

ABSTRACT
The B-cell receptor (BCR) expressed by a clonal B cell tumor is a tumor specific antigen (idiotype). However, the T-cell epitopes within human BCRs which stimulate protective immunity still lack detailed characterization. In this study, we identified 17 BCR peptide-specific CD4+ T-cell epitopes derived from BCR heavy and light chain variable region sequences. Detailed analysis revealed these CD4+ T-cell epitopes stimulated normal donors' and patients' Th1 CD4+ T cells to directly recognize the autologous tumors by secretion of IFNγ, indicating the epitopes are processed and presented by tumor cells. One BCR peptide-specific CD4+ T cell line was also cytotoxic and lysed autologous tumor cells through the perforin pathway. Sequence analysis of the epitopes revealed that 10 were shared by multiple primary patients' tumors, and 16 had the capacity to bind to more than one HLA DRB1 allele. T cells stimulated by shared epitopes recognized primary tumors expressing the same sequences on multiple HLA DRB1 alleles. In conclusion, we identified 17 BCR-derived CD4+ T-cell epitopes with promiscuous HLA DRB1 binding affinity that are shared by up to 36% of patients, suggesting a strategy to overcome the requirement for individual preparation of therapeutic agents targeting idiotype.
KEYWORDS: B-cell receptor, idiotype, immunotherapy, T cells
Introduction
In 2016, it is predicted that 130,370 new cases of B-cell malignancies including chronic lymphocytic leukemia (CLL), multiple myeloma and lymphoma will be diagnosed and 38,580 people will die of these diseases.1 The mainstay of therapy for these diseases has been chemotherapy with or without rituximab. Despite improvements, only a minority of patients achieve long-term disease control.2 Thus, novel strategies, such as immunotherapy, are still needed to eliminate the residual disease and prolong the disease-free survival of patients after standard treatment. Current tumor immunotherapy has predominantly focused on the induction of CD8+ T cells in response to a tumor vaccine, or adoptive transfer of tumor antigen-specific CD8+ T cells.3 However, tumor cells have developed complicated mechanisms to evade the immune surveillance by CD8+ T cells.4 Clinical trials with tumor antigen-specific CD8+ T cells have had limited success, indicating that CD8+ T cells alone are not enough to eliminate the tumor cells, and the success of tumor immunotherapy requires new strategies to orchestrate the targeting of the tumor cells by different immune cells.5
The B-cell receptor (BCR) expressed by a clonal B cell tumor is a tumor specific antigen.6 Idiotype (Id) refers to the highly variable region of the BCR that is expressed by clonal malignant B cells. Active vaccination with idiotype proteins has been shown to induce protective immune responses in many human and animal models.7,8 In a recently completed randomized, double blind, multicenter phase III clinical trial, idiotype vaccination administered to patients assessed as having minimal residual disease (MRD), improved disease-free survival of follicular lymphoma (FL) patients, providing proof of the principle that therapeutic vaccines against Id targets can improve clinical outcomes.9 However, a major limitation of idiotype vaccines is the requirement of a custom-made product for each patient, making the production of the vaccine laborious and time-consuming. One strategy to circumvent this requirement would be to fully characterize T-cell epitopes among tumor-derived idiotype or BCR sequences to identify potentially shared epitopes expressed by multiple patients.
In this study, we used peptide strategy and identified 17 CD4+ T-cell epitopes from BCR heavy and light chains. T cells stimulated by these epitopes specifically recognized the autologous tumors expressing the same sequences by secreting a large amount of interferon gamma (IFNγ), indicating the epitopes are processed and presented by tumors. One BCR peptide-specific CD4+ T cell line directly lysed the tumor cells though the perforin pathways, indicating it is cytotoxic. Sequence analysis revealed 10 CD4+ T-cell epitopes we identified are shared by patients and 16 of the BCR peptide-specific CD4+ T-cell epitopes have the capacity to bind to more than one HLA DRB1 allele. T cells stimulated by shared T-cell epitopes recognized the tumor expressing the same sequences on several HLA DRB1 alleles. Our study revealed a strategy to overcome the limitation of individual preparation of idiotype vaccines and provided a basis for the future development of novel clinical immunotherapies against B-cell malignancies through CD4+ T-cell therapy.
Materials and methods
Normal donors and patient samples
The Institutional Review Board of The University of Texas MD Anderson Cancer Center approved the study. Peripheral blood mononuclear cells (PBMCs) of normal donors were obtained from the Gulf Coast Regional Blood Center near the Texas Medical Center. An informed consent was obtained prior to the collection of blood and tissue samples, in accordance with the Declaration of Helsinki. The PBMCs were isolated from blood samples by density gradient separation. Tissue samples from lymphoma patients were processed into single-cell suspension and cryopreserved in aliquots. Tumor cells were isolated with B cell negative isolation kit from Miltenyi Biotech, Inc., sorted out with anti-Lambda or anti-Kappa antibodies and labeled as tumor Ig light chain (+) B cells.10,11 The normal B cells were sorted as the tumor Ig light chain (−) B cells at same time. Monocytes were isolated with CD14 and MHC class II antibody. Tumor-free PBMCs were prepared by CD19 beads depletion. The purity was confirmed by flow cytometry.
