline cycling (Supplementary Figure 7). As TCDD exposure duration increased, the doses necessary for NAFLD pathology progression decreased suggesting a putative role for anthropogenic, dietary, and endogenous AhR ligands in NAFLD, particularly for compounds with long half-lives. However, ascorbic acid synthesis is not conserved and therefore the relevance of these results in human disease remains to be determined. Nevertheless, TCDD-elicited fibrosis involves complex interactions between diverse cell types and signaling pathways involving metabolic reprogramming to support fibrogenesis. The energetic costs of metabolic reprogramming and mechanism for sustaining ATP levels in response to TCDD exposure warrants further investigation, particularly the use of collagen as a proline source and the consumption of glycogen for ascorbic acid synthesis. Further elucidation of the roles of these pathways in pathology progression may identify novel targets for the treatment of fibrotic diseases.
SUPPLEMENTARY DATA
Supplementary data are available online at http://toxsci.oxfordjournals.org/.
ACKNOWLEDGMENTS
The authors would like to thank Drs Agnes Forgacs and Anna Kopec for their assistance in the animal experiments.
FUNDING
This work was supported by the National Institute of Environmental Health Sciences Superfund Research Program (NIEHS SRP P42ES04911). T.R.Z. is supported by AgBioResearch at MSU. R.N. is supported by the MSU Barnett Rosenberg Endowed Assistantship and Integrative Training in the Pharmacological Sciences grant (NIH 5T32GM092715). S.Y.L. is supported by the 2016 AACR-Incyte Corporation NextGen Grant for Transformative Cancer Research, Grant Number 16-20-46-LUNT, and the Office of the Assistant Secretary of Defense for Health Affairs, through the Breast Cancer Research Program, under Award No. W81XWH-15-1-0453. Opinions, interpretations, conclusions, and recommendations are those of the author and are not necessarily endorsed by the Department of Defense. J.M. is supported by the Throne Holst Foundation and the University of Oslo. K.A.F. is supported by the Canadian Institutes of Health Research (CIHR) Doctoral Foreign Study Award (DFS-140386). P.D. is supported by the MSU National Institute of Environmental Health Sciences Training Grant (T32 ES007255).
REFERENCES
- Ahmed S., Bott D., Gomez A., Tamblyn L., Rasheed A., Cho T., MacPherson L., Sugamori K. S., Yang Y., Grant D. M., et al. (2015). Loss of the mono-ADP-ribosyltransferase, Tiparp, increases sensitivity to dioxin-induced steatohepatitis and lethality. J. Biol. Chem. 290, 16824–16840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baffy G., Brunt E. M., Caldwell S. H. (2012). Hepatocellular carcinoma in non-alcoholic fatty liver disease: An emerging menace. J. Hepatol. 56, 1384–1391. [DOI] [PubMed] [Google Scholar]
- Barbul A. (2008). Proline precursors to sustain Mammalian collagen synthesis. J. Nutr. 138, 2021S–2024S. [DOI] [PubMed] [Google Scholar]
- Braun L., Csala M., Poussu A., Garzo T., Mandl J., Banhegyi G. (1996). Glutathione depletion induces glycogenolysis dependent ascorbate synthesis in isolated murine hepatocytes. FEBS Lett. 388, 173–176. [DOI] [PubMed] [Google Scholar]
- Brenner D. A., Kisseleva T., Scholten D., Paik Y. H., Iwaisako K., Inokuchi S., Schnabl B., Seki E., De Minicis S., Oesterreicher C., et al. (2012). Origin of myofibroblasts in liver fibrosis. Fibrogenesis Tissue Repair 5(Suppl 1), S17.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner D. A., Paik Y. H., Schnabl B. (2015). Role of gut microbiota in liver disease. J. Clinical Gastroenterol. 49(Suppl 1), S25–S27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Choi S. S., Michelotti G. A., Chan I. S., Swiderska-Syn M., Karaca G. F., Xie G., Moylan C. A., Garibaldi F., Premont R., et al. (2012). Hedgehog controls hepatic stellate cell fate by regulating metabolism. Gastroenterology 143, 1319–1329.e1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X., Vispute S. G., Liu J., Cheng C., Kharitonenkov A., Klaassen C. D. (2014). Fibroblast growth factor (Fgf) 21 is a novel target gene of the aryl hydrocarbon receptor (AhR). Toxicol. Appl. Pharmacol. 278(1), 65–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clasquin M. F., Melamud E., Rabinowitz J. D. (2012). LC-MS data processing with MAVEN: A metabolomic analysis and visualization engine. Curr. Protoc. Bioinformatics. Chapter 14, Unit14 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Tullio M. C. (2012). Beyond the antioxidant: The double life of vitamin C. Subcell. Biochem. 56, 49–65. [DOI] [PubMed] [Google Scholar]
