

Abstract
Idiopathic multifocal choroiditis (MFC) and/or punctate inner choroidopathy (PIC) describe a chronic progressive bilateral inflammatory chorioretinopathy that predominantly affect healthy myopic white women with no known associated systemic or ocular diseases. The principal sites of involvement are the retinal pigment epithelium (RPE) and outer retinal spaces; the choroid is not affected during the active phase of the disease. Idiopathic MFC with atrophy is a recently described variant. Although there is no generally accepted standard treatment, anti-inflammatory and anti-VEGF (vascular endothelial growth factor) agents are necessary in the acute stage to control the inflammation and choroidal neovascularization (CNV).
Keywords: Idiopathic Multifocal Choroiditis, Panuveitis, Punctate Inner Choroidopathy
INTRODUCTION
Alex Krill and co-authors in 1969,[1,2] were the first to use “multifocal choroiditis” (MFC) to describe a non-infectious, idiopathic ocular disorder resembling the “presumed ocular histoplasmosis syndrome” (POHS).[3] Nozik and Dorsch in 1973[4] described a similar chorioretinopathy associated with anterior uveitis in two patients with fundus lesions similar to POHS, but with the presence of vitreous and anterior chamber inflammation, but no evidence of infectious disease.[4] These patients, were categorized as MFC with panuveitis (MFCPU).
Dreyer and Gass in 1984[5] were the first to report MFCPU in a larger series of 28 cases confirming it as a distinct clinical entity from POHS. In the same year, Watzke et al[6] described ten myopic women with MFC but with smaller chorioretinal lesions and called these cases “punctate inner choroidopathy” (PIC). These authors believed that PIC is a distinctive entity in which small yellow-gray dots in the posterior pole with no signs of ocular inflammation develop into atrophic chorioretinal scars and become progressively more pigmented with time.[6] Joondeph and Tessler in 1990[7] were the first to use the terms MFC and PIC as inflammatory disorders distinct from POHS, which predominantly affect young myopic females.[3] They described MFC as a chronic bilateral disease that was variably associated with vitritis, and often with anterior chamber inflammation which may also show progressive subretinal fibrosis and macular choroidal neovascularization.[3] Since then, there has been no evidence to support MFC and PIC as separate entities: Their clinical course and treatments are similar, and both conditions are idiopathic.[3]
CLASSIFICATION
Causes of MFC lesions can be classified into infectious (POHS, tuberculosis, brucellosis, coccidiomycosis, candidiasis and other fungal septicemias, syphilis, West Nile virus, etc.), noninfectious [sarcoidosis and other granulomatous diseases (e.g., Blau syndrome)], and idiopathic [without uveitis, or with uveitis (MFCPU)].[3,8]
Demographics, Signs and Symptoms
Idiopathic MFC generally describes a relatively uncommon, chronic, inflammatory chorioretinopathy that predominantly affects young (median age of 30 years)[9] healthy myopic white women with no known associated systemic disease or other recognized ocular syndromes, and a high risk of secondary CNV, circumscribed areas of chorioretinal atrophy and subretinal pigmented fibrotic scars. Although idiopathic MFC may represent an ocular manifestation of an autoimmune disease in genetically susceptible patients, the precise etiopathogenesis still remains unclear.[10,11,12,13]
Clinically, patients may complain of a temporal scotoma, metamorphopsia, floaters, photopsias, photophobia, and decreased vision. Visual acuity is often good at presentation.[9] Affected eyes typically show multiple punched-out chorioretinal lesions ranging from 50 to 350 μm in size, both the posterior pole and periphery, often with clustering of lesions nasal to the disc, with minimal or no anterior uveitis or vitritis and no signs of progressive or diffuse subretinal fibrosis.[8] Other findings include peripapillary atrophy, scarring, and curvilinear chorioretinal streaks (Schlaegel lines) in the far periphery [Figure 1].[13,14,15] Patients have negative or nonreactive results on relevant diagnostic modalities; such as histoplasmin skin reaction, syphilis (non-treponemal and treponemal) serology, tests for tuberculosis (tuberculin skin test or interferon-γ release assays and chest radiographs), tests for sarcoidosis (serum angiotensin-converting enzyme and chest radiograph or computed tomography scan) and neoplastic/non-neoplastic anti-retinal antibodies.