Reagents
The complete T-cell medium included RPMI 1640 medium supplemented with 10% fetal bovine serum, 10 mM HEPES, 1× Glutamax, 50 μM mercaptoethanol, 1 mM sodium pyruvate, and 10 μg/mL gentamicin (all reagents from Invitrogen, Carlsbad, CA). Mouse anti-human CD4+ (APC-H7), CD8+ (PE-H7), CD14 (APC), MHC II (FITC), IFNγ (AF 700), TNFα (FITC), GM-CSF (PerCP-Cy5.5), IL-4 (APC), IL-9 (PE), IL-10 (Texas-red),and IL-17 (Pacific blue) were obtained from BD Biosciences, San Jose, CA. Anti-human Ig Lambda (V450), Ig Kappa (PE cy7) were from eBiosciences. Enzyme-linked immunosorbent assay (ELISA) kits for IFNγ were obtained from R&D Systems, Minneapolis, MN. BCR framework region peptides or 15-mer overlapping peptides were synthesized by NEO Group, Inc. or Sigma-Aldrich, St. Louis, MO to greater than 90% purity and dissolved in dimethyl sulfoxide (Sigma-Aldrich). Anti-human HLA-ABC (clone W6/32, eBioscience, San Diego, CA); anti-HLA-DP, DQ, DR (clone T⇐39, BD Biosciences); anti-HLA DR (clone G46-6, BD Biosciences), anti-HLA DP (Cat# SC-53308, Santa cruz), anti-HLA DQ (clone Tu169, BD Biosciences); and mouse IgG2a isotype control (eBioscience, Cat# 16-4724-81) were used for HLA-blocking studies as described before.12
Generation of BCR peptide-specific CD4+ T cells
BCR peptide-specific T-cell lines were generated from normal donors or patients, using methods previously reported.12 Briefly, PBMCs (1 × 105 cells/well) were stimulated with 10 μg/mL of each peptide in quadruplicate in a 96-well, U-bottom-microculture plate (Corning Inc., Lowell, MA) in 200 μL of culture medium. The culture medium consisted of 50% AIM-V medium (Invitrogen), 50% RPMI 1640 medium (Invitrogen), 10% human AB serum (Valley Biomedical, Winchester, VA), and 100 IU/mL of interleukin-2 (IL-2). Cells were restimulated with the corresponding peptide every 3 d. After five stimulations, T cells from each well were washed and incubated with autologous tumor free PBMCs in the presence or absence of the corresponding peptide. After 18 h, the production of IFNγ in the supernatants was determined by ELISA. T cells that secreted large amounts of IFNγ were further expanded by Rapid Expansion Protocol (REP) to obtain sufficient number of cells for additional functional assays.13
Intracellular cytokine staining assay
Effector T cells were mixed with autologous antigen-presenting cells (APCs, CD3-depleted PBMCs) loaded with 10 µg/mL of peptides at a 1:1 ratio. Two hours later, 5 µg/mL of Brefeldin A (Sigma) was added to block the transfer of Golgi part. The detection of the intracellular cytokine was performed with a BD Cytofix/CytopermTM Plus Fixation/Permeabilization kit 12 h later. Once staining with CD4+ or CD8+ antibodies on the cell surface was completed, the T cells were washed, fixed and permeabilized and stained with 10 µL of mouse anti-human IFNγ, TNFα, GM-CSF, IL-4, IL-10, IL-17, and IL-9 for 30 min at 4°C in the dark. After washing two times in 1× Perm buffer, the samples were analyzed by flow cytometry and the data was analyzed with cell Quest software or Flowjo.
Mapping of the binding affinity of BCR peptides to MHC alleles
Epstein–Barr virus (EBV)-transformed lymphoblastoid B-cell lines (BLCLs) were obtained from International Histocompatibility Working Group (IHWG). To map the binding affinity of peptides, the BLCLs of different HLA DRB1 alleles were pulsed, or not-pulsed with BCR peptides for 2 h and then incubated with peptide-specific T cells for overnight. After 18 h, the production of IFNγ was determined in the supernatants by ELISA. The information of HLA DRB1 alleles on the BLCLs is listed in Table S1.
Cytotoxicity assay
APCs were cultured overnight with 40 µg/mL of peptides in the presence of 3 µg/mL of β2-microglobulin, washed twice, labeled with chromium-51 (51Cr) for 1 h at 37°C, and used as targets in a cytotoxicity assay. Primary tumor cells were isolated from PBMCs or biopsy samples of lymphoma patients with a B-cell isolation kit prior to labeling with 51Cr. Tumor free PBMCs were collected after the CD19+ cells depletion. Target cells (1×104 cells/well) were then incubated with the effector T cells at the indicated ratios in 96-well round-bottom plates at 37°C for 4 h, and target cell lysis was determined by chromium release into the supernatants. Spontaneous lysis was determined from the supernatant of target cells cultured without effector cells, and the maximal lysis was determined from the supernatant of target cells cultured with 1% Triton X-100. The specific lysis of target cells was calculated as follows: (Experimental lysis−Spontaneous lysis)×100/(Maximal lysis−Spontaneous lysis). For HLA-blocking studies, target cells were incubated for 30 min with 20 µg/mL of anti-HLA blocking antibodies prior to co-culturing with the effector T cells. All assays were performed in triplicate wells and repeated at least three times.
Statistical analysis
Student's t-test was used to compare various experimental groups. p values <0.05 were considered statistically significant. Unless otherwise indicated, means and standard deviations are shown.
Results
Generation of Th1 CD4+ T cell lines against the BCR with peptides from patients
In order to see if BCR T-cell epitopes can stimulate the proliferation of CD4+ T cells from patients' autologous PBMCs, we synthesized 134 15-mer overlapping peptides that corresponded to the heavy and light chains of three lymphoma patients' BCRs with known HLA DR alleles (Table S2). Then we used these peptides to stimulate the patients' autologous PBMCs. We succeeded in generating five BCR peptide-specific T cells from the three patients that specifically secreted a large amount of IFNγ upon incubation with autologous tumor-free PBMCs pulsed with peptides (Fig. 1A). An intracellular staining assay revealed Th1 CD4+ T cells specifically secreted a lot of IFNγ, TNFα, GM-CSF, but not IL-4, IL-10, IL-17, IL-9 (Figs. 1B and S1).