- Dere E., Lo R., Celius T., Matthews J., Zacharewski T. R. (2011). Integration of genome-wide computation DRE search, AhR ChIP-chip and gene expression analyses of TCDD-elicited responses in the mouse liver. BMC Genomics 12, 365.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dranoff J. A., Wells R. G. (2010). Portal fibroblasts: Underappreciated mediators of biliary fibrosis. Hepatology 51, 1438–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fader K. A., Nault R., Ammendolia D. A., Harkema J. R., Williams K. J., Crawford R. B., Kaminski N. E., Potter D., Sharratt B., Zacharewski T. R. (2015). 2,3,7,8-Tetrachlorodibenzo-p-dioxin alters lipid metabolism and depletes immune cell populations in the Jejunum of C57BL/6 mice. Toxicol. Sci. 148, 567–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Salguero P. M., Hilbert D. M., Rudikoff S., Ward J. M., Gonzalez F. J. (1996). Aryl-hydrocarbon receptor-deficient mice are resistant to 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced toxicity. Toxicol. Appl. Pharmacol. 140, 173–179. [DOI] [PubMed] [Google Scholar]
- Frantz C., Stewart K. M., Weaver V. M. (2010). The extracellular matrix at a glance. J. Cell Sci. 123, 4195–4200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girer N. G., Murray I. A., Omiecinski C. J., Perdew G. H. (2016). Hepatic aryl hydrocarbon receptor attenuates fibroblast growth factor 21 expression. J. Biol. Chem. 291(29), 15378–15387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser S. S., Gaudio E., Miller T., Alvaro D., Alpini G. (2009). Cholangiocyte proliferation and liver fibrosis. Expert Rev. Mol. Med. 11, e7.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey W. A., Jurgensen K., Pu X., Lamb C. L., Cornell K. A., Clark R. J., Klocke C., Mitchell K. A. (2016). Exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) increases human hepatic stellate cell activation. Toxicology 344–346, 26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan M. Q., Mohammadpour H., Hermansky S. J., Murray W. J., Stohs S. J. (1987). Comparative effects of BHA and ascorbic acid on the toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in rats. Gen. Pharmacol. 18, 547–550. [DOI] [PubMed] [Google Scholar]
- He J., Hu B., Shi X., Weidert E. R., Lu P., Xu M., Huang M., Kelley E. E., Xie W. (2013). Activation of the aryl hydrocarbon receptor sensitizes mice to nonalcoholic steatohepatitis by deactivating mitochondrial sirtuin deacetylase Sirt3. Mol. Cell. Biol. 33, 2047–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang G., Elferink C. J. (2012). A novel nonconsensus xenobiotic response element capable of mediating aryl hydrocarbon receptor-dependent gene expression. Mol. Pharmacol. 81, 338–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J., Johnson L. C., Knight J., Callahan M. F., Riedel T. J., Holmes R. P., Lowther W. T. (2012). Metabolism of [13C5]hydroxyproline in vitro and in vivo: Implications for primary hyperoxaluria. Am. J. Physiol. Gastrointest. Liver pPhysiol. 302, G637–G643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R., Weinberg R. A. (2009). The basics of epithelial-mesenchymal transition. J. Clin. Invest. 119, 1420–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama A., Nakatsuka A., Eguchi J., Murakami K., Teshigawara S., Kanzaki M., Nunoue T., Hida K., Wada N., Yasunaka T., et al. (2015). Beneficial impact of Gpnmb and its significance as a biomarker in nonalcoholic steatohepatitis. Sci. Rep. 5, 16920.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keppler D., Decker K. (1974). Glycogen determination with amyloglucosidase In Methods of enzymatic analysis, pp. 1127–1131. Academic Press, New York. [Google Scholar]
