

Abstract
Flavonoids, the compounds that impart color to fruits, flowers, and seeds, are the most widespread secondary metabolites in plants. However, a systematic analysis of these loci has not been performed in Brassicaceae. In this study, we isolated 649 nucleotide sequences related to flavonoid biosynthesis, i.e., the Transparent Testa (TT) genes, and their associated amino acid sequences in 17 Brassicaceae species, grouped into Arabidopsis or Brassicaceae subgroups. Moreover, 36 copies of 21 genes of the flavonoid biosynthesis pathway were identified in Arabidopsis thaliana, 53 were identified in Brassica rapa, 50 in Brassica oleracea, and 95 in B. napus, followed the genomic distribution, collinearity analysis and genes triplication of them among Brassicaceae species. The results showed that the extensive gene loss, whole genome triplication, and diploidization that occurred after divergence from the common ancestor. Using qRT-PCR methods, we analyzed the expression of 18 flavonoid biosynthesis genes in 6 yellow- and black-seeded B. napus inbred lines with different genetic background, found that 12 of which were preferentially expressed during seed development, whereas the remaining genes were expressed in all B. napus tissues examined. Moreover, 14 of these genes showed significant differences in expression level during seed development, and all but four of these (i.e., BnTT5, BnTT7, BnTT10, and BnTTG1) had similar expression patterns among the yellow- and black-seeded B. napus. Results showed that the structural genes (BnTT3, BnTT18, and BnBAN), regulatory genes (BnTTG2 and BnTT16) and three encoding transfer proteins (BnTT12, BnTT19, and BnAHA10) might play an crucial roles in the formation of different seed coat colors in B. napus. These data will be helpful for illustrating the molecular mechanisms of flavonoid biosynthesis in Brassicaceae species.
Keywords: Brassica napus L., Brassicaceae species, expression profile, flavonoid biosynthesis pathway, phylogenetic analysis, seed coat color
Introduction
Rapeseed (Brassica napus) is the most important source of edible vegetable oil and protein-rich meal in the world diet, and the seeds, which store protein and oil, are the major harvested plant organ (Nesi et al., 2008). However, the quality of rapeseed oil and meal is greatly affected by the pigments and polyphenols derived from flavonoid biosynthesis that remain after oil extraction. Rapeseed use is limited by the concentration of anti-nutritional factors, including phenolic compounds, lignin, tannins, and proanthocyanidins, it contains. Previous research showed that yellow-seeded B. napus has a thinner seed coat, less pigmentation, and higher protein and oil contents than does black-seeded B. napus in the same background, rendering it a more nutritional feed for livestock (Chen and Heneen, 1992; Tang et al., 1997; Meng et al., 1998). Thus, selecting lines with a stable yellow-seed trait is one of the most important breeding aims for B. napus.
In plant kingdom, flavonoid biosynthesis pathway play important roles in the coloration of fruits, flowers, and seeds, and numerous evidences had showed that TT-type genes and their homologs are crucial for the accumulation of flavonoids and their derivatives (Nesi et al., 2001; Winkel-Shirley, 2002; Xie et al., 2003; Baudry et al., 2004; Hoffmann et al., 2006; Lepiniec et al., 2006; Kasai et al., 2007). In the model plant Arabidopsis thaliana, the formation of transparent and colorless testa (seed coat) were associated with tt loci that are disrupted the flavonoid synthesis pathway in the loss-of-function mutations (Wan et al., 2002; Winkel-Shirley, 2002; Baudry et al., 2004; Lepiniec et al., 2006). To date, 17 genes involved in this pathway have been cloned and functionally characterized, including eight structural genes (i.e., TT3, TT4, TT5, TT6, TT7, FLS1, LDOX, and BAN; Albert et al., 1997; Devic et al., 1999; Xie et al., 2003; Routaboul et al., 2006; Chiu et al., 2010), six regulatory genes (TT1, TT2, TT8, TTG1, TTG2, and TT16; Nesi et al., 2000, 2001; Baudry et al., 2006; Routaboul et al., 2006), and three encoding transfer proteins (TT12, TT19, and AHA10; Debeaujon et al., 2001; Baxter et al., 2005), which were also classified as Early Biosynthetic Genes (CHS, CHI, and F3H etc.) and Late Biosynthetic Genes (BAN, DFR, and TTG1, etc.) (Nesi et al., 2000, 2001; Winkel-Shirley, 2001; Lepiniec et al., 2006). Homologs of some of these genes, named TT–type genes, have also been identified and shown to be involved in the flavonoid biosynthetic pathway. These genes are thus candidate genes for the molecular basis of seed color manifestation (Supplementary Table S1). TTG1 in Brassica rapa has the same gene function as its orthologs in A. thaliana, i.e., it influences root hairiness and the color of the seed coat (Zhang et al., 2009). BrTT8 was recently shown to regulate the accumulation of proanthocyanidins (PAs) in the seed coat and to regulate the expression of the late biosynthetic genes (LBGs) of the flavonoid pathway in B. rapa, and an analysis in the “sarson” line of B. rapa showed that the yellow-seeded trait was caused by loss of BrTT8 function (Li X. et al., 2012). In addition, BjuA.TT8 and BjuB.TT8 co-segregated perfectly with the seed coat color phenotype in allotetraploid Brassica juncea (Padmaja et al., 2014). However, the inheritance of seed coat color is complex in B. napus. In previous studies, a stable major quantitative trait locus (QTL) for seed coat color of B. napus was detected in different generations and environments, and TT10 was considered as a candidate gene involved in seed coat color, based on microsynteny of this QTL with Arabidopsis genome sequences (Fu et al., 2007). The following findings showed that BnTT10 functions in proanthocyanidin polymerization and lignin biosynthesis, as well as seed coat pigmentation in B. napus (Zhang et al., 2013). Additionally, Chai et al. (2009) found that TT12 was also a candidate gene for seed coat color in B. napus. Moreover, several key loci isolated from B. napus by our group, such as F3'H, PAL1, TTG1, and TT2, showed no or limited down-regulation in the yellow-seeded lines (Wei et al., 2007; Xu et al., 2007; Ni et al., 2008; Lu et al., 2009). Based on a marker closely linked with a major QTL for seed fiber and color in B. napus, Stein et al. proposed that the transparent testa gene AHA10 has a strong effect on both seed color and lignin content (Stein et al., 2013). Undoubtedly, the inheritance of seed color in B. napus is also sensitive to environmental influences, such as lighting, temperature, maturity, and harvest time (Chen and Heneen, 1992; Deynze et al., 1995). Therefore, the molecular mechanism underlying the yellow seed coat trait is unclear in Brassica species.
The family Brassicaceae is well known for its large variation in chromosome numbers, common occurrence of polyploids and many reports of interspecific gene flow (Marhold and Lihová, 2006). Moreover, Brassicaceae plants arose form a common ancestor, of which B. napus (AACC, genome size ~849.7 Mb) was allotetraploid species formed ~7500 years ago by hybridization between B. rapa (AA, genome size ~312 Mb) and Brassica oleracea (CC, genome size ~540 Mb), followed by genome duplications and mergers during the evolutionary process (Chalhoub et al., 2014). Therefore, Brassica is an ideal model to increase knowledge of polyploid evolution (Parkin et al., 2005; Albertin et al., 2006), which is usually assumed that the physiology and developmental biology of TT genes in A. thaliana are highly similar to those of other Brassicaceae plants. Although dozens of genes involved in the flavonoid biosynthesis pathway of A. thaliana were identified based on tt mutations (Holton and Cornish, 1995; Devic et al., 1999; Wan et al., 2002; Xie et al., 2003; Baudry et al., 2006; Lepiniec et al., 2006; Routaboul et al., 2006; Saito et al., 2013), only some of these have been characterized in other Brassicaceae plants, and to date no comprehensive study of these genes has been reported. We previously conducted a systematic study of the expression profiles of related genes in B. napus seeds at different stages of development (Qu et al., 2013).
In this study, we identified 21 genes involved in the flavonoid biosynthesis pathway in 17 sequenced Brassicaceae species. We systematically analyzed the phylogenetic relationships and triplication events of these genes among the Brassicaceae plants. Additionally, the inheritance of flavonoid biosynthesis pathway in B. napus is quite complicated, and the regulatory mechanisms underlying the biosynthesis of the relevant genes were not well understood. Hence, using quantitative real-time PCR (qRT-PCR) analysis, we identified significant differences (Student's t-test, P < 0.05 or 0.01) in the expression patterns of 18 genes associated with the flavonoid biosynthesis pathway in the stems, leaves, buds, flowers, siliques, and pericarps, and at five different stages of seed development (10, 20, 30, 40, and 50 DAP) in six inbred rapeseed lines, which were used to represent typical yellow- and black-seeded genotypes of B. napus, have different genetic backgrounds (Figure 1, Table 1). These results provide useful information for identifying key genes or regulatory nodes that control yellow seed coat formation, and provide insight into the inheritance of qualitative differences between the yellow- and black-seeded B. napus.
Figure 1.
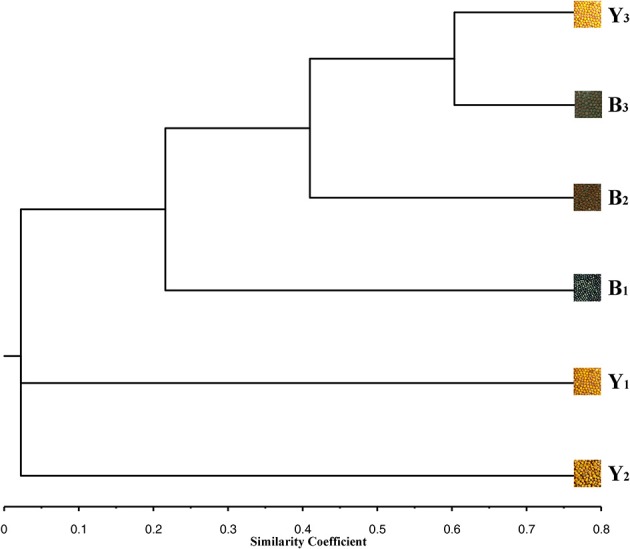
Phenogram showing Jaccard's genetic similarity coefficients for 6 inbred lines revealed by UPGMA clustering based on genetic fingerprints calculated from 2300 SNP markers. The phenotypic characteristics of 6 inbred lines were represented by seeds.