Figure 1.

Generation of Th1 CD4+ T-cell lines against BCR overlapping peptides from autologous lymphoma patients. (A) IFNγ ELISA assay of autologous CD4+ T cells stimulated with autologous PBMCs, pulsed or nonpulsed with patient-derived 15-mer BCR overlapping peptides. Briefly, PBMCs (1 × 105 cells/well) were stimulated with 10 μg/mL of each peptide in a 96-well, U-bottom-microculture plate every 3 d. After five stimulations, T cells from each well were washed and incubated with PBMCs in the presence or absence of the corresponding peptide. The production of interferon (IFN)γ was determined in the supernatants by ELISA after 18 h. (B) Intracellular cytokine staining of autologous BCR peptide-specific CD4+ T cells stimulated by APCs, pulsed or nonpulsed with peptides. (C) Blocking of IFNγ production by autologous BCR peptide-specific CD4+ T cells by HLA antibodies. (D) Recognition of autologous tumor by BCR peptide-specific CD4+ T cells. Data are representative of three individual experiments. FL, follicular lymphoma; SMZL, splenic marginal zone B-cell lymphoma.
Figure 1.

(Continued)
We also performed an HLA antibody blocking assay and found that anti-HLA DR, but not anti-HLA DQ, and DP antibodies can successfully block the recognition of BCR peptides by T cells, indicating the peptides bind to HLA DR alleles (Fig. 1C). In order to see if the epitopes we identified are processed and presented by autologous tumor cells, we incubated the autologous BCR peptide-specific CD4+ T cells with autologous tumors. We found that these BCR peptide-reactive CD4+ T cells secreted a large amount of IFNγ upon incubation with the autologous tumor Ig light chain (+) cells, whereas the response to the autologous tumor Ig light chain (−) normal B cells or monocytes was lower. This indicates that the BCR T-cell epitopes can stimulate CD4+ T cells that recognize the autologous tumor cells more efficiently than normal cells (Figs. 1D and S2).
Generation of one cytotoxic CD4+ T cell line against the BCR with peptide from a PL patient
In one plasma cell leukemia (PL) patient, we stimulated the autologous patients' PBMCs with the 9-mer or 10-mer peptides, previously designed to stimulate MHC class I restricted T cells (Table S3). We generated one T cell line that specifically secreted a large amount of IFNγ when cultured with peptide-pulsed PBMCs (Fig. 2A). However, the intracellular cytokine staining assay revealed, that the peptide (PL1VK12: YLAWYQQKPG)–stimulated T cells are CD4+, but not CD8+, T cells that specifically secreted the IFNγ (Fig. 2B). The CD4+ T cells specifically secreted a lot of IFNγ, TNFα, and GM-CSF, but not IL-4, IL-10, IL-17, or IL-9 (Fig. 2C). The recognition of peptides by the T cells was also blocked by HLA DR and HLA class II, but not HLA DP, and DQ antibodies, indicating the peptide binds to HLA DR allele (Fig. 2D). Intracellular staining revealed that the PL1VK12 peptide-specific CD4+ T cells expressed high levels of perforin, and granzyme B and A, indicating they are cytotoxic (Fig. 2E). Incubation of the CD4+ T cells with APCs pulsed with peptides revealed that the CD4+ T cells exerted a specific cytotoxic function against the PL1VK12 peptide-pulsed APCs, but not APCs pulsed with control peptide or APCs alone, confirming that the CD4+ T cells are cytotoxic (Fig. 2F). To analyze the cytotoxic mechanism of the CD4+ T cells, we performed an HLA antibody inhibition assay. We found that HLA class II and HLA DR, but not HLA class I, antibodies significantly blocked the lysis of PL1VK12 peptide-pulsed APCs by the CD4+ T cells (Fig. 2G). Incubation of T cells with perforin inhibitor concanamycin A (CMA) also significantly inhibited the killing capacity of T cells, indicating that the CD4+ T cells mediated the killing through the perforin signaling pathway. Finally, we incubated the T cells with the tumor cells derived from the autologous patient. We found that the CD4+ T cells specifically lysed the autologous tumor Ig light chain (+) cells, but not autologous tumor-free PBMCs, indicating that the CD4+ T cell epitope was processed and presented by the tumor cells (Fig. 2H). Altogether, these results indicate that we have identified a T-cell epitope in the BCR that can stimulate cytotoxic CD4+ T cells, which specifically target the autologous tumor through the perforin and HLA DR signaling pathways.
Figure 2.

Generation of one cytotoxic CD4+ T cell line against the BCR peptide from a PL patient. (A) IFNγ ELISA assay of autologous CD4+ T cells stimulated with autologous PBMCs, pulsed or nonpulsed with patient-derived peptide. (B, C) Intracellular staining of cytokines secretion by autologous cytotoxic CD4+ T cells versus APCs, pulsed or nonpulsed with peptide. (D) Blocking of IFNγ production by autologous BCR peptide-specific CD4+ T cells with HLA antibodies. (E) Intracellular staining of Granzyme A, B, Perforin, and FasL of autologous cytotoxic CD4+ T cells. (F) Cytotoxicity of autologous CD4+ T cells against APCs pulsed with PL1VK12, control peptide, or APCs alone, and (G) in the presence or absence of HLA class I and II, HLA DR antibodies, and CMA inhibitor. (H) Cytotoxicity of autologous CD4+ T cells against autologous tumor and tumor-free PBMCs as negative controls. PL, plasma cell leukemia.