- Kopec A. K., Boverhof D. R., Nault R., Harkema J. R., Tashiro C., Potter D., Sharratt B., Chittim B., Zacharewski T. R. (2013). Toxicogenomic evaluation of long-term hepatic effects of TCDD in immature, ovariectomized C57BL/6 mice. Toxicol. Sci. 135, 465–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamouille S., Xu J., Derynck R. (2014). Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 15, 178–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. H., Wada T., Febbraio M., He J., Matsubara T., Lee M. J., Gonzalez F. J., Xie W. (2010). A novel role for the dioxin receptor in fatty acid metabolism and hepatic steatosis. Gastroenterology 139, 653–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Z., Tian H., Lam K. S., Lin S., Hoo R. C., Konishi M., Itoh N., Wang Y., Bornstein S. R., Xu A., et al. (2013). Adiponectin mediates the metabolic effects of FGF21 on glucose homeostasis and insulin sensitivity in mice. Cell Metabolism 17, 779–789. [DOI] [PubMed] [Google Scholar]
- Loskutoff D. J., Quigley J. P. (2000). PAI-1, fibrosis, and the elusive provisional fibrin matrix. J. Clin. Invest. 106, 1441–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu P., Yan J., Liu K., Garbacz W. G., Wang P., Xu M., Ma X., Xie W. (2015). Activation of aryl hydrocarbon receptor dissociates fatty liver from insulin resistance by inducing fibroblast growth factor 21. Hepatology 61, 1908–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunt S. Y., Muralidhar V., Hosios A. M., Israelsen W. J., Gui D. Y., Newhouse L., Ogrodzinski M., Hecht V., Xu K., Acevedo P. N., et al. (2015). Pyruvate kinase isoform expression alters nucleotide synthesis to impact cell proliferation. Mol. Cell. 57, 95–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris S. M., Jr. (2009). Recent advances in arginine metabolism: Roles and regulation of the arginases. Br. J. Pharmacol. 157, 922–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naba A., Clauser K. R., Ding H., Whittaker C. A., Carr S. A., Hynes R. O. (2016). The extracellular matrix: Tools and insights for the “omics” era. Matrix Biol. 49, 10–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nault R., Colbry D., Brandenberger C., Harkema J. R., Zacharewski T. R. (2015a). Development of a computational high-throughput tool for the quantitative examination of dose-dependent histological features. Toxicol. Pathol. 43, 366–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nault R., Fader K. A., Kirby M. P., Ahmed S., Matthews J., Jones A. D., Lunt S. Y., Zacharewski T. R. (2016). Pyruvate kinase isoform switching and hepatic metabolic reprogramming by the environmental contaminant 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol. Sci. 149, 358–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nault R., Fader K. A., Zacharewski T. (2015b). RNA-Seq versus oligonucleotide array assessment of dose-dependent TCDD-elicited hepatic gene expression in mice. BMC Genomics 16, 373.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omenetti A., Syn W. K., Jung Y., Francis H., Porrello A., Witek R. P., Choi S. S., Yang L., Mayo M. J., Gershwin M. E., et al. (2009). Repair-related activation of hedgehog signaling promotes cholangiocyte chemokine production. Hepatology 50, 518–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phang J. M., Liu W., Hancock C. N., Fischer J. W. (2015). Proline metabolism and cancer: Emerging links to glutamine and collagen. Curr. Opin. Clin. Nutr. Metab. Care 18, 71–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierre S., Chevallier A., Teixeira-Clerc F., Ambolet-Camoit A., Bui L. C., Bats A. S., Fournet J. C., Fernandez-Salguero P., Aggerbeck M., Lotersztajn S., et al. (2014). Aryl hydrocarbon receptor-dependent induction of liver fibrosis by dioxin. Toxicol. Sci. 137, 114–124. [DOI] [PubMed] [Google Scholar]