Figure 1.
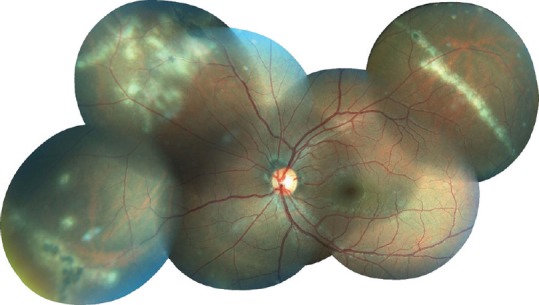
Color fundus photograph of a 21-year-old man, presenting with blurred vision and floaters in both eyes over several months. Note multiple, diffuse and discrete nummular outer retinal/choroidal white spots and equatorial linear/curvilinear streak lesions (Schlaegel lines) in the left eye.
Primary vitreoretinal lymphoma (also known as primary central nervous system lymphoma with ocular involvement), causes subretinal pigment epithelium (sub-RPE) nodular elevations about 100 μm in diameter which demostrate autofluorescence and fluorescein angiographic findings similar to those seen in MFC.[16] Usually idiopathic MFC can be distinguished from masqueraders based on clinical manifestations, fundus imaging, and clinical course.[13]
Peripheral MFC is a non-infectious type of the disease and strongly associated with sarcoidosis, characterized by a rather poor visual outcome, macular edema and relatively high prevalence of complications.[17]
MFCPU is a variant of idiopathic MFC that has been described in association with anterior uveitis and/or vitritis, multiple punched-out atrophic chorioretinal lesions of variable size (typically 500–1000 μm in size) in the posterior pole and midperiphery of the retina, older age and more visual impairment at presentation. This type of the disease is more frequently associated with a non-remitting clinical course and higher frequency of structural complications such as cataract, cystoid macular edema (CME), and epiretinal membrane (ERM) related to intraocular inflammation. MFCPU has a poor visual prognosis for both eyes, and normally requires higher levels of maintenance immunosuppression.[12,18]
Haplotyping of elderly patients with MFCPU has shown significantly reduced frequency of HLA B-7, HLA- DR1, and HLA- DR15.[19]
Many patients exhibit idiopathic MFC lesions without inflammation. Some authors have used the term PIC to designate the non-inflammatory aspect of the disease,[3,6] but, most authors believe that idiopathic MFC and PIC represent the same underlying disease entity[3,20] and both are associated with identical interleukin-10 and tumor necrosis factor (TNF) haplotypes.[21]
NATURAL COURSE
Idiopathic MFC is a chronic progressive bilateral disease and one of the most common causes of visual loss in this entity is related to development of CNV.[12,18] The rate of increase in the size and number of lesions and development of new or recurrent CME and/or CNV is also high despite the relative absence of anterior uveitis or vitritis. This observation reflects the need for long term follow-up and lends further support to the proposition that inflammation in idiopathic MFC can be confined to the outer retina/RPE. Despite these and other complications, visual acuity can be maintained over many years and the visual prognosis is good in most cases.[8]
According to Spaide et al[20] the principal sites involved appear to be the sub-RPE and outer retinal spaces. Acute lesions are often associated with more widespread involvement of the ellipsoid zone (EZ) extending beyond the zones of RPE involvement, as discontinuity or elevation due to deposition of material, which may be homogenous or heterogenous. Active lesions show a consistent appearance of RPE elevation due to deposition of homogenous material of medium reflectivity on SD-OCT examination. The RPE elevations are usually conical although larger ones are broader in the lateral dimension but similar in height. Some of the solid RPE detachments appear to rupture through the RPE resulting in an outpouring of infiltrates into the outer retina. With separation or attenuation of the RPE line, there is increased light penetration into the choroid and deeper structures. EDI-OCT imaging of the choroid beneath the active lesions show a small increase in choroidal thickness as compared with the surrounding choroid in some eyes. Other than these rather subtle findings, there do not appear to be any other choroidal perturbations. By autofluorescent imaging, RPE elevations are round areas of minimal hyper-autofluorescence. However, if there is dehiscence of the RPE, a corresponding spot of absent autofluorescence is observed. The infiltrative changes in the outer retina extend both above and laterally from the apex of the ruptured RPE elevation.[20]