Table 1.
List of B. napus genotypes used in this study.
| No. | Genetic background | Yellow seed degree | Location |
|---|---|---|---|
| B1 | ZY821 | 22.23 | Chongqing, China |
| Y1 | GH06 | 126.43 | Chongqing, China |
| B2 | ZS9 | 63.00 | Chongqing, China |
| Y2 | 05E258-1 | 121.46 | Chongqing, China |
| B3 | ZS9/06E241(BC2F6) | 54.21 | Chongqing, China |
| Y3 | ZS9/06E241(BC2F6) | 131.68 | Chongqing, China |
Materials and methods
Plant materials
Plant materials were collected from yellow-seeded and black-seeded B. napus in different genetic backgrounds (Table 1, Figure 1). The yellow seed degree are calculated using the published reports (Li et al., 2012). The B1 and B2 were inbred lines ZY821 and ZS11, and Y1 and Y2 were GH06 and 05E258-1, which were used to represent typical black-seeded and yellow-seeded genetic types of B. napus, respectively. B3 and Y3 were represent two near isogenic lines of black- and yellow-seeded B. napus, selected from the successive backcross of the sixth generation with parent ZS11 and recurrent parent 06E241 (Qu et al., 2015). The plants were grown under normal field conditions at Chongqing Rapeseed Technology Research Center (CRTRC) in 2014. Field management essentially followed normal agronomic procedures. Various tissue organs, including stems (St), leaves (Le), flowers (Fl), buds (Bu), silique pericarps (SP), and seeds harvested at 10, 20, 30, 40, and 50 days after pollination (DAP) were sampled and stored at −80°C until used.
Identification of flavonoid biosynthesis pathway gene members in brassicaceae species
The full genome sequences of Brassicaceae were downloaded from multiple databases, including the BRAD database (http://brassicadb.org/brad/ftpTrans.php; Cheng et al., 2011), the Phytozome database (http://phytozome.jgi.doe.gov/pz/portal.html#!search; Goodstein et al., 2012), PGDD (PLANT GENOME DUPLICATION DATABASE; http://chibba.agtec.uga.edu/duplication/index/files; Lee et al., 2013), the Raphanus sativus Genome DataBase (http://radish.kazusa.or.jp/; Kitashiba et al., 2014), and the B. napus database (http://www.genoscope.cns.fr/brassicanapus/; Chalhoub et al., 2014). From amongst the species with full genome sequences, the following 17 species were selected: Aethionema arabicum (Aa), Arabidopsis halleri (Ah), Arabidopsis lyrata (Al), A. thaliana (At), Brassica napus L. (Bn), B. oleracea (Bo), B. rapa (Br), Boechera stricta (Bs), Capsella grandiflora (Cg), Capsella rubella (Cr), Camelina sativa (Cs), Leavenworthia alabamica (La), Raphanus sativus L. (Rs), Sisymbrium irio (Si), Schrenkiella parvula (Sp), Thellungiella halophile (Th), and Thellungiella salsuginea (Ts). All coding sequences (CDSs) and amino acid sequences were stored in a local Brassicaceae database using Geneious Pro 4.8.5 software (http://www.geneious.com/; Biomatters, Auckland, New Zealand). To identify the flavonoid biosynthesis pathway genes and their homologous genes, all amino acid sequences of flavonoid biosynthesis pathway genes in A. thaliana retrieved from TAIR 10 (http://www.arabidopsis.org/; Lamesch et al., 2012) were used as queries to search against the Brassicaceae protein models with HMMER3 (version 3.1b2 with Pfam HMM library Pfam 28.0; Finn et al., 2011). To identify the flavonoid biosynthesis genes, unique protein sequences of these genes from the A. thaliana genome were used as query using BLASTP program (Altschul et al., 1997) in the local Brassicaceae database developed in this research. All taxa were named using two-letter acronyms and gene type was used as the species gene name. Briefly, the first uppercase letter represents the genus, the second the species, and the following the gene name. A number at the end indicates the copy number. For example, A. thaliana phenylalanine ammonia lyase 1, which has only one copy in A. thaliana, is indicated by AtPAL1.
Mapping of flavonoid biosynthesis pathway genes among brassicaceae species
To assign the location of flavonoid biosynthesis pathway genes in the Brassicaceae species genomes, the GFF genome files were downloaded from the aforementioned databases. Then, MapChart 2.0 was used to draw graphic representations of their corresponding physical position on pseudo-molecular chromosomes of Brassica crops.
Phylogenetic analysis
Based on previously described methods, all sequence alignments for each flavonoid biosynthesis pathway gene superfamily were performed using ClustalW2 software (Larkin et al., 2007), and phylogenetic analysis was carried out using Molecular Evolutionary Genetics Analysis (MEGA) 6.0 (Tamura et al., 2013) with a maximum likelihood (ML). In the ML method, phylogenetic trees were constructed using the JTT+I+G substitution model in PhyML version 3.0.1. To ensure the accuracy of the phylogenetic tree, each tree was subjected to bootstrap analysis with 1000 replicates (Guindon et al., 2010). Finally, all the phylogenetic trees were visualized using FigTree v1.4.2 (http://tree.bio.ed.ac.uk/software/figtree/).
Expression pattern analysis of TT-type genes in B. napus
To characterize differences in expression of the 18 genes associated with the flavonoid biosynthesis pathway between the yellow- and black-seeded varieties, we designed the primers in consensus region based on alignment the gene sequence (Table 3). Then the total RNA was extracted from various tissues using the RNAprep Pure Plant Kit (TIANGEN BIOTECH, Beijing, China) according to manufacturer's instructions. Then, 1 μg RNA sample was reverse transcribed with the Oligo dT-Adaptor Primer using the RNA PCR Kit (AMV) Ver. 3.0 (TaKaRa, http://www.takara.com.cn). To monitor sample uniformity of initial RNA input and RT efficiency, Brassica napus 26S rRNA was used as the internal control according to the previously described method (Qu et al., 2013).
Table 3.
Primers of the flavonoid biosynthesis pathway genes and housekeeping gene used for qRT-PCR.
| Target gene | Forward sequence (5′–3′) | Reverse sequence (5′–3′) | GenBank | AGI number |
|---|---|---|---|---|
| BnTT4 | GACTACTACTTCCGCATCACCAACAG | GCCTAGCTTAGGGACTTCAACAACC | AF076335 | AT5G13930 |
| BnTT5 | CTTCCTCGGTGGCGCAGGTG | ACACAGTTCTCCGTTACTTTCTCTGA | EU402417 | AT3G55120 |
| BnTT6 | TGGGTGAAAGTGACGGAGGAGT | TGGTTCCAGGGTCAGTGTGACG | DQ513329 | AT3G51240 |
| BnTT7 | GCCATAGCCCGTGACCCGGA | GCTTCTCCGGCGTAACTCCTCC | DQ324379 | AT5G07990 |
| BnTT3 | AGACCGTGTGCGTAACCGGC | AGGATCGCGAACAGTGGCACG | DQ767950 | AT5G42800 |
| BnTT18 | GGCTTAGAGCCTGACCGTCTAGAGAA | TGAGCTTCCACGCCAAGTGCT | GQ120562 | AT4G22880 |
| BnBAN | GGACTTGTGATGACCGAAGAAAACTG | ATGTAGCGACCAGAAGCTGTTTCTTT | FJ938339 | AT1G61720 |
| BnTT12 | GCTCCACAGAGACATACGAGCCG | ACGGTGACGAAGCTGAGCATGTA | EU818785 | AT3G59030 |
| BnTT19 | ACATCTTCTTCGTCAGCCATTTGGTCA | GGTCCACGATGGCTCGGTGC | AB117793 | AT5G17220 |
| BnTT10 | GCGACTGTGCCAAGAAACGGT | CCCCACGTGAGATGTCTATCAAAGTG | HM805059 | AT5G48100 |
| BnAHA10 | ACCCATTGCCATGCCCACTGT | GCTCGGCCTGCAAGCAACAA | NM_101587 | AT1G17260 |
| BnTT2 | AGCTGGTCTCAAGAGGTGTGGCA | AGCCTCCCAGCTATCAACGACC | DQ778647 | AT5G35550 |
| BnTT8 | GGCTGAAGAGGCTGCGTCGG | GTGCTGTGCAAGCCCTCGCT | EU192027 | AT4G09820 |
| BnTTG1 | TCCTCCGGCGACTTCCTCCG | GCTGCGTCTCCACCACGGAC | EF175930 | AT5G24520 |
| BnTT16 | TGCTCACATCGGTCTCATCGTCT | GCTCGTGTGGAGGAATGGAGGC | EU192028 | AT5G23260 |
| BnTTG2 | AAACCTAAAGCAAAGCTTGTCTCCCA | ACTTCCTTTGACTTGCTTCTGTCCGT | FJ012168 | AT2G37260 |
| BnTT1 | TCGCTACAACAATCTTCAGATGCACA | TCCTGCACCCTTCAACGCAGC | AF190298 | AT1G34790 |
| BnTT15 | ACAAAATGACGGGACAGTGGAAGTT | GGCTGCACATCGCCTCGAGTT | BT005834 | AT1G43620 |
| BnACTIN7 | TGGGTTTGCTGGTGACGAT | TGCCTAGGACGACCAACAATACT | EV116054 | AT5g09810 |
| BnUBC21 | CCTCTGCAGCCTCCTCAAGT | CATATCTCCCCTGTCTTGAAATGC | EV086936 | AT5g25760 |
Primers for amplifying partial sequences of flavonoid biosynthesis genes were designed from conserved nucleotide regions identified by multiple alignments of sequences.