Generation of Th1 CD4+ T cell lines against BCR framework regions with peptides from normal donors
The framework region of BCRs are shared by the idiotype antigens.14,15 In order to see if the framework regions contained CD4+ T-cell epitopes, we first synthesized 326 peptides (Table S4) that correspond to the whole sequence of FR1(framework region 1), FR2, and FR3 of B-cell receptor subfamilies, based on data from the international ImMuno GeneTics information system (IMGT, http://www.imgt.org/IMGTrepertoire/Protein/index.php). Then, we used these peptides to stimulate four normal donors' PBMCs with known HLA DRB1 alleles (Table S1). We succeeded in generating five T-cell lines that are BCR framework region-specific. The stimulated T cells specifically secreted a large amount of IFNγ when incubated with normal donors' PBMCs pre-loaded with BCR framework region peptides (Fig. 3A). An intracellular staining assay revealed that the BCR framework region-specific T cells are Th1 CD4+ T cells, which specifically secreted a large amount of IFNγ, TNFα, and GM-CSF, but not IL-4, IL-10, IL-9, or IL-17 (Figs. 3B S3). The HLA antibody blocking assay revealed that HLA class II and HLA DR, but not HLA class I, HLA DQ, or HLA DP antibodies can successfully block the activity of T cells, indicating that these peptides are bound to HLA DR alleles (Fig. 3C).
Figure 3.

Generation of Th1 CD4+ T-cell lines against BCR framework region peptides from normal donors. (A) IFNγ ELISA assay of donor T cells stimulated with PBMC which have known HLA DRB1 alleles as antigen-presenting cells (APCs), pulsed or nonpulsed with BCR framework region peptides. (B) Intracellular cytokine staining of BCR framework region-specific CD4+ T cells stimulated by normal donor APCs, pulsed or nonpulsed with peptides. (C) Blocking of IFNγ production by BCR framework region-specific CD4+ T cells by HLA antibodies. (D) Identification of 15-mer minimal T-cell epitopes by BCR framework region-specific CD4+ T cells by IFNγ ELISA assay. Data are shown as mean value of triplicate wells ± SEM and are representative of three individual experiments.
Figure 3.

(Continued)
Since the peptides derived from the BCR framework regions (FR1, FR2, and FR3) are 17-38 amino acids-long, to map the minimal epitopes, we put the sequences of the five positive BCR framework region peptides into the Immune Epitope Database Analysis Resource (IEDB, http://tools.immuneepitope.org/analyze/html/mhc_II_binding.html) and predicted 58 15-mer peptides to bind to the donors' HLA DRB1 alleles (Table S5). Then, we incubated the T cells with the donors' PBMCs pulsed with these 15-mer minimal peptides. We found the D173 VH P106 T cells recognized four 15-mer peptides (VH P106-2, 4, 8, 11) derived from the VH P106 peptides, whereas D30 VK P14 T cells recognized one 15-mer peptide (VK P14-5); D126 VH P121 T cells recognized two 15-mer peptides (VH P121-3,10); D126 VH P125 T cells recognized one 15-mer peptide (VH P125-13); and the D30 VH P60 T cells recognized three 15-mer peptides ( VH P60-1,2,3) (Fig. 3D). In total, we have identified 11 15-mer peptides derived from the BCR framework region that can be recognized by CD4+ T cells we generated from normal donors, indicating that they are immunogenic.
Recognition of primary human tumors expressing “shared” T-cell epitopes by BCR peptide-specific CD4+ T cells
To determine whether the CD4+ T-cell epitopes we have identified may be shared among different B-cell tumors, we blasted these 17 CD4+ T-cell epitopes with the FL BCR sequence database.12 We found that VHP106-2 and 4 are shared by 2 out of 64 patients with BCR heavy chain (HC); VHP106-8 is shared by 4 out of 64 patients (6.3%) with HC; VHP106-11 is shared by 3 out of 64 (4.6%) patients with HC; VHP121-10 is shared by 5 out of 64 (6.3%) patients with HC; SMZL1VL1 is shared by 7 out of 90 patients (7.8%) with lambda chain (LC); and PL1VK12 is shared by 20 out of 123 (16.3%) patients with kappa chain (KC). In total, the 17 CD4+ T-cell epitopes we have identified can be shared by 36% of patients with BCR HCs, 16.3% of patients with KC, and 7.8% of patients with LC (Table 1).
Table 1.
Shared specificity and characteristics of BCR CD4+ T-cells epitopes.