NEW VARIANT OF MULTIFOCAL CHOROIDITIS
In some patients as the inflammatory episodes continue to recur, additional heterogeneous zonal, multizonal or diffuse outer retinal atrophy may appear similar to the development of chorioretinal scars, and some may even progress to end-stage patterns of chorioretinal atrophy (a new variant).[13]
This variant of “idiopathic MFC with atrophy”, involving the posterior pole and peripheral fundus, may demonstrate chorioretinal lesions and a spectrum of zonal, multizonal or diffuse outer retinal, RPE, and/or chorioretinal atrophy. There may be foveal sparing until late into the disease course in the involved eyes. This phenomenon is distinct from focal lesions seen in eyes with typical MFC. Within the chorioretinal atrophic zones, spectral domain optical coherence tomography (SD-OCT) and fundus autofluorescence (FAF) demonstrate geographic areas of both photoreceptor and RPE disruption. More advanced zones show corresponding atrophy of the inner choroid. Inflammation may initially affect only the outer parts of photoreceptors and cause an associated visual field scotoma. As bouts of inflammation recur, continued progression to multizonal or diffuse disease may occur. Eventually, involvement of the RPE and choroid leads to end-stage disease, which causes severe chorioretinal atrophy.[13] Foveal sparing is common until late in the disease course in these patients, and the majority of patients maintain good visual acuity. Munk et al,[22] however, reported eight patients with idiopathic MFC with atrophy associated with adjacent photoreceptor attenuation, an acute/subacute onset of decreased visual acuity and scotomata with vision loss as severe as counting fingers/hand motion. Visual acuity fully recovered in only three of the eight cases after treatment with corticosteroids and/or systemic mycophenolate mofetil (MMF) but no other agents such as cyclosporine, methotrexate, azathioprine, infliximab, or tacrolimus were used.[22]
This variant of idiopathic MFC with atrophy, unlike classic acute zonal occult outer retinopathy, does not demonstrate the pathognomonic orange to-gray demarcating line on ophthalmoscopic examination and the typical trizonal degeneration, visualized on FAF, indocyanine green angiography (ICG), and OCT images.[23]
Interestingly, genetic factors including inflammatory complement factor H have a strong association with MFC and age-related macular degeneration.[24] Having an underlying genetic susceptibility may predispose a single patient to more than one inflammatory disease. The occurrence of MFC in siblings also raises the possibility of a familial association in the pathogenesis of the disease.[25]
TREATMENT
Currently, when there is no evidence of CNV, there is no universally accepted standard treatment for the majority of patients with idiopathic MFC as the visual prognosis is excellent. The only exception to this would be those patients with inflammatory lesions very close to fovea in whom medical treatment should be considered.[9] Systemic corticosteroid therapy is typically the first-line treatment,[26] and in conjunction with immunomodulatory therapy, has shown to reduce the amount of inflammatory infiltration of the subretinal space and outer retina, and reduce damage seen on multimodal imaging.
Some authors[27] believe that single-agent immunosuppression is able to achieve treatment success in the majority of patients, but dose escalation of mycophenolate mofetil (MMF) is typical, and a second immunosuppressive agent is needed in about one-fifth of the patients.[27]
From the authors’ point of view, both anti-inflammatory and anti-VEGF therapy (in the presence of CNV) are necessary in the acute stage to control the inflammation and CNV. However, when neovascularization has subsided there is no standard method of management based on clinical trials for long-term immunosuppression therapy of the quiescent stage in young female patients of child-bearing age.
Financial Support and Sponsorship
Nil.
Conflicts of Interest
There are no conflicts of interest.