Real-time PCR was performed using SYBR® Premix Ex Taq™ II (Perfect Real Time) (TaKaRa, China) in a 20 μl volume that included 10 μl of SYBR® Premix Ex Taq™ II, 2 μl (100 ng) of template cDNA, and 0.4 μM of each PCR primer. All primer sequences used for the qRT-PCR are listed in Table 3, designed according to the methods described in our previous research (Qu et al., 2013). Then the specific primers used in this study and cycling conditions were 95°C for 2 min, followed by 40 cycles at 95°C for 10 s (denaturation) and 60°C for 20 s (annealing and extension). The melting curves of each PCR application were obtained using the following cycling conditions: 95°C for 10 s followed by a constant increase in temperature between 65 and 95°C at an increment of 0.5°C/cycle, and samples were run on the Bio-Rad CFX96 Real Time System (USA). The relative expression of the target genes was analyzed using the 2−ΔΔCt method with BnACTIN7 (EV116054) and BnUBC21 (EV086936) as internal controls (Wu et al., 2010). Three biological replicates for each sample were used for real-time PCR analysis and three technical replicates were analyzed for each biological replicate. Then the values represent the average ± SD of three biological replicates with three technical replicates of each tissue and organ. Relative gene expression levels were normalized according to the expression values in black-seeded at 10 DAP.
Results
Characterization of flavonoid biosynthesis pathway gene members in brassicaceae species
To identify members of the Brassicaceae gene subfamily that are involved in the flavonoid biosynthesis pathway, the whole genome sequences of 17 species were downloaded from multiple public databases (Materials and Methods). In total, 649 nucleotide sequences of 21 genes and their associated amino acid sequences were respectively were identified using the HMM profile (Finn et al., 2011). In addition, we used the nucleotide and encoded amino acid sequences of four genes from B. juncea, Bj_PAL1 (ACX31148.1), Bj_TT3 (ADB45307.1), Bj_TT5 (ADB45305.1), and Bj_TT18 (ACH58397.1), that we identified and aligned previously (Qu et al., 2013). The copy numbers of each gene varied from 20 to 91, and the genes were widely distributed in the 17 Brassicaceae species examined. The encoded amino acid sequences ranged from 84 aa (Bn_UGT2d) to 1676 aa (Sp_TT10; Supplementary Table S2). In addition, the number of gene families also varied by species; for example, only 10 copies of PAL3 were found in 6 species, but 43 C4H gene copies occurred in 17 species with as many as 10 orthologs (BnC4H) in B. napus (Supplementary Table S2). However, TT4, TT6, and AHA10 were not identified in all Brassicaceae species. For example, TT4 was not found in A. halleri and C. rubella, TT6 was absent from A. halleri, and AHA10 was not present in A. lyrata (Supplementary Table S2). We used the sequences of flavonoid biosynthesis genes from A. thaliana as query to search genome databases, and identified highly conserved sequences, including LESS ADHESIVE POLLEN5/6 (LAP5/6) and TT4 in A. halleri and C. rubella, DMR6-LIKE OXYGENASE1/2 (DLO1/2) and TT6 in A. halleri, and members of the Autoinhibited H(+)-ATPase (AHA) superfamily, which might be homologs of TT4, TT6, and AHA10 and function in flavonoid biosynthesis in A. lyrata. Here, 36 copies of 21 genes of the flavonoid biosynthesis pathway were identified in A. thaliana, but 53 were identified in B. rapa, 50 in B. oleracea, and 95 in B. napus, in accordance with the fact that sequences present as a single copy in the A. thaliana were present in 2–8 copies in B. napus (Cavell et al., 1998), and with the observation that excessive gene loss is typical after polyploidization in eukaryotes (Sankoff et al., 2010; Wang et al., 2011).
Genomic distribution on chromosomes of brassicaceae species
All of the flavonoid biosynthesis pathway genes in 17 Brassicaceae species were mapped onto pseudo-molecules or chromosomes using GFF files of their nucleotide sequences (Supplementary Table S3). Brassica crops are the ideal model for studying genome evolution (Wang et al., 2011). To intuitively assign the physical position to the chromosomes of B. rapa, B. oleracea, and B. napus, all gene members of flavonoid biosynthesis pathway were mapped to their chromosomes [52 (98.1%) gene copies in B. rapa, 39 (78.0%) gene copies in B. oleracea, and 84 (88.4%) gene copies in B. napus] and 22 (1 in B. rapa, 11 in B. oleracea, and 11 in B. napus) were distributed on the unanchored scaffolds (Figure 2, Supplementary Table S3), which showed strong collinearity between the A subgenomes from B. rapa and B. napus and the C subgenomes from B. oleracea and B. napus (Figure 2). This result indicates that the gene copies are distributed in orthologous blocks in each genome, and that substantial genome reshuffling had occurred. For example, copies of TT10 and TT16 were not identified on B. napus chromosome A02, and the differential gene copies located in orthologous blocks on chromosome C06 differed greatly between B. oleracea and B. napus (Figure 2). These findings are consistent with the fact that Brassicaceae genomes underwent Brassicaceae-lineage-specific whole genome triplication, followed by diploidization after divergence from their common ancestor (Lysak et al., 2005; Town et al., 2006; Mun et al., 2009; Wang et al., 2011; Cheng et al., 2013).
Figure 2.

Genomic distribution of the flavonoid biosynthesis pathway genes on B. rapa, B. oleracea, and B. napus chromosomes. The dissociation fraction of chromosomes indicates the normal single-copy locus mapped to the random chromosome; Ann and Cnn are the indeterminate Chromosome A and C. Numbers in parentheses are the physical distance of the scaffold for each gene. The collinearity of homologous genes were indicated by the ligatures among Brassica species. The homologous genes of flavonoid biosynthesis pathway were indicated by the same colors of the exact same fonts among different chromosomes of Brassica species.
Phylogenetic analysis and the triplication of the flavonoid biosynthesis pathway gene superfamily
Despite different flavonoid biosynthesis pathway genes having similar functions in Brassicaceae species, the phylogenetic relationships of members of each superfamily have not been comprehensively studied. To gain insight into the functional roles of flavonoid biosynthesis pathway genes that may have arisen during evolution, we performed a phylogenetic analysis using the putative amino acid sequence based on each member of the flavonoid biosynthesis pathway superfamily in Brassicaceae species.
Initially, we conducted a BLASTP analysis against the local database, which included 17 Brassicaceae species, followed by HMM searching (Finn et al., 2011). We identified 649 sequences of 21 flavonoid biosynthesis pathway gene family members, and found that the number of gene copies varied from 1 to 10 in each family (Supplementary Table S2), which were mapped onto pseudo-molecules or chromosomes using GFF files of their nucleotide sequences (Supplementary Table S3). To intuitively assign the physical position to the chromosomes of B. rapa, B. oleracea, and B. napus, all gene members of flavonoid biosynthesis pathway were mapped to their chromosomes [52 (98.1%) gene copies in B. rapa, 39 (78.0%) gene copies in B. oleracea, and 84 (88.4%) gene copies in B. napus] and 22 (1 in B. rapa, 11 in B. oleracea, and 11 in B. napus) were distributed on the unanchored scaffolds (Figure 2, Supplementary Table S3), which showed strong collinearity between the A subgenomes from B. rapa and B. napus and the C subgenomes from B. oleracea and B. napus (Figure 2). This result indicates that the gene copies are distributed in orthologous blocks in each genome, and that substantial genome reshuffling had occurred. For example, copies of TT10 and TT16 were not identified on B. napus chromosome A02, and the differential gene copies located in orthologous blocks on chromosome C06 differed greatly between B. oleracea and B. napus (Figure 2). These findings are consistent with the fact that Brassicaceae genomes underwent Brassicaceae-lineage-specific whole genome triplication, followed by diploidization after divergence from their common ancestor (Lysak et al., 2005; Town et al., 2006; Mun et al., 2009; Wang et al., 2011; Cheng et al., 2013).
In addition, we performed a phylogenetic analysis to identify each flavonoid biosynthesis pathway gene type, and constructed their corresponding phylogenetic trees using MEGA 6.0 with the ML method and modified the tree using FigTree v1.4.2. According to the phylogeny generated using the ML method, all representative sequences for each gene member from the Brassicaceae species formed a well-supported clade, which was classified into the Arabidopsis or Brassicaceae subgroups, but each gene was assigned to monophyletic clades using other substitution models (Figures 3–5, and Supplementary Figures S1–S10). For example, we identified 4 members of PAL, which encodes key enzymes of the phenylpropanoid pathway, that were clearly grouped into four subclades (Figure 3). The gene members, Bn_PAL1b and Si_PAL3 belonged to the same PAL2 subcategories, and Aa_PAL4 and Cs_PAL4c clustered alone or in the vicinity of the PAL3 and PAL4 group (Figure 3; PAL1, Gray; PAL2, Light blue; PAL3, Pink; PAL4, Green). We could thus predict the functions of specific flavonoid biosynthesis pathway genes in Brassicaceae species, because genes with similar functions tend to be retained with orthologous genes (Koonin, 2005). In addition, Brassica species are an ideal model for systematically studying polyploidy genome evolution. We identified one clade of genes of the flavonoid biosynthesis pathway, and the copies in B. rapa, B. oleracea, and B. napus (e.g., C4H, TT4, TT6, TT18, TT19, and UGT2) were divided into different subclades or monophyletic subclades by phylogenetic analysis (Figures 4A–F). However, multiple gene copies of TT12, TTG2, and BAN were present in the subclades in B. rapa, B. oleracea, and B. napus, which have high levels of divergence from their common ancestor with A. thaliana (Figures 5A–C).
Figure 3.

Phylogenetic relationships of PAL gene family of Brassicaceae species. PAL1, PAL2, PAL3, and PAL4 were indicated by gray, light blue, purple and green color. The Bn_PAL1b and Si_PAL3 were denoted by red font that maybe the synonymous of PAL2. The Red color diamond, triangle, circle, and rectangle were denoted the gene copies in A. thaliana, B. rapa, B. oleracea, and B. napus, respectively. Scale bar (the numbers) indicates the estimated number of amino acid substitutions per site.
Figure 5.