| Types | Peptides | Subfamily and regiona | Sequence | Length | HLA DR binding | Sharedb | Shared (%) | Total shared (%) |
|---|---|---|---|---|---|---|---|---|
| Normal donors | VH P60-1 | IGHV3-16FR2 | MNWARKAPGKGLEWV | 15 | 1101, 0301 | 0/64 | 0.0 | VH 23/64 (36%) |
| VH P60-2 | IGHV3-16FR2 | NWARKAPGKGLEWVS | 15 | 1101, 0301 | 0/64 | 0.0 | ||
| VH P60-3 | IGHV3-16FR2 | WARKAPGKGLEWVSG | 15 | 1101, 0301 | 0/64 | 0.0 | ||
| VH P106-2 | IGHV3-11FR3 | KNSLYLQMNSLRAED | 15 | 0401, 1101, 0701 | 2/64(VH) | 3.1 | ||
| VH P106-4 | IGHV3-11FR3 | SLYLQMNSLRAEDTA | 15 | 0401, 1101, 0701 | 2/64(VH) | 3.1 | ||
| VH P106-8 | IGHV3-11FR3 | GRFTISRDNAKNSLY | 15 | 0401, 1101, 0701 | 4/64(VH) | 6.3 | ||
| VH P106-11 | IGHV3-11FR3 | QMNSLRAEDTAVYYC | 15 | 0401, 1101, 0701 | 3/64(VH) | 4.6 | ||
| VH P121-3 | IGHV3-74FR3 | AKNTLYLQMNSLRAE | 15 | 0301, 1101 | 0/64 | 0.0 | ||
| VHP121-10 | IGHV3-74FR3 | DSVKGRFTISRDNAK | 15 | 0301, 1101 | 5/64(VH) | 7.8 | ||
| VHP125-13 | IGHV4-30-2FR3 | FSLKLSSVTAADTAV | 15 | 0101, 0301 | 2/64(VH) | 3.1 | ||
| Patients | FL1VH17 | IGHV4-34FR3 | SRLTISVDTSKNQFS | 15 | 0701, 1501, 0801,101 | 0/64 | 0.0 | |
| FL2VH3 | IGHV4 FR1 | PGLVKPSETLSLTCT | 15 | 1101, 0701, 0801,0101,1 501,1301 | 3/64(VH) | 4.7 | ||
| FL2VH4 | IGHV4CDR1 | KPSETLSLTCTVSGG | 15 | 1101, 0701,0801,0101, 1501,1401 | 2/64 (VH) | 3.1 | ||
| FL1VL5 | IGLV1CDR1 | RVTISCSGSTSNIGS | 15 | 0701 | 0/90 | 0.0 | VL 7/90 (7.8%) | |
| SMZL1VL1 | IGLV1FR1 | QSVLTQPPSASGTPGQ | 16 | 0301, 0401 | 7/90(VL) | 7.8 | ||
| PL1VK12 | IGKV3 CDR1 | YLAWYQQKPG | 10 | 0701,1401,0801 | 20/123 (VK) | 16.3 | VK 20/123 (16.3%) | |
| Normal donors | VK P14-5 | IGKV2-24FR1 | QTPLSSPVTLGQPAS | 15 | 1101, 0801, 0701,1301 | 0/123 | 0.0 |
Analysis and blast by IMGT Web site (http://imgt.cines.fr/IMGT_vquest/vquest?livret=0&Option=humanIg).
Individual peptide sequences were compared with Ig V region sequences available from either 64 FL patients with VH chain, 90 FL patients with l light chain, or 123 patients with k light chain isotype, respectively.
To see if the T cells generated against the shared CD4+ T-cell epitopes can recognize the tumors expressing the “shared” sequence, we incubated the BCR peptide-specific CD4+ T cells with tumors expressing the “shared” Id sequence and HLA DRB1 alleles (D173 VH P106 T cells against MCL1-Tu, HLA DRB1: 0401/0101; D126 VH P121 T cells against MCL2-Tu HLA DRB1: 101/1501). We found that these BCR peptide-reactive CD4+ T cells secreted a large amount of IFNγ upon incubation with the autologous tumor Ig light chain (+) cells, whereas the response to the autologous tumor Ig light chain (−) normal B cells or monocytes was lower. This indicates that the BCR T-cell epitopes can stimulate CD4+ T cells that recognize the autologous tumor cells more efficiently than normal cells (Figs. 4A and S4). The same was found for the autologous CD4+ T cell generated from patients (FL2VH4 T cells against MCL3-Tu, HLA DRB1 1101/0401; FL1VH17 T cells against MCL4-Tu, HLA DRB1: 0701/1501; PL1 VK12 T cells against MCL5-Tu, HLA DRB1: 0401/0701, MCL6-Tu, HLA DRB1: 0701/1501) (Fig. 4B and C).
Figure 4.
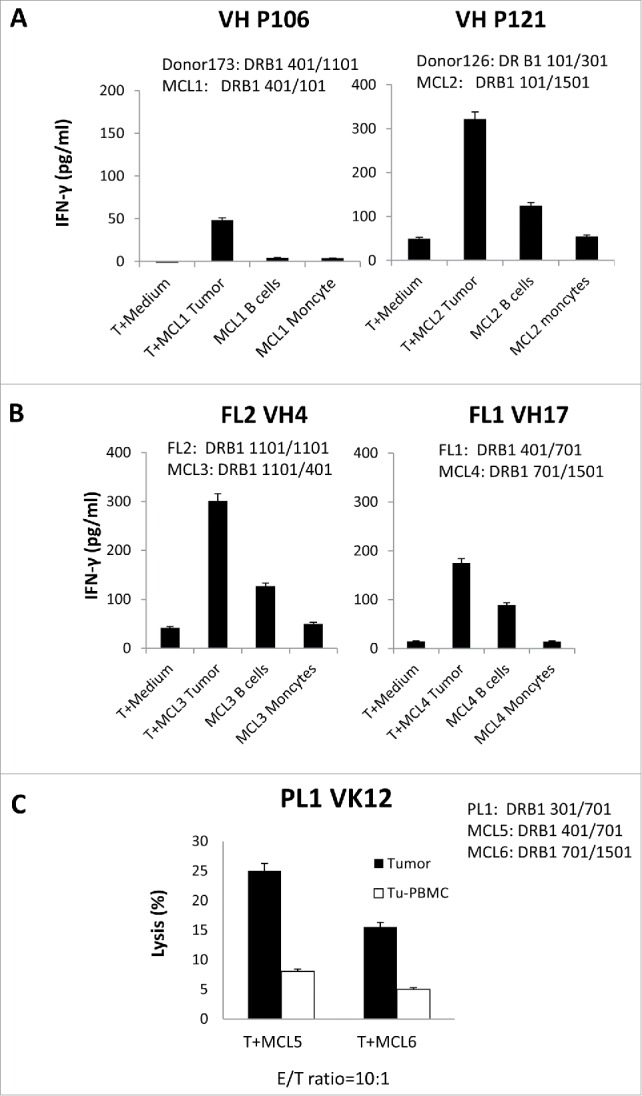
Recognition of primary human tumors expressing shared T-cell epitopes by BCR peptide-specific CD4+ Tcells. (A) IFNγ ELISA assay of allogeneic or (B) autologous BCR peptide-specific CD4+ T cells stimulated with mantle cell lymphomas (MCL) expressing the same epitopes and HLA DRB1 alleles. (C) Cytotoxic assay of autologous BCR peptide-specific CD4+ T cells stimulated with mantle cell lymphomas (MCL) expressing the same epitopes and HLA DRB1 alleles. Data are representative of three individual experiments.