REFERENCES
- 1.Krill AE, Chishti MI, Klien BA, Newell FW, Potts AM. Multifocal inner choroiditis. Trans Am Acad Ophthalmol Otolaryngol. 1969;73:222–245. [PubMed] [Google Scholar]
- 2.Krill AE, Archer D. Choroidal neovascularization in multifocal (presumed histoplasmin) choroiditis. Arch Ophthalmol. 1970;84:595–604. doi: 10.1001/archopht.1970.00990040597007. [DOI] [PubMed] [Google Scholar]
- 3.Essex RW, Wong J, Jampol LM, Dowler J, Bird AC. Idiopathic multifocal choroiditis: A comment on present and past nomenclature. Retina. 2013;33:1–4. doi: 10.1097/IAE.0b013e3182641860. [DOI] [PubMed] [Google Scholar]
- 4.Nozik RZ, Dorsch W. A new chorioretinopathy associated with anterior uveitis. Am J Ophthalmol. 1973;76:758–762. doi: 10.1016/0002-9394(73)90573-4. [DOI] [PubMed] [Google Scholar]
- 5.Dreyer RF, Gass JD. Multifocal choroiditis and panuveitis. A syndrome that mimics ocular histoplasmosis. Arch Ophthalmol. 1984;102:1776–1784. doi: 10.1001/archopht.1984.01040031440019. [DOI] [PubMed] [Google Scholar]
- 6.Watzke RC, Packer AJ, Folk JC, Benson WE, Burgess D, Ober RR. Punctate inner choroidopathy. Am J Ophthalmol. 1984;98:572–584. doi: 10.1016/0002-9394(84)90243-5. [DOI] [PubMed] [Google Scholar]
- 7.Joondeph B, Tessler H. Multifocal choroiditis. Int Ophthalmol Clin. 1990;30:286–290. doi: 10.1097/00004397-199030040-00015. [DOI] [PubMed] [Google Scholar]
- 8.Fung AT, Pal S, Yannuzzi NA, Christos P, Cooney M, Slakter JS, et al. Multifocal choroiditis without panuveitis: Clinical characteristics and progression. Retina. 2014;34:98–107. doi: 10.1097/IAE.0b013e31829234cb. [DOI] [PubMed] [Google Scholar]
- 9.Amer R, Lois N. Punctate inner choroidopathy. Surv Ophthalmol. 2011;56:36–53. doi: 10.1016/j.survophthal.2010.03.009. [DOI] [PubMed] [Google Scholar]
- 10.Jampol LM, Becker KG. White spot syndromes of the retina: A hypothesis based on the common genetic hypothesis of autoimmune/inflammatory disease. Am J Ophthalmol. 2003;135:376–379. doi: 10.1016/s0002-9394(02)02088-3. [DOI] [PubMed] [Google Scholar]
- 11.Fine HF, Zhitomirsky I, Freund KB, Barile GR, Shirkey BL, Samson CM, et al. Bevacizumab (avastin) and ranibizumab (lucentis) for choroidal neovascularization in multifocal choroiditis. Retina. 2009;29:8–12. doi: 10.1097/IAE.0b013e318187aff9. [DOI] [PubMed] [Google Scholar]
- 12.Thorne JE, Wittenberg S, Jabs DA, Peters GB, Reed TL, Kedhar SR, et al. Multifocal choroiditis with panuveitis: Incidence of ocular complications and of loss of visual acuity. Ophthalmology. 2006;113:2310–2316. doi: 10.1016/j.ophtha.2006.05.067. [DOI] [PubMed] [Google Scholar]
- 13.Jung JJ, Khan S, Mrejen S, Gallego-Pinazo R, Cunningham ET, Freund KB, et al. Idiopathic multifocal choroiditis with outer retinal or chorioretinal atrophy. Retina. 2014;34:1439–1450. doi: 10.1097/IAE.0000000000000079. [DOI] [PubMed] [Google Scholar]
- 14.Matsumoto Y, Francis JH, Yannuzzi LA. Curvilinear streaks in multifocal choroiditis. Eur J Ophthalmol. 2007;17:448–450. doi: 10.1177/112067210701700332. [DOI] [PubMed] [Google Scholar]
- 15.Somkijrungroj T, Pearlman JA, Gonzales JA. Multifocal choroiditis and panuveitis presenting with progressive equatorial linear streaks: Evolution of schlaegel lines documented with multimodal imaging. Retinal Cases Brief Rep. 2015;9:214–217. doi: 10.1097/ICB.0000000000000141. [DOI] [PubMed] [Google Scholar]