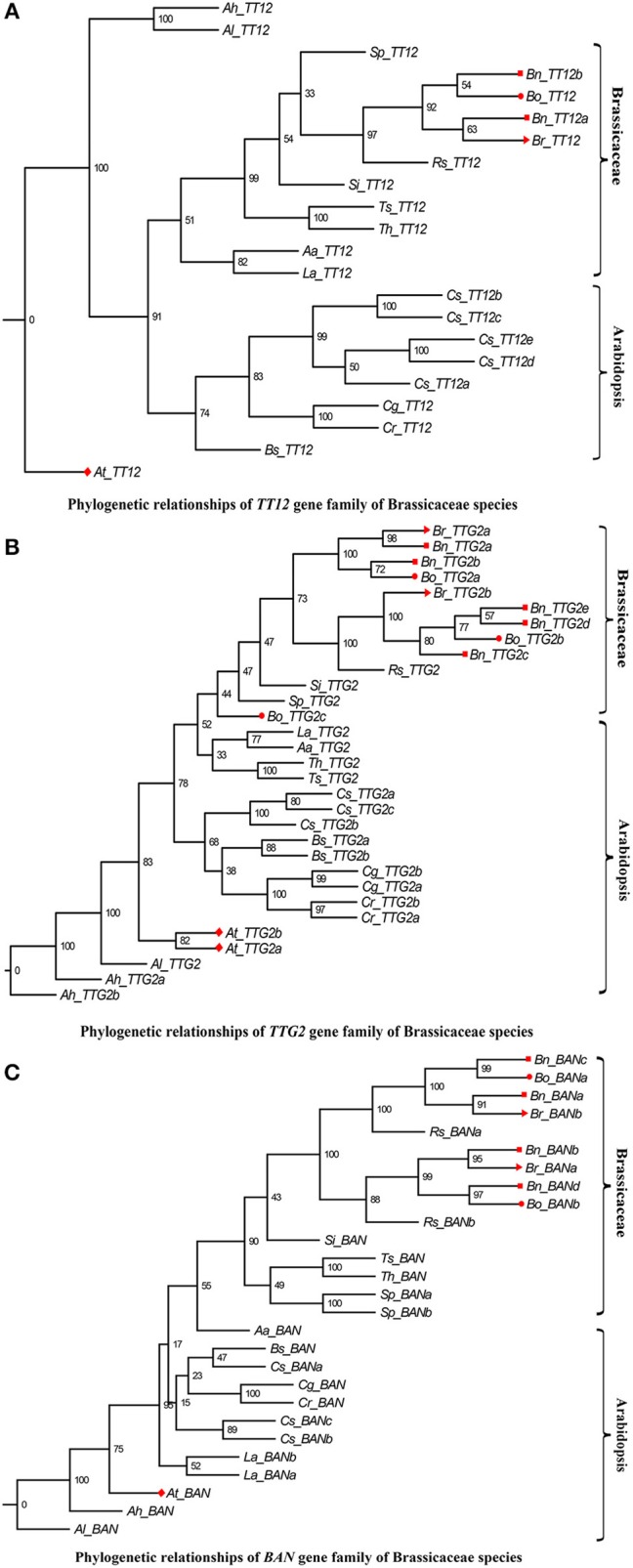
Phylogenetic relationships of TT12, TTG2, and BAN gene family of Brassicaceae species. The Red color diamond, triangle, circle, and rectangle were denoted the gene copies in A. thaliana, B. rapa, B. oleracea, and B. napus, respectively. Scale bar (the numbers) indicates the estimated number of amino acid substitutions per site. (A–C) were indicated TT12, TTG2, and BAN gene family, respectively.
Figure 4.

Phylogenetic relationships of C4H, TT4, TT6, TT18, TT19, and UGT2gene family of Brassicaceae species. The Red color diamond, triangle, circle, and rectangle were denoted the gene copies in A. thaliana, B. rapa, B. oleracea, and B. napus, respectively. Scale bar (the numbers) indicates the estimated number of amino acid substitutions per site. (A–F) were indicated C4H, TT4, TT6, TT18, TT19, and UGT2 gene family, respectively.
Based on the collinearity of orthologs, we identified syntenic orthologous genes of the flavonoid biosynthesis pathway among the genomes of the Brassicaceae species (Supplementary Table S4). In the genomes of B. rapa, B. oleracea, and B. napus, the copies of C4H were distributed in the LF, MF1, and MF2 subgenomes. Besides, the copies of C4H were also identified outside the genome triplication segments in the genomes of Brassica crops. However, 10 of 25 gene members had a single copy in the B. rapa and B. oleracea genomes, but multiple copies in B. napus. Moreover, we identified 10 pseudo-copies of five genes in seven species, i.e., 3 of which in C. rubella (C4H, TT10, and UGT2), 2 copies of TT1 in L. alabamica and S. irio, and 5 copies of TT6 in A. arabicum, B. rapa, B. oleracea, and B. napus (Supplementary Table S4). These findings can be used to systematically study gene retention in the triplicated genomes of Brassicaceae species, as well as to understand the evolutionary history of these orthologous genes among Brassicaceae species.
Development- and tissue-specific expression of TT-type genes in B. napus
In this study, we analyzed the expression patterns of 18 TT-type genes involved in flavonoid biosynthesis by qRT-PCR in various tissues of black- and yellow-seeded lines of B. napus with different genetic backgrounds. Except for BnTT4, BnTT5, BnTT6, and BnTT7, the expression level of 11 structural genes was much higher in developing seeds than in other tissues in B. napus (Figure 6). The expression level of BnBAN, BnTT12, BnTT19, and BnAHA10 peaked during the early stages of seed development, which were the EBGs (Figures 6G–I,K, Table 4). BnTT3, and BnTT18 were expressed in a similar pattern as BnBAN, BnTT12, BnTT19, and BnAHA10, but peaked later in development, which were the LBGs (Figures 6E,F,J, Table 4). Thus, these genes may be regulated by the same upstream gene or they may have a synergistic effect on the flavonoid biosynthetic pathway during seed development. In addition, the expression levels of BnTT5 and BnTT7 did not differ significantly (Student's t-test, P > 0.05) among the rapeseed tissues, whereas BnTT4, BnTT5, BnTT6, and BnTT7 expression was higher in the buds and flowers of B. napus (Figures 6A–D, Table 4). Furthermore, the expression level of five regulatory genes, BnTT2, BnTT8, BnTT16, BnTTG2, and BnTT1, was much higher in developing B. napus seeds, and peaked during the early and middle stages of seed development, which may be the EBGs (Figures 6L,M,O,P,Q, Table 4). However, BnTT15 was expressed at higher levels in flowers than in other organs, indicating that this gene may play an important role in flower formation (Figure 6R).
Figure 6.

Comparison expression analysis of genes involved in flavonoid biosynthesis in different tissues and organs between the yellow- and black-seeded B. napus. Three biological replicates for each sample were used for real-time PCR analysis and three technical replicates were analyzed for each biological replicate. Values represent the average ± SD of three biological replicates with three technical replicates of each tissue and organ. 10–50, Different stages of seed development; Fl, Flowers; SP, Silique pericarps; St, Stems; Bu, Buds; Le, Leaves. Red indicates high expression and blue indicates low expression. (A–R) were indicated the genes of BnTT4, BnTT5, BnTT6, BnTT7, BnTT3, BnTT18, BnBAN, BnTT12, BnTT19, BnTT10, BnAHA10, BnTT2, BnTT8, BnTTG1, BnTT16, BnTTG2, BnTT1, and BnTT15, respectively.
Table 4.
The tissue specificity and expression stages of flavonoid biosynthesis pathway genes in B. napus and A. thaliana.
| Name | Tissue specificity | Expressed during | ||
|---|---|---|---|---|
| B. napus | A. thaliana | B. napus | A. thaliana | |
| TT4 | Except Silique pericarps | Whole plant | EBGs | EBGs |
| TT5 | Whole plant | Whole plant | NA | EBGs |
| TT6 | Whole plant | Whole plant | LBGs | LBGs |
| TT7 | Whole plant | Whole plant | NA | LBGs |
| TT3 | Development seeds | Whole plant | EBGs | EBGs |
| TT18 | Development seeds | Development seeds | LBGs | LBGs |
| BAN | Development seeds | Development seeds | EBGs | EBGs |
| TT12 | Development seeds | Development seeds | EBGs | EBGs |
| TT19 | whole plant | Whole Plant | EBGs | EBGs |
| TT10 | Development seeds | Development seeds and flarol organs | NA | LBGs |
| AHA10 | Development seeds and stem | Development seeds and stem | EBGs | EBGs |
| TT2 | Development seeds | Development seeds | EBGs | EBGs |
| TT8 | Development seeds | Development seeds | EBGs | EBGs |
| TTG1 | whole plant | whole plant | LBGs | LBGs |
| TT16 | Development seeds and flowers | Development seeds and flowers | EBGs | EBGs |
| TTG2 | Whole plant | Whole plant | LBGs | LBGs |
| TT1 | Development seeds and floral organs | Development seeds | EBGs | EBGs |
| TT15 | Whole plant | Whole plant | LBGs | LBGs |
EBGs and LBGs means the Early Biosynthetic Genes and the Late Biosynthetic Genes, respectively. NA indicates that the expression stage of genes were inconclusive during the developmental seeds of B. napus.