Promiscuous binding of BCR CD4+ T-cells epitopes to HLA DRB1 alleles
It is known that HLA-DRB1 alleles have overlapping binding capacity.16 In order to see if the BCR CD4+ T-cell epitopes we identified can bind to other HLA DRB1 alleles, we put the 17 BCR CD4+ T-cell epitopes into the online MHC class II binding prediction program from IEDB analysis Resources (http://tools.immuneepitope.org/analyze/html/mhc_II_binding.html) and selected the 10 most frequent HLA DRB1 alleles (HLA DRB1 0101–HLA DRB1 1501). We found that 16 of these CD4+ T-cell epitopes were predicted to bind to more than one HLA DRB1 allele with high to modest affinity (low percentile rank) (Table 2). The PL1VK12 peptide, a 10-mer, was not predicted by the program due to its short length.
Table 2.
Predicted binding affinity of 16 CD4+ T-cell epitopes to 10 HLA DRB1 alleles.
| Name | Subfamilies | Sequence | HLA-DRB1*01:01 (18.5%) | HLA-DRB1*15:01 (19.9%) | HLA-DRB1*03:01 (17.7%) | HLA-DRB1*04:01 (23.6%) | HLA-DRB1*07:01 (26.2%) | HLA-DRB1*08:01 (5.5%) | HLA-DRB1*09:01 (3.6%) | HLA-DRB1*11:01 (17.0%) | HLA-DRB1*12:01 (2.8%) | HLA-DRB1*13:01 (21.7%) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| VH P60-1 | IGHV3-16FR2 | MNWARKAPGKGLEWV | 38.88 | 47.97 | 25.24 | 49.18 | 36.71 | 0.89 | 9.48 | 8.05 | 74 | 3.23 |
| VH P60-2 | IGHV3-16FR2 | NWARKAPGKGLEWVS | 50.11 | 60.23 | 70.94 | 71.64 | 36.71 | 0.89 | 13.88 | 13.01 | 83.25 | 18.13 |
| VH P60-3 | IGHV3-16FR2 | WARKAPGKGLEWVSG | 61.86 | 68.73 | 70.94 | 62.65 | 36.71 | 0.89 | 27.91 | 20.27 | 85.79 | 18.13 |
| VH P106-2 | IGHV3-11FR3 | KNSLYLQMNSLRAED | 0.39 | 2.16 | 7.67 | 0.1 | 37.22 | 1.96 | 7.39 | 0.38 | 7.3 | 0.71 |
| VH P106-4 | IGHV3-11FR3 | SLYLQMNSLRAEDTA | 0.19 | 2.71 | 7.67 | 0.11 | 37.22 | 1.96 | 11.81 | 0.38 | 13 | 0.71 |
| VH P106-8 | IGHV3-11FR3 | GRFTISRDNAKNSLY | 65.22 | 34.5 | 2.99 | 3.53 | 52.05 | 23.02 | 43 | 19.36 | 46.16 | 16.46 |
| VH P106-11 | IGHV3-11FR3 | QMNSLRAEDTAVYYC | 20.92 | 40.81 | 5.24 | 6.19 | 63.43 | 5.97 | 61.16 | 20.55 | 50.67 | 4.25 |
| VH P121-3 | IGHV3-74FR3 | DSVKGRFTISRDNAK | 54.79 | 54.79 | 39.34 | 6.71 | 7.12 | 16.35 | 4.16 | 59.2 | 7.59 | 53.21 |
| VHP121-10 | IGHV3-74FR3 | AKNTLYLQMNSLRAE | 0.39 | 2.11 | 7.67 | 0.11 | 26.46 | 1.96 | 8.48 | 0.38 | 5.21 | 0.71 |
| VHP125-13 | IGHV4-30-2FR3 | FSLKLSSVTAADTAV | 8.78 | 14.94 | 3.66 | 2.04 | 5.34 | 5.55 | 8.2 | 7.1 | 28.52 | 5.9 |
| FL1VH17 | IGHV4-34FR3 | SRLTISVDTSKNQFS | 68.73 | 36.88 | 10.92 | 3.07 | 37.98 | 21.21 | 54.55 | 22.98 | 55.13 | 26.85 |
| FL2VH3 | IGHV4 FR1 | PGLVKPSETLSLTCT | 51.33 | 39.3 | 2.68 | 8.64 | 16.9 | 12.2 | 7.7 | 15.22 | 51.87 | 5.4 |
| FL2VH4 | IGHV4CDR1 | KPSETLSLTCTVSGG | 43.33 | 62.53 | 27.58 | 5.15 | 52 | 19.9 | 51.3 | 18.71 | 71.13 | 12.28 |
| FL1VL5 | IGLV1CDR1 | RVTISCSGSTSNIGS | 59.47 | 42.81 | 13.15 | 10.42 | 32.15 | 21.21 | 47.18 | 19.99 | 75.94 | 25.15 |
| SMZL1VL1 | IGLV1FR1 | QSVLTQPPSASGTPG | 14.48 | 46.26 | 9.46 | 4.02 | 56.29 | 18.52 | 43.5 | 14.02 | 46.16 | 3.87 |
| VK P14-5 | IGKV2-24FR1 | QTPLSSPVTLGQPAS | 46.41 | 33.88 | 22.89 | 9.98 | 47.24 | 14.03 | 61.16 | 16.7 | 76.48 | 21.91 |
Underlined: The scores of predicted binding affinity of peptides that were significantly low (<10) were underlined.