- 16.Chan CC, Rubenstein JL, Coupland SE, Davis JL, Harbour JW, Johnston PB, et al. Primary vitreoretinal lymphoma: A report from an International Primary Central Nervous System Lymphoma Collaborative Group symposium. Oncologist. 2011;16:1589–1599. doi: 10.1634/theoncologist.2011-0210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koop A, Ossewaarde A, Rothova A. Peripheral multifocal chorioretinitis. Acta Ophthalmol. 2013;91:492–497. doi: 10.1111/j.1755-3768.2012.02483.x. [DOI] [PubMed] [Google Scholar]
- 18.Kedhar SR, Thorne JE, Wittenberg S, Dunn JP, Jabs DA. Multifocal choroiditis with panuveitis and punctate inner choroidopathy: Comparison of clinical characteristics at presentation. Retina. 2007;27:1174–1179. doi: 10.1097/IAE.0b013e318068de72. [DOI] [PubMed] [Google Scholar]
- 19.Nolle B, Faul S, Jenisch S, Westphal E. Peripheral multifocal chorioretinitis with panuveitis: Clinical and immunogenetic characterization in older patients. Graefes Arch Clin Exp Ophthalmol. 1998;236:451–460. doi: 10.1007/s004170050105. [DOI] [PubMed] [Google Scholar]
- 20.Spaide RF, Goldberg N, Freund KB. Redefining multifocal choroiditis and panuveitis and punctate inner choroidopathy through multimodal imaging. Retina. 2013;33:1315–1324. doi: 10.1097/IAE.0b013e318286cc77. [DOI] [PubMed] [Google Scholar]
- 21.Atan D, Fraser-Bell S, Plskova J, Kuffová L, Hogan A, Tufail A, et al. Punctate inner choroidopathy and multifocal choroiditis with panuveitis share haplotypic associations with IL10 and TNF loci. Invest Ophthalmol Vis Sci. 2011;52:3573–3581. doi: 10.1167/iovs.10-6743. [DOI] [PubMed] [Google Scholar]
- 22.Munk MR, Jung JJ, Biggee K, Tucker WR, Sen HN, Schmidt-Erfurth U, et al. Idiopathic multifocal choroiditis/punctate inner choroidopathy with acute photoreceptor loss or dysfunction out of proportion to clinically visible lesions. Retina. 2015;35:334–343. doi: 10.1097/IAE.0000000000000370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mrejen S, Khan S, Gallego-Pinazo R, Jampol LM, Yannuzzi LA. Acute zonal occult outer retinopathy: A classification based on multimodal imaging. JAMA Ophthalmol. 2014;132:1089–1098. doi: 10.1001/jamaophthalmol.2014.1683. [DOI] [PubMed] [Google Scholar]
- 24.Ferrara DC, Merriam JE, Freund KB, Spaide RF, Takahashi BS, Zhitomirsky I, et al. Analysis of major alleles associated with age-related macular degeneration in patients with multifocal choroiditis: Strong association with complement factor H. Arch Ophthalmol. 2008;126:1562–1566. doi: 10.1001/archopht.126.11.1562. [DOI] [PubMed] [Google Scholar]
- 25.Levine JP, Freund KB, Cooney MJ, Klancnik JM, Jr, Shirkey BL, Yannuzzi LA. Multifocal choroiditis in siblings. Retinal Cases Brief Rep. 2008;2:151–153. doi: 10.1097/ICB.0b013e3180590c55. [DOI] [PubMed] [Google Scholar]
- 26.Brueggeman RM, Noffke AS, Jampol LM. Resolution of punctate inner choroidopathy lesions with oral prednisone therapy (Photo Essay) Arch Ophthalmol. 2002;120:996. doi: 10.1001/archopht.120.7.996. [DOI] [PubMed] [Google Scholar]
- 27.Goldberg NR, Lyu T, Moshier E, Godbold J, Jabs DA. Success with single-agent immunosuppression for multifocal choroidopathies. Am J Ophthalmol. 2014;158:1310–1317. doi: 10.1016/j.ajo.2014.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]