Differential expression patterns of TT-type genes in B. napus
In this study, most of the key genes involved in flavonoid biosynthesis were found to exhibit similar expression patterns, and the expression levels of genes varied greatly between the yellow- and black-seeded lines of B. napus (Figure 7). The structural genes involved in flavonoid biosynthesis, i.e., BnTT3, BnTT18, BnBAN, BnTT12, BnTT19, and BnAHA10, had similar expression patterns in the developing seeds of different genetic backgrounds, but had significant differences (Student's t-test, P < 0.01) in expression level between the black- and yellow-seeded lines (Figures 7C,E–I,K, Table 4). BnTT4, BnBAN, BnTT12, BnTT19, and BnAHA10 expression peaked at 20 DAP, which were EBGs (Figures 7A,G–I,K, Table 4), and BnTT6, BnTT3, and BnTT18 expression peaked at 40 DAP, were belong the LBGs (Figures 7E,F, Table 4). These genes were expressed at higher levels in the black-seeded than in the yellow-seeded lines, especially at 20 DAP (Figures 7A,H,K, Table 4), 30 DAP (Figures 7G,I), and 40 DAP (Figures 7B,E,F, Table 4), but the expression patterns of these genes were hardly affected by genetic background. Additionally, BnTT7, BnTT5, and BnTT10 expression peaked at different development stages both in the black- and yellow-seeded lines (Figures 7B,D,J, Table 4, and Supplementary Table S5), indicating that the expression of these genes was affected by the genetic background. As for the expression patterns of structural genes, regulatory genes, such as BnTT2, BnTT8, BnTT16, BnTTG2, BnTT1, and BnTT15, had similar expression patterns in the black- and yellow-seeded B. napus, with the genetic background having a minimal effect, and the expression of these genes peaked during the early and middle developmental stages (20 and 30 DAP, EBGs; Figures 7L,M,O–R, Table 4). In addition, the expression patterns and levels of BnTTG1 showed obvious variations during seed development of different lines of B. napus, suggesting that they were significantly affected by genetic background (Figure 7N).
Figure 7.

Expression pattern analysis of genes involved in flavonoid biosynthesis in the developing seeds of different B. napus lines by qRT-PCR. The yellow-seeded lines include Y1, Y2, and Y3, and the black-seeded lines include B1, B2, and B3 (A–R). Expression was monitored at five stages of seed development (10, 20, 30, 40, and 50 DAP). Values represent the average ± SD of three biological replicates with three technical replicates of each developmental stage. Error bars denote standard error of the mean (SEM) of three experiments. Relative gene expression levels were normalized according to the expression values in black-seeded B. napus at 10 DAP.
Although the expression patterns of most of the genes involved in flavonoid biosynthesis did not significantly differ (Student's t-test, P > 0.05) between the developing seeds of the black- and yellow-seeded B. napus, the expression levels of these genes did differ and peaked at different developmental stages (Figure 7), indicating that these genes can also be classified as Early Biosynthetic Genes and Late Biosynthetic Genes, as in Arabidopsis (Lepiniec et al., 2006). The expression of genes that function upstream in the flavonoid biosynthesis pathway peaked before those that functioned later, suggesting that the downstream genes were influenced by the upstream genes. For example, the expression of BnTT4, which encodes a protein that catalyzes the first committed step of flavonoid biosynthesis (Albert et al., 1997; Tang et al., 1997), peaked at 20 DAP, which was before the expression of the downstream genes BnTT5, BnTT6, and BnTT7 peaked (Figure 7). All flavonoids and isoflavonoids are derived from the nargingenin chalcone generated by this first enzyme. Moreover, the expression of most of the regulatory genes also peaked at 20 DAP (Figure 7), suggesting that they may be essential for regulating the expression of genes involved in flavonoid biosynthesis during the early stages of seed development. Although, the expression of BnTTG2 peaked later than did that of BnTT2 and BnTT8, the expression of BnTT2 was greater in yellow- than in black-seeded B. napus, and BnTTG1 expression was also largely affected by the genetic background (Figures 7L–N,P), indicating that the mechanism of BnTTG2 is different from that of its orthologs in A. thaliana (Lepiniec et al., 2006). In addition, the expression of BnTT6, BnTT18, and BnTT2 peaked sooner in the yellow-seeded than in the black-seeded lines (Figures 7C,F,L).
Discussion
Flavonoids are secondary metabolites that are extensively distributed in the plant kingdom. They not only play an important role in color formation in fruits and flowers, but are also well-known for their positive effect on health, due to their antioxidant and antitumor properties (Winkel-Shirley, 2001, 2002; Lepiniec et al., 2006; Routaboul et al., 2006). Genome-wide analyses of gene superfamilies have been widely performed after the completion of numerous plant genome projects. For instance, numerous genome-wide studies of gene superfamilies have been performed in the diploid species, B. rapa and B. oleracea (Song et al., 2013; Duan et al., 2014; Li et al., 2014; Diehn et al., 2015; Lu et al., 2015). Several gene superfamilies have also been reported in the allotetraploid species B. napus (Sun et al., 2014; Raboanatahiry et al., 2015) However, a comprehensive analysis of the superfamily of genes underlying the flavonoid biosynthesis pathway in Brassica has not been reported to date. Here, we identified 649 nucleotide sequences of 21 flavonoid biosynthesis pathway genes and their corresponding amino acid sequences in 17 Brassicaceae species (Table 2, Supplementary Table S2). The gene copy numbers differed between species; for example, 10 copies of PAL3 were found in 6 species, whereas 43 copies of C4H were identified in 17 species (Table 2, Supplementary Table S2). Although we did not detect orthologs for some genes involved in flavonoid biosynthesis (e.g., TT4, TT6, and AHA10), in accordance with the fact that the excessive gene loss is typical after polyploidy formation in eukaryotes (Sankoff et al., 2010; Wang et al., 2011), we identified orthologs of many of these genes by querying the Brassica genome databases with the sequences of A. thaliana genes involved in flavonoid biosynthesis. In addition, it has been widely suggested that the genome structures are highly conserved among Brassica species (Krishnamurthy et al., 2014; Thamilarasan et al., 2014; Dong et al., 2016). Each of the gene copies was found to be distributed in orthologous blocks by collinearity analysis between the A and C subgenomes (Figure 2). Not all gene members could be accurately annotated on chromosomes and the number of gene copies varied greatly in the orthologous blocks (Figure 2, Supplementary Table S3), indicating that may be associated with Brassicaceae-lineage-specific whole genome triplication, followed by diploidization after divergence from the common ancestor (Lysak et al., 2005; Town et al., 2006; Mun et al., 2009; Wang et al., 2011; Cheng et al., 2013). Although, subgenome sequences present higher levels of conservation in extensive collinear genome blocks among Brassicaceae species, we found that all genes of the flavonoid biosynthesis pathway were phylogenetically classified into two major subcategories (Arabidopsis and Brassicaceae species; Figures 3–5, Supplementary Figures S1–S11), consistent with the functional divergence of orthologous gene groups between Arabidopsis and Brassicaceae species during evolution. These results revealed that diversification occurred among the flavonoid biosynthesis pathway gene family members, likely indicating that functional divergence of orthologous gene groups occurred between Arabidopsis and Brassicaceae species during evolution. These findings provide insight into the functional divergence of these genes among Brassicaceae species. In addition, the identification of conserved genomic blocks will provide useful phylogenetic, polyploidization, and comparative genomics information (Schranz et al., 2006; Cheng et al., 2013). Subgenomes can be classified based on gene density into the following three groups: least fractionated (LF), medium fractionated (MF1), and most fractionated (MF2) (Wang et al., 2011; Cheng et al., 2013). We then performed the triplication of flavonoid genes in the whole gennomes of Brassicaceae species. Furthermore, 10 pseudo-copies of five flavonoid biosynthesis pathway genes were identified, such as C4H, TT10, UGT2, TT1, and TT6 (Supplementary Table S4). These results will provide detailed information for systematic studies of the functions and roles of these genes in flavonoid biosynthesis pathway at the molecular level.
Table 2.
Characterization of genes involved in the phenylpropane-flavonoid biosynthesis pathway of Brassicaceae species.
| Gene name | Length (aa) | No. of Brassicaceae Species | A. thaliana | B. rapa | B. oleracea | B. napus | Total No. |
|---|---|---|---|---|---|---|---|
| PAL1 | 263–728 | 18 | 1 | 2 | 2 | 4 | 28 |
| PAL2 | 377–725 | 16 | 1 | 3 | 4 | 5 | 31 |
| PAL3 | 107–698 | 6 | 1 | 2 | 2 | 2 | 10 |
| PAL4 | 344–709 | 16 | 1 | 1 | 1 | 2 | 22 |
| C4H | 468–1197 | 17 | 1 | 5 | 5 | 10 | 43 |
| TT1 | 287–323 | 17 | 1 | 1 | 1 | 2 | 20 |
| TT2 | 156–265 | 17 | 1 | 1 | 1 | 2 | 20 |
| TT3 | 244–387 | 18 | 1 | 1 | 1 | 3 | 24 |
| TT4 | 393–734 | 15 | 1 | 5 | 3 | 9 | 39 |
| TT5 | 197–254 | 18 | 1 | 3 | 3 | 4 | 30 |
| TT6 | 274–548 | 16 | 1 | 4 | 3 | 6 | 34 |
| TT7 | 364–687 | 17 | 1 | 1 | 1 | 2 | 21 |
| TT8 | 497–613 | 17 | 1 | 1 | 1 | 2 | 23 |
| TT10 | 489–1676 | 17 | 1 | 2 | 1 | 3 | 22 |
| TT12 | 507–560 | 17 | 1 | 1 | 1 | 2 | 22 |
| TT15 | 473–864 | 17 | 1 | 1 | 1 | 2 | 24 |
| TT16 | 183–318 | 17 | 1 | 3 | 3 | 6 | 33 |
| TT18 | 351–416 | 18 | 1 | 2 | 2 | 4 | 28 |
| TT19 | 213–439 | 17 | 1 | 2 | 2 | 5 | 27 |
| TTG1 | 120–521 | 17 | 1 | 2 | 1 | 3 | 24 |
| TTG2 | 114–431 | 17 | 1 | 2 | 3 | 4 | 31 |
| AHA10 | 918–985 | 16 | 1 | 1 | 1 | 2 | 20 |
| BAN | 216–1041 | 17 | 1 | 2 | 2 | 4 | 27 |
| UGT2a | 422–566 | 12 | 1 | 1 | 2 | 2 | 14 |
| UGT2b | 84–562 | 17 | 1 | 4 | 3 | 5 | 35 |
Seed coat color was previously reported to involve a similar mechanism in Brassica and Arabidopsis species (Marles and Gruber, 2004). Hence, identifying candidate genes by cloning Brassica TT genes involved in the flavonoid biosynthetic pathway and conducting comparative studies of these genes is a reasonable approach, and many homologs of these genes have also been identified in B. napus (Wei et al., 2007; Xu et al., 2007; Ni et al., 2008; Chai et al., 2009; Lu et al., 2009; Chen et al., 2013). However, little is known about the mechanism underlying seed color formation in B. napus. Using three groups of B. napus plants in different genetic backgrounds, we showed that the 12 genes (BnTT3, BnTT18, BnBAN, BnTT12, BnTT19, BnTT10, BnAHA10, BnTT2, BnTT8, BnTT16, BnTTG2, and BnTT1) investigated in this study were highly expressed and showed clear divergence in organ specificity in the developing seed (Figure 6), suggesting that these genes play an important role in seed development and are involved in the accumulation of seed pigmentation. Furthermore, BnTT15 was highly expressed in flowers (Figure 6R). By contrast, BnTT5, BnTT7, and BnTTG1 expression did not differ significantly (Student's t-test, P > 0.05) among B. napus organs (Figures 6B,D,N). Similar expression profiles were observed for orthologs of these genes in A. thaliana (Schmid et al., 2005), suggesting evolutionary conservation of the regulatory mechanism governing flavonoid accumulation. Our study lays the foundation for future research aimed at deciphering the expression profiles of different gene copies in B. napus. These findings provide insight into the characteristics and functions of flavonoid pathway genes in B. napus.