To confirm our results, we incubated the BCR peptide-specific CD4+ T cells with EBV-transformed lymphoblastoid BLCLs of different HLA DRB1 alleles, pulsed or nonpulsed with BCR peptides. We found that all the BCR CD4+ T-cell epitopes can induce the IFNγ secretion of T cells with multiple peptide-pulsed BLCLs of different HLA DRB1 alleles, except the FL1 VL5 T-cell epitope, which only induced the IFNγ secretion of T cells with HLA DRB1 0701-expressing BLCLs (Fig. 5A and B). This indicates that the epitopes can bind to different HLA DRB1 alleles. Finally, we incubated the T cells with the tumors expressing the “shared” Id sequence on different HLA DR alleles. We found that these BCR peptide-reactive CD4+ T cells secreted a large amount of IFNγ upon incubation with the autologous tumor Ig light chain (+) cells, whereas the response to the autologous tumor Ig light chain (−) normal B cells or monocytes was lower (D126 VH P125 T cells against MCL3-Tu, HLA DRB1: 1101/401; PL1 VK12 T cells against MCL1-Tu, HLA DRB1: 0301/0501). This indicates that the BCRs contain the “shared” T-cell epitopes and CD4+ T cells can recognize them on promiscuous HLA DRB1 alleles (Fig. 5C and D).
Figure 5.
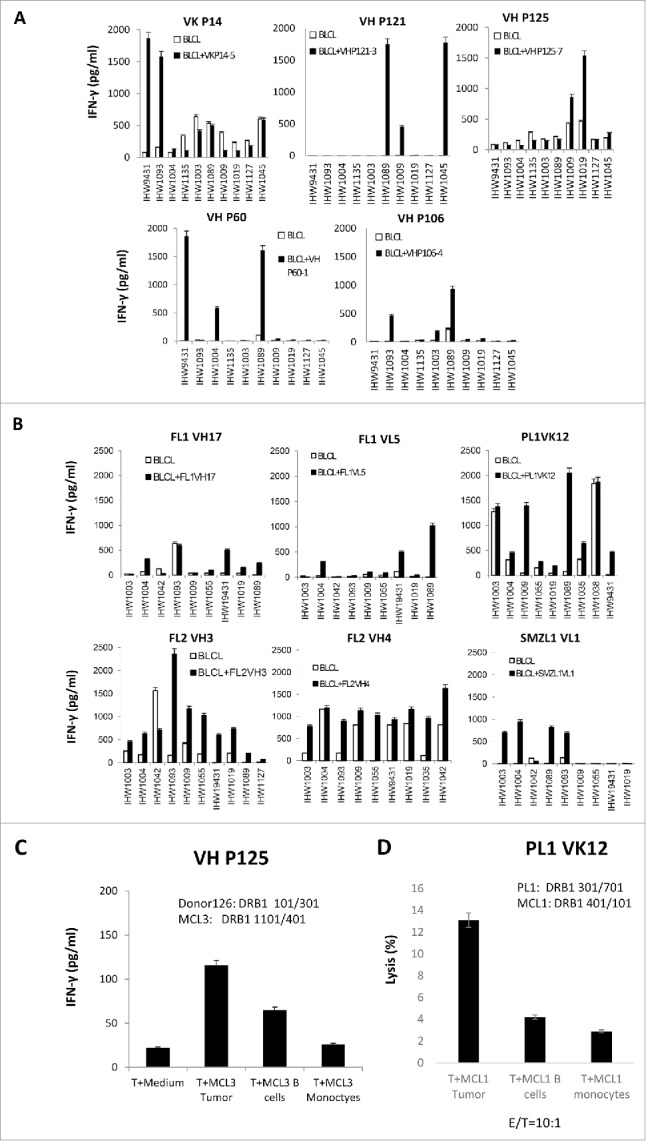
Promiscuous binding of BCR peptides to HLA DRB1 alleles. (A) IFNγ ELISA assay of allogeneic (normal donors) and (B) autologous CD4+ T cells stimulated with B lymphoblastoid cell lines (BLCLs), pulsed or nonpulsed with BCR peptides. (C) IFNγ ELISA assay of BCR framework region-specific CD4+ T cells stimulated with lymphoma expressing the same epitope and mismatched HLA DRB1 alleles. (D) Cytotoxicity (E/T ratio=10:1) of autologous BCR peptide-specific CD4+ T cells against lymphomas or tumor-free PBMCs expressing the same epitope and mismatched HLA DRB1 alleles. Data are representative of three individual experiments.
Discussion
The BCR, also called idiotype in tumors, expressed by a clonal B-cell tumor is a tumor-specific antigen and can stimulate antitumor immune response in both human and animal models.6-8 However, the individual preparation of idiotype tumor vaccine is time-consuming and costly. In this study, we identified 17 BCR peptide-specific CD4+ T cell epitopes from BCR heavy and light chains. T cells stimulated by these epitopes recognized the autologous tumors by IFNγ secretion or cytotoxic activity against tumors, indicating that the epitopes are processed and presented by tumors. Moreover, 10 CD4+ T-cell epitopes that we identified can be “shared” by patients and 16 BCR epitopes that we identified can bind to multiple HLA DRB1 alleles. T cells stimulated by the “shared” epitopes recognized the tumors expressing the same sequence on the same or mismatched HLA DRB1 alleles. Although some of the CD4+ T cells recognizing the epitopes derived from the BCR framework also recognized normal B cells to some extent, our study revealed a strategy to overcome the limitation of individual preparation of idiotype vaccines and provided a basis for the development of a more generic CD4+ T cell immunotherapy in the clinic.