- Ramezani-Moghadam M., Wang J., Ho V., Iseli T. J., Alzahrani B., Xu A., Van der Poorten D., Qiao L., George J., Hebbard L. (2015). Adiponectin reduces hepatic stellate cell migration by promoting tissue inhibitor of metalloproteinase-1 (TIMP-1) secretion. J. Biol. Chem. 290, 5533–5542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts A. B., Kim S. J., Noma T., Glick A. B., Lafyatis R., Lechleider R., Jakowlew S. B., Geiser A., O'Reilly M. A., Danielpour D. and., et al. (1991). Multiple forms of TGF-beta: Distinct promoters and differential expression. Ciba Found. Symp. 157, 7–15; discussion 15–28. [DOI] [PubMed] [Google Scholar]
- Roca H., Hernandez J., Weidner S., McEachin R. C., Fuller D., Sud S., Schumann T., Wilkinson J. E., Zaslavsky A., Li H., et al. (2013). Transcription factors OVol15402 and OVol15402 induce the mesenchymal to epithelial transition in human cancer. PLoS One 8, e76773.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syn W. K., Jung Y., Omenetti A., Abdelmalek M., Guy C. D., Yang L., Wang J., Witek R. P., Fearing C. M., Pereira T. A., et al. (2009). Hedgehog-mediated epithelial-to-mesenchymal transition and fibrogenic repair in nonalcoholic fatty liver disease. Gastroenterology 137, 1478–1488.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takuma M., Ushijima K., Kumazaki M., Ando H., Fujimura A. (2015). Influence of dioxin on the daily variation of insulin sensitivity in mice. Environ. Toxicol. Pharmacol. 40, 349–351. [DOI] [PubMed] [Google Scholar]
- Taylor K. W., Novak R. F., Anderson H. A., Birnbaum L. S., Blystone C., Devito M., Jacobs D., Kohrle J., Lee D. H., Rylander L., et al. (2013). Evaluation of the association between persistent organic pollutants (POPs) and diabetes in epidemiological studies: A national toxicology program workshop review. Environ. Health Perspect. 121, 774–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas T., MacKenzie S. A., Gallo M. A. (1990). Regulation of polyamine biosynthesis by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Toxicol. Lett. 53, 315–325. [DOI] [PubMed] [Google Scholar]
- Van Gool B., Dedieu S., Emonard H., Roebroek A. J. (2015). The matricellular receptor LRP1 forms an interface for signaling and endocytosis in modulation of the extracellular tumor environment. Front. Pharmacol. 6, 271.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng H., Mertens P. R., Gressner A. M., Dooley S. (2007). IFN-gamma abrogates profibrogenic TGF-beta signaling in liver by targeting expression of inhibitory and receptor Smads. J. Hepatol. 46, 295–303. [DOI] [PubMed] [Google Scholar]
- Wight T. N., Potter-Perigo S. (2011). The extracellular matrix: An active or passive player in fibrosis? Am. J. Physiol. Gastrointest. Liver Physiol. 301, G950–G955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J., Wishart D. S. (2010). MetPA: A web-based metabolomics tool for pathway analysis and visualization. Bioinformatics 26, 2342–2344. [DOI] [PubMed] [Google Scholar]
- Yao L., Wang C., Zhang X., Peng L., Liu W., Liu Y., He J., Jiang C., Ai D., Zhu Y. (2016). Hyperhomocysteinemia activates the aryl hydrocarbon receptor/CD36 pathway to promote hepatic steatosis in mice. Hepatology 64, 92–105. [DOI] [PubMed] [Google Scholar]
- Yeager R. L., Reisman S. A., Aleksunes L. M., Klaassen C. D. (2009). Introducing the “TCDD-inducible AhR-Nrf2 gene battery”. Toxicol. Sci. 111, 238–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshiji H., Kuriyama S., Miyamoto Y., Thorgeirsson U. P., Gomez D. E., Kawata M., Yoshii J., Ikenaka Y., Noguchi R., Tsujinoue H., et al. (2000). Tissue inhibitor of metalloproteinases-1 promotes liver fibrosis development in a transgenic mouse model. Hepatology 32, 1248–1254. [DOI] [PubMed] [Google Scholar]
- Zhang L., Nichols R. G., Correll J., Murray I. A., Tanaka N., Smith P. B., Hubbard T. D., Sebastian A., Albert I., Hatzakis E., et al. (2015). Persistent organic pollutants modify gut microbiota-host metabolic homeostasis in mice through aryl hydrocarbon receptor activation. Environ. Health Perspect. 123, 679–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.