As in A. thaliana, the flavonoid biosynthesis pathway has been characterized mainly using tt mutants that exhibited a transparent and colorless testa (seed coat; Yu, 2013). Moreover, much research has focused on identifying the seed pigments involved in the formation of seed coat color in B. napus (Theander et al., 1977; Marles and Gruber, 2004; Akhov et al., 2009; Qu et al., 2013). Homologous genes in the B. napus flavonoid biosynthesis pathway have also been cloned and characterized (Wei et al., 2007; Xu et al., 2007; Ni et al., 2008; Akhov et al., 2009; Auger et al., 2009; Chai et al., 2009; Lu et al., 2009; Chen et al., 2013). However, a few of these genes was comprehensively functionalized in B. napus. Using qRT-PCR analysis, we now examined the temporal and spatial expression patterns of 18 flavonoid biosynthesis genes in the developing seeds of black- and yellow-seeded B. napus, sourced from different backgrounds. We found that the majority of genes had similar expression patterns in the developing seed, suggesting that these genes not only participated in the flavonoid pathway, but also might be regulated by an upstream regulatory gene involved in seed coat color formation in B. napus. In addition, we classified these genes based on the time at which their expression peaked. The first group of genes with similar expression patterns in black- and yellow-seeded lines (BnTT4, BnBAN, BnTT12, BnTT19, BnAHA10, BnTT8, BnTT16, BnTTG2, and BnTT15) was expressed at different levels in different tissues and showed the highest expression levels at 20 DAP, and was considered as the early biosynthetic genes (Figures 7, 8). By contrast, BnTT4, BnTT12, and BnAHA10 have higher expression levels in black-seeded than in yellow-seeded lines (Figures 7A,H,K). The first dedicated step for flavonoid biosynthesis of plant is catalyzed by TT4, which produces naringenin chalcone, and thus TT4 critically influences many important flavonoid-related characteristics, such as seed coat color, flower color, and pigmentation of the stem and leaf surface (Hoffmann et al., 2006; Kasai et al., 2007). Chai et al. (2009) proposed BnTT12 a potential candidate gene for seed coat color formation in B. napus. Stein et al. (2013) found that the transparent testa gene AHA10 strongly affected both seed color and lignin content using a marker that was closely linked to a major QTL for seed fiber and color in B. napus. Moreover, TT12 and AHA10 in Arabidopsis were both found to be related to the vacuolar transport of proanthocyanidin in seed coats (Debeaujon et al., 2001; Baxter et al., 2005). Thus, the difference in seed coat color between black- and yellow-seeded rapes seems to be related to the reduction in precursor accumulation following down-regulation of the encoding gene (Figure 8).
Figure 8.

The pathway of flavonoid biosynthesis, modification, and transport in B. napus. Bn, Brassca napus; BAN, BANYULS; AHA10, Autoinhibited H+-ATPase isoform 10; TT(G), TRANSPARENT TESTA (GLABRA); UGT, UDP flavonoid glucosyl transferase. The the Early Biosynthetic Genes (EBGs) and the Late Biosynthetic Genes (LBGs) red and blue colors, respectively. The bold means that genes might play an crucial roles in the formation of different seed coat colors in B. napus.
Another group of genes (BnTT5, BnTT7, BnTT18, BnBAN, BnTT19, BnTT2, and BnTTG2) exhibited drastic differences during the middle to late stages of seed coat development between the black- and yellow-seeded B. napus (Figures 7B,D,F,G,I,L,P). Additionally, TTG1, TT2, and TT8 were found to modulate the activity of proteins encoding TT3 (DFR), TT18 (LDOX), BAN, and TT12 in the proanthocyanidin subpathway, respectively. Moreover, we previously found that PAs and polyphenol compounds gradually increase during seed maturation and result in significant differences (Student's t-test, P < 0.05 or 0.01) in the colors of black- and yellow-seeded B. napus seed coats (Qu et al., 2013). However, BnTT5 and BnTTG1 were found to possess different expression patterns among the rapeseed seeds, suggesting that they were largely influenced by the genetic background (Figures 7B,N). These data suggest that numerous compounds accumulate in the seed coat during later development stage and confer color to the mature seed. Therefore, inhibiting the expression of genes involved in their biosynthesis can lead to lighter color seeds during the middle and late development stages in B. napus, and improve the nutritional quality of rapeseed oil and meal. Moreover, this can also explain why the expression of upstream genes peaked sooner than or simultaneously with the downstream genes in the developing seeds (Figures 7, 8), indicating that the upstream genes not only control the downstream genes, but also that the upstream genes cooperate in the flavonoid biosynthesis pathway. Together, the flavonoid biosynthetic pathway of Brassica species is much more complex than that in A. thaliana, with the former not only having more synthesis-related genes, but also exhibiting interactions with other genes involved in flavonoid biosynthesis at multiple loci (Figure 8). These findings provide insight into the molecular and biochemical mechanism of seed coat color development in B. napus.
Author contributions
CQ and FF designed and wrote the manuscript. HZ and ZW performed the data mining and gene expression analysis. YZ and XW collected the flavonoid gene sequences and bioinformatics. XX and ZT carried out reagents and the field experiments. KZ and RW analyzed the accuracy data and edited Figures. KL and JL contributed to interpretation and modification of the data and manuscript. All authors read and approved the final manuscript.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the National Science Foundation of China (31401412, U1302266, 31571701), the utilization of heterosis and selection of strong advantage of hybrid (2016YFD0101300), Projects in the National Science and Technology Pillar Program (2013BAD01B03-12), the Chongqing Project of Main Crop Variety Innovation (cstc2012ggB80008), the 111 Project (B12006), Chongqing Basic Scientific and advanced technology Research (cstc2015jcyjBX0001), and Fundamental Research Funds for the Central Universities (XDJK2016B030).
Glossary
Abbreviations
- TT:
Transparent Testa
- Bn:
Brassica napus L.
- TTG:
TRANSPARENT TEST AGLABRA
- PAL:
Phenylalanine ammonia-lyase
- C4H:
cinnamate 4-hydroxylase
- FLS:
flavonol synthase
- LDOX:
leucoanthocyanidin dioxygenase
- BAN:
BANYULS
- AHA10:
H+-ATPase isoform 10
- PAs:
proanthocyanidins
- Bj:
Brassica juncea
- BLAST:
basic local alignment search tool
- BRAD:
Brassica Database
- PGDD:
PLANT GENOME DUPLICATION DATABASE
- DAP:
days after pollination.
Supplementary material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2016.01755/full#supplementary-material
References
- Akhov L., Ashe P., Tan Y., Datla R., Selvaraj G. (2009). Proanthocyanidin biosynthesis in the seed coat of yellow-seeded, canola quality Brassica napus YN01-429 is constrained at the committed step catalyzed by dihydroflavonol 4-reductase. Botany 87, 616–625. 10.1139/B09-036 [DOI] [Google Scholar]
- Albert S., Delseny M., Devic M. (1997). BANYULS, a novel negative regulator of flavonoid biosynthesis in the Arabidopsis seed coat. Plant J. 11, 289–299. 10.1046/j.1365-313X.1997.11020289.x [DOI] [PubMed] [Google Scholar]
- Albertin W., Balliau T., Brabant P., Chèvre A.-M., Eber F., Malosse C., et al. (2006). Numerous and rapid nonstochastic modifications of gene products in newly synthesized Brassica napus allotetraploids. Genetics 173, 1101–1113. 10.1534/genetics.106.057554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. 10.1093/nar/25.17.3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auger B., Baron C., Lucas M. O., Vautrin S., Bergès H., Chalhoub B., et al. (2009). Brassica orthologs from BANYULS belong to a small multigene family, which is involved in procyanidin accumulation in the seed. Planta 230, 1167–1183. 10.1007/s00425-009-1017-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudry A., Caboche M., Lepiniec L. (2006). TT8 controls its own expression in a feedback regulation involving TTG1 and homologous MYB and bHLH factors, allowing a strong and cell specific accumulation of flavonoids in Arabidopsis thaliana. Plant J. 46, 768–779. 10.1111/j.1365-313X.2006.02733.x [DOI] [PubMed] [Google Scholar]
- Baudry A., Heim M. A., Dubreucq B., Caboche M., Weisshaar B., Lepiniec L. (2004). TT2, TT8, and TTG1 synergistically specify the expression of BANYULS and proanthocyanidin biosynthesis in Arabidopsis thaliana. Plant J. 39, 366–380. 10.1111/j.1365-313X.2004.02138.x [DOI] [PubMed] [Google Scholar]
- Baxter I. R., Young J. C., Armstrong G., Foster N., Bogenschutz N., Cordova T., et al. (2005). A plasma membrane H+ - ATPase is required for the formation of proanthocyanidins in the seed coat endothelium of Arabidopsis thaliana. Proc. Natl. Acad. Sci. U.S.A. 102, 2649–2654. 10.1073/pnas.0406377102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavell A. C., Lydiate D. J., Parkin I. A., Dean C., Trick M. (1998). Collinearity between a 30-centimorgan segment of Arabidopsis thaliana chromosome 4 and duplicated regions within the Brassica napus genome. Genome 41, 62–69. 10.1139/g97-097 [DOI] [PubMed] [Google Scholar]
- Chai Y. R., Lei B., Huang H. L., Li J. N., Yin J. M., Tang Z. L., et al. (2009). TRANSPARENT TESTA12 genes from Brassica napus and parental species: cloning, evolution, and differential involvement in yellow seed trait. Mol. Genet. Genomics 281, 109–123. 10.1007/s00438-008-0399-1 [DOI] [PubMed] [Google Scholar]
- Chalhoub B., Denoeud F., Liu S., Parkin I. A., Tang H., Wang X., et al. (2014). Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 345, 950–953. 10.1126/science.1253435 [DOI] [PubMed] [Google Scholar]
- Chen B., Heneen W. (1992). Inheritance of seed colour in Brassica campestris L. and breeding for yellow-seeded B. napus L. Euphytica 59, 157–163. 10.1007/BF00041268 [DOI] [Google Scholar]
- Chen G., Deng W., Peng F., Truksa M., Singer S., Snyder C. L., et al. (2013). Brassica napus TT16 homologs with different genomic origins and expression levels encode proteins that regulate a broad range of endothelium-associated genes at the transcriptional level. Plant J. 74, 663–677. 10.1111/tpj.12151 [DOI] [PubMed] [Google Scholar]
- Cheng F., Liu S., Wu J., Fang L., Sun S., Liu B., et al. (2011). BRAD, the genetics and genomics database for Brassica plants. BMC Plant Biol. 11:136. 10.1186/1471-2229-11-136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng F., Mandáková T., Wu J., Xie Q., Lysak M. A., Wang X. (2013). Deciphering the diploid ancestral genome of the mesohexaploid Brassica rapa. Plant Cell 25, 1541–1554. 10.1105/tpc.113.110486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu L. W., Zhou X., Burke S., Wu X., Prior R. L., Li L. (2010). The purple cauliflower arises from activation of a MYB transcription factor. Plant Physiol. 154, 1470–1480. 10.1104/pp.110.164160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debeaujon I., Peeters A. J. M., Léon-Kloosterziel K. M., Koornneef M. (2001). The TRANSPARENT TESTA12 gene of Arabidopsis encodes a multidrug secondary transporter-like protein required for flavonoid sequestration in vacuoles of the seed coat endothelium. Plant Cell Online 13:853. 10.1105/tpc.13.4.853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devic M., Guilleminot J., Debeaujon I., Bechtold N., Bensaude E., Koornneef M., et al. (1999). The BANYULS gene encodes a DFR-like protein and is a marker of early seed coat development. Plant J. 19, 387–398. 10.1046/j.1365-313X.1999.00529.x [DOI] [PubMed] [Google Scholar]
- Deynze A. E., Landry B. S, Pauls, K. P. (1995). The identification of restriction fragment length polymorphisms linked to seed colour genes in Brassica napus. Genome 38, 534–542. 10.1139/g95-069 [DOI] [PubMed] [Google Scholar]
- Diehn T. A., Pommerrenig B., Bernhardt N., Hartmann A., Bienert G. P. (2015). Genome-wide identification of aquaporin encoding genes in Brassica oleracea and their phylogenetic sequence comparison to Brassica crops and Arabidopsis. Front. Plant Sci. 6:166. 10.3389/fpls.2015.00166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X., Yi H., Han C.-T., Nou I.-S., Hur Y. (2016). GDSL esterase/lipase genes in Brassica rapa L.: genome-wide identification and expression analysis. Mol. Genet. Genomics 291, 531–542. 10.1007/s00438-015-1123-6 [DOI] [PubMed] [Google Scholar]
- Duan W., Song X., Liu T., Huang Z., Ren J., Hou X., et al. (2014). Genome-wide analysis of the MADS-box gene family in Brassica rapa (Chinese cabbage). Mol. Genet. Genomics 290, 239–255. 10.1007/s00438-014-0912-7 [DOI] [PubMed] [Google Scholar]
- Finn R. D., Clements J., Eddy S. R. (2011). HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37. 10.1093/nar/gkr367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu F. Y., Liu L. Z., Chai Y. R., Chen L., Yang T., Jin M. Y., et al. (2007). Localization of QTLs for seed color using recombinant inbred lines of Brassica napus in different environments. Genome 50, 840–854. 10.1139/g07-068 [DOI] [PubMed] [Google Scholar]
- Goodstein D. M., Shu S., Howson R., Neupane R., Hayes R. D., Fazo J., et al. (2012). Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 40, D1178–D1186. 10.1093/nar/gkr944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S., Dufayard J.-F., Lefort V., Anisimova M., Hordijk W., Gascuel O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Sys. Biol. 59, 307–321. 10.1093/sysbio/syq010 [DOI] [PubMed] [Google Scholar]
- Hoffmann T., Kalinowski G., Schwab W. (2006). RNAi-induced silencing of gene expression in strawberry fruit (Fragaria × ananassa) by agroinfiltration: a rapid assay for gene function analysis. Plant J. 48, 818–826. 10.1111/j.1365-313X.2006.02913.x [DOI] [PubMed] [Google Scholar]
- Holton T. A, Cornish, E. C. (1995). Genetics and biochemistry of anthocyanin biosynthesis. Plant Cell 7, 1071–1083. 10.1105/tpc.7.7.1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai A., Kasai K., Yumoto S., Senda M. (2007). Structural features of GmIRCHS, candidate of the I gene inhibiting seed coat pigmentation in soybean: implications for inducing endogenous RNA silencing of chalcone synthase genes. Plant Mol. Biol. 64, 467–479. 10.1007/s11103-007-9169-4 [DOI] [PubMed] [Google Scholar]
- Kitashiba H., Li F., Hirakawa H., Kawanabe T., Zou Z., Hasegawa Y., et al. (2014). Draft sequences of the radish (Raphanus sativus L.) genome. DNA Res. 21, 481–490. 10.1093/dnares/dsu014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin E. V. (2005). Orthologs, paralogs, and evolutionary genomics 1. Annu. Rev. Genet. 39, 309–338. 10.1146/annurev.genet.39.073003.114725 [DOI] [PubMed] [Google Scholar]
- Krishnamurthy P., Hong J. K., Kim J. A., Jeong M.-J., Lee Y.-H., Lee S. I. (2014). Genome-wide analysis of the expansin gene superfamily reveals Brassica rapa-specific evolutionary dynamics upon whole genome triplication. Mol. Genet. Genomics 290, 521–530. 10.1007/s00438-014-0935-0 [DOI] [PubMed] [Google Scholar]
- Lamesch P., Berardini T. Z., Li D., Swarbreck D., Wilks C., Sasidharan R., et al. (2012). The Arabidopsis Information Resource (TAIR): improved gene annotation and new tools. Nucleic Acids Res. 40, D1202–D1210. 10.1093/nar/gkr1090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., et al. (2007). Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948. 10.1093/bioinformatics/btm404 [DOI] [PubMed] [Google Scholar]
- Lee T.-H., Tang H., Wang X., Paterson A. H. (2013). PGDD: a database of gene and genome duplication in plants. Nucleic Acids Res. 41, D1152–D1158. 10.1093/nar/gks1104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepiniec L., Debeaujon I., Routaboul J.-M., Baudry A., Pourcel L., Nesi N., et al. (2006). Genetics and biochemistry of seed flavonoids. Annu. Rev. Plant Biol. 57, 405–430. 10.1146/annurev.arplant.57.032905.105252 [DOI] [PubMed] [Google Scholar]
- Li J., Gao G., Xu K., Chen B., Yan G., Li F., et al. (2014). Genome-wide survey and expression analysis of the putative non-specific lipid transfer proteins in Brassica rapa L. PLoS ONE 9:e84556. 10.1371/journal.pone.0084556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Chen L., Hong M., Zhang Y., Zu F., Wen J., et al. (2012). A large insertion in bHLH transcription factor BrTT8 resulting in yellow seed coat in Brassica rapa. PLoS ONE 7:e44145. 10.1371/journal.pone.0044145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Liu X., Li J., Yin J., Xu X. (2012). Construction of near – infrared reflectance spectroscopy model for seed color of rapeseed. Chin. J. Oil Crop Sci. 34, 533–536. [Google Scholar]
- Lu J., Li J., Lei B., Wang S., Chai Y. (2009). Molecular cloning and characterization of two Brassica napus TTG1 genes reveal genus-specific nucleotide preference, extreme protein-level conservation and fast divergence of organ-specificity. Genes Genomics 31, 129–142. 10.1007/BF03191146 [DOI] [Google Scholar]
- Lu K., Guo W., Lu J., Yu H., Qu C., Tang Z., et al. (2015). Genome-wide survey and expression profile analysis of the Mitogen-Activated Protein Kinase (MAPK) gene family in Brassica rapa. PLoS ONE 10:e0132051. 10.1371/journal.pone.0132051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lysak M. A., Koch M. A., Pecinka A., Schubert I. (2005). Chromosome triplication found across the tribe Brassiceae. Genome Res. 15, 516–525. 10.1101/gr.3531105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marhold K., Lihová J. (2006). Polyploidy, hybridization and reticulate evolution: lessons from the Brassicaceae. Plant Sys. Evol. 259, 143–174. 10.1007/s00606-006-0417-x [DOI] [Google Scholar]
- Marles M., Gruber M. Y. (2004). Histochemical characterisation of unextractable seed coat pigments and quantification of extractable lignin in the Brassicaceae. J. Sci. Food Agric. 84, 251–262. 10.1002/jsfa.1621 [DOI] [Google Scholar]
- Meng J., Shi S., Gan L., Li Z., Qu X. (1998). The production of yellow-seeded Brassica napus (AACC) through crossing interspecific hybrids of B. campestris (AA) and B. carinata (BBCC) with B. napus. Euphytica 103, 329–333. 10.1023/A:1018646223643 [DOI] [Google Scholar]
- Mun J. H., Kwon S. J., Yang T. J., Seol Y. J., Jin M., Kim J. A., et al. (2009). Genome-wide comparative analysis of the Brassica rapa gene space reveals genome shrinkage and differential loss of duplicated genes after whole genome triplication. Genome Biol 10:R111. 10.1186/gb-2009-10-10-r111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesi N., Debeaujon I., Jond C., Pelletier G., Caboche M., Lepiniec L. (2000). The TT8 gene encodes a basic helix-loop-helix domain protein required for expression of DFR and BAN genes in Arabidopsis siliques. Plant Cell Online 12, 1863–1878. 10.1105/tpc.12.10.1863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesi N., Delourme R., Brégeon M., Falentin C., Renard M. (2008). Genetic and molecular approaches to improve nutritional value of Brassica napus L. seed. Compt. Rendus Biol. 331, 763–771. 10.1016/j.crvi.2008.07.018 [DOI] [PubMed] [Google Scholar]
- Nesi N., Jond C., Debeaujon I., Caboche M., Lepiniec L. (2001). The Arabidopsis TT2 gene encodes an R2R3 MYB domain protein that acts as a key determinant for proanthocyanidin accumulation in developing seed. Plant Cell 13, 2099–2114. 10.1105/TPC.010098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni Y., Jiang H., Lei B., Li J., Chai Y. (2008). Molecular cloning, characterization and expression of two rapeseed (Brassica napus L.) cDNAs orthologous to Arabidopsis thaliana phenylalanine ammonia-lyase 1. Euphytica 159, 1–16. 10.1007/s10681-007-9448-9 [DOI] [Google Scholar]
- Padmaja L. K., Agarwal P., Gupta V., Mukhopadhyay A., Sodhi Y. S., Pental D., et al. (2014). Natural mutations in two homoeologous TT8 genes control yellow seed coat trait in allotetraploid Brassica juncea (AABB). Theor. Appl. Genet. 127, 339–347. 10.1007/s00122-013-2222-6 [DOI] [PubMed] [Google Scholar]
- Parkin I. A., Gulden S. M., Sharpe A. G., Lukens L., Trick M., Osborn T. C., et al. (2005). Segmental structure of the Brassica napus genome based on comparative analysis with Arabidopsis thaliana. Genetics 171, 765–781. 10.1534/genetics.105.042093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu C., Fu F., Lu K., Zhang K., Wang R., Xu X., et al. (2013). Differential accumulation of phenolic compounds and expression of related genes in black-and yellow-seeded Brassica napus. J. Exp. Bot. 64, 2885–2898. 10.1093/jxb/ert148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu C.-M., Li S.-M., Duan X.-J., Fan J.-H., Jia L.-D., Zhao H.-Y., et al. (2015). Identification of candidate genes for seed glucosinolate content using association mapping in brassica napus L. Genes. 6, 1215–1229. 10.3390/genes6041215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raboanatahiry N. H., Yin Y., Chen L., Li M. (2015). Genome-wide identification and Phylogenic analysis of kelch motif containing ACBP in Brassica napus. BMC Genomics 16:512. 10.1186/s12864-015-1735-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Routaboul J. M., Kerhoas L., Debeaujon I., Pourcel L., Caboche M., Einhorn J., et al. (2006). Flavonoid diversity and biosynthesis in seed of Arabidopsis thaliana. Planta 224, 96–107. 10.1007/s00425-005-0197-5 [DOI] [PubMed] [Google Scholar]
- Saito K., Yonekura-Sakakibara K., Nakabayashi R., Higashi Y., Yamazaki M., Tohge T., et al. (2013). The flavonoid biosynthetic pathway in Arabidopsis: structural and genetic diversity. Plant Physiol. Biochem. 72, 21–34. 10.1016/j.plaphy.2013.02.001 [DOI] [PubMed] [Google Scholar]
- Sankoff D., Zheng C., Zhu Q. (2010). The collapse of gene complement following whole genome duplication. BMC Genomics 11:313. 10.1186/1471-2164-11-313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid M., Davison T. S., Henz S. R., Pape U. J., Demar M., Vingron M., et al. (2005). A gene expression map of Arabidopsis thaliana development. Nat. Genet. 37, 501–506. 10.1038/ng1543 [DOI] [PubMed] [Google Scholar]
- Schranz M. E., Lysak M. A., Mitchell-Olds T. (2006). The ABC's of comparative genomics in the Brassicaceae: building blocks of crucifer genomes. Trends Plant Sci. 11, 535–542. 10.1016/j.tplants.2006.09.002 [DOI] [PubMed] [Google Scholar]
- Song X., Li Y., Hou X. (2013). Genome-wide analysis of the AP2/ERF transcription factor superfamily in Chinese cabbage (Brassica rapa ssp. pekinensis). BMC genomics 14:573. 10.1186/1471-2164-14-573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein A., Wittkop B., Liu L., Obermeier C., Friedt W., Snowdon R. J. (2013). Dissection of a major QTL for seed colour and fibre content in Brassica napus reveals colocalization with candidate genes for phenylpropanoid biosynthesis and flavonoid deposition. Plant Breed. 132, 382–389. 10.1111/pbr.12073 [DOI] [Google Scholar]
- Sun Y., Wang C., Yang B., Wu F., Hao X., Liang W., et al. (2014). Identification and functional analysis of mitogen-activated protein kinase kinase kinase (MAPKKK) genes in canola (Brassica napus L.). J. Exp. Bot. 65, 2171–2188. 10.1093/jxb/eru092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K., Stecher G., Peterson D., Filipski A., Kumar S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. 10.1093/molbev/mst197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z., Li J., Zhang X., Chen L., Wang R. (1997). Genetic variation of yellow-seeded rapeseed lines (Brassica napus L.) from different genetic sources. Plant Breed. 116, 471–474. 10.1111/j.1439-0523.1997.tb01033.x [DOI] [Google Scholar]
- Thamilarasan S. K., Park J.-I., Jung H.-J., Nou I.-S. (2014). Genome-wide analysis of the distribution of AP2/ERF transcription factors reveals duplication and CBFs genes elucidate their potential function in Brassica oleracea. BMC Genomics 15:422. 10.1186/1471-2164-15-422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theander O., Aman P., Miksche G. E., Yasuda S. (1977). Carbohydrates, polyphenols, and lignin in seed hulls of different colors from turnip rapeseed. J. Agric. Food Chem. 25, 270–273. 10.1021/jf60210a042 [DOI] [Google Scholar]
- Town C. D., Cheung F., Maiti R., Crabtree J., Haas B. J., Wortman J. R., et al. (2006). Comparative genomics of Brassica oleracea and Arabidopsis thaliana reveal gene loss, fragmentation, and dispersal after polyploidy. Plant Cell Online 18, 1348–1359. 10.1105/tpc.106.041665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan L., Xia Q., Qiu X., Selvaraj G. (2002). Early stages of seed development in Brassica napus: a seed coat-specific cysteine proteinase associated with programmed cell death of the inner integument. Plant J. 30, 1–10. 10.1046/j.1365-313X.2002.01262.x [DOI] [PubMed] [Google Scholar]
- Wang X., Wang H., Wang J., Sun R., Wu J., Liu S., et al. (2011). The genome of the mesopolyploid crop species Brassica rapa. Nat. Genet. 43, 1035–1039. 10.1038/ng.919 [DOI] [PubMed] [Google Scholar]
- Wei Y. L., Li J. N., Lu J., Tang Z. L., Pu D. C., Chai Y. R. (2007). Molecular cloning of Brassica napus TRANSPARENT TESTA 2 gene family encoding potential MYB regulatory proteins of proanthocyanidin biosynthesis. Mol. Biol. Reports 34, 105–120. 10.1007/s11033-006-9024-8 [DOI] [PubMed] [Google Scholar]
- Winkel-Shirley B. (2001). Flavonoid biosynthesis. A colorful model for genetics, biochemistry, cell biology, and biotechnology. Plant Physiol. 126, 485–493. 10.1104/pp.126.2.485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkel-Shirley B. (2002). Biosynthesis of flavonoids and effects of stress. Cur. Opin. Plant Biol. 5, 218–223. 10.1016/S1369-5266(02)00256-X [DOI] [PubMed] [Google Scholar]
- Wu G., Zhang L., Wu Y., Cao Y., Lu C. (2010). Comparison of five endogenous reference genes for specific PCR detection and quantification of Brassica napus. J. Agric. Food Chem. 58, 2812–2817. 10.1021/jf904255b [DOI] [PubMed] [Google Scholar]
- Xie D. Y., Sharma S. B., Paiva N. L., Ferreira D., Dixon R. A. (2003). Role of anthocyanidin reductase, encoded by BANYULS in plant flavonoid biosynthesis. Science 299, 396. 10.1126/science.1078540 [DOI] [PubMed] [Google Scholar]
- Xu B. B., Li J. N., Zhang X. K., Wang R., Xie L. L., Chai Y. R. (2007). Cloning and molecular characterization of a functional flavonoid 3′-hydroxylase gene from Brassica napus. J. Plant Physiol. 164, 350–363. 10.1016/j.jplph.2006.03.001 [DOI] [PubMed] [Google Scholar]
- Yu C. Y. (2013). Molecular mechanism of manipulating seed coat coloration in oilseed Brassica species. J. Appl. Genet. 54, 135–145. 10.1007/s13353-012-0132-y [DOI] [PubMed] [Google Scholar]
- Zhang J., Lu Y., Yuan Y., Zhang X., Geng J., Chen Y., et al. (2009). Map-based cloning and characterization of a gene controlling hairiness and seed coat color traits in Brassica rapa. Plant Mol. Biol. 69, 553–563. 10.1007/s11103-008-9437-y [DOI] [PubMed] [Google Scholar]
- Zhang K., Lu K., Qu C., Liang Y., Wang R., Chai Y., et al. (2013). Gene Silencing of BnTT10 family genes causes retarded pigmentation and lignin reduction in the seed coat of Brassica napus. PLoS ONE 8:e61247. 10.1371/journal.pone.0061247 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.