About 50% of lymphomas and myelomas expressed low MHC class I alleles and 20% of them completely lost the expression of MHC class I alleles.17 Thus, immunotherapy with tumor antigen-specific CD8+ T cells alone is not enough and the elimination of the residual tumor cells depends on the orchestration of different immune cells through different strategies. Tumor antigen-specific CD4+ T cells have specific value in this regard.18 Besides Th1 and Th2 T cells, recent studies have identified several new subsets of CD4+ T cells, such as Th17, Treg, Th9, Tfh, and Th22 T cells.19 Tregs are immune-suppressive CD4+ T cells and inhibit antitumor immunity and autoimmunity.20 The Tfh and Th22 tissue-specific CD4+ T cells involved tumor development and are located in the lymphoma node and colon tissue.21,22 The Th9 and Th17 are newly discovered CD4+ T cell subsets that have been reported to play a role in antitumor effects.23,24 Interestingly, the antitumor effects of Th17 and Th9 are heavily dependent on their capacity to secret IFNγ.25,26 Moreover, it is reported that Th17 cells have to convert to Th1 cells to be able to exert an antitumor effect in mice.27 This indicated that the Th1 T cell is ultimately important for the antitumor immunity. There are several mechanisms of Th1 T cells which contribute to the antitumor effects.28 First, Th1 T cells can increase the expression of co-activators on the surface of antigen-presenting cells(APCs) (28). Second, the cytokines secreted by Th1 CD4+ T cells can attract other immune cells to the tumor sites.29 Also, the cytokines secreted by Th1 T cells can target the tumor environment and eliminate antigen-loss tumor variants.30-32 Finally, tumor antigen-specific Th1 T cells can also directly target tumor cells through cytotoxicity, FASL, or senescence induction.33-36 Therefore, the percentage of Th1 CD4+ T cells in the stromal environment is directly correlated with a cancer patient's prognosis.37 Adoptive transfer of tumor antigen-specific Th1 CD4+ T cells mediates a durable tumor remission in humans and provides better protection in mice.38,39 All of these indicate the BCR peptide-specific CD4+ T cells generated in this study may mediate a protective antitumor immunity in the clinical setting.
The framework region of B-cell receptors is “shared” by idiotype antigens.15 To study the immunogenicity of the framework regions, we synthesized framework region peptides that completely cover the FR1, FR2, and FR3 regions and used them to stimulate the T cells from normal donors. Our strategy is based on studies that indicated long peptides are more efficiently endocytosed, processed, and presented by APCs; thus, they are more immunogenic.40-42 Using this strategy, we have identified 11 T-cell epitopes from the BCR framework region that are immunogenic. By incubating the BCR peptide-specific T cells with tumors expressing the T-cell epitopes and HLA DRB1 alleles, we found that the T cells secreted a lot of IFNγ, indicating the epitopes were processed and presented by the tumor cells. Interestingly, the CD4+ T cells we generated in this study can recognize the epitopes on BLCLs of different HLA DRB1 alleles, indicating the promiscuous binding capacity of BCR peptides. It is reported that the anchoring sites of different HLA DRB1 alleles shared a high homology, and peptides with conserved anchoring amino acids can bind to more than one HLA DR allele.43 Our studies of the promiscuous binding capacity of BCR peptides indicate the HLA DRB1-restricted CD4+ T-cell epitopes may have a broader usage in clinic.
CD4+ T cells that possess cytotoxic functions have been described for some time in viral infections, autoimmune disease, and vascular damage.35,36 Recent studies have identified several tumor antigen-specific cytotoxic CD4+ T cells in cancer patients, indicating the possible antitumor function of this subtype of T cells.44,45 It was previously suspected that cytotoxic CD4+ T cells are end terminally differentiated Th1 cells.46 However, recent studies found cytotoxic CD4+ T cells may represent a separate polarized functional subset of CD4+ T cells.36,47-49 The cytotoxic activity of the CD4+ T cells we identified was blocked by MHC class II antibody and CMA, indicating that the cytotoxic CD4+ T cells used the perforin pathway to kill tumors.50 Since lymphoma and myeloma are B-cell tumors, which express both MHC class I and II alleles on the cell surface, the cytotoxic CD4+ T cells we generated may directly target lymphoma and myeloma cells, providing an additional effector mechanism against primary tumors. Our study of BCR peptide-specific CD4+ epitopes, including potentially shared epitopes, may provide a basis for future development of therapeutic strategies against B-cell tumors.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This study was support by the Leukemia and Lymphoma Society Specialized Center of Research Grant #7262-08(LWK), Myeloma SPORE Grant P50CA142509, Myeloma SPORE Career Program (JW), the Brian D. Novis research grant from the International Myeloma Foundation (JW), and the Lady Leukemia League Research Grant(JW), and the National Natural Science Foundation of China Grant No. 81570189 (JW), and Guangzhou Department of Science and Information Technology, People's Republic of China Grant No 2014Y2-00092.
Author contributions
J. W. and L.W. K. designed experiments; J.W, B.F, Z.L, D.G, K.M, M.S, and H.W performed experiments; S.N, L.C, H.T, S.M, and S.C provided critical reagents or suggestions; J. W. and L.W.K analyzed data and wrote the paper.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA 2016; 66(1):7-30; PMID:26742998; http://dx.doi.org/ 10.3322/caac.21332 [DOI] [PubMed] [Google Scholar]
- 2.Gangatharan S, Kuruvilla J. Relapsed and refractory aggressive NHL: time for a change. Transfus Apher Sci 2013; 49(1):72-9; PMID:23835116; http://dx.doi.org/ 10.1016/j.transci.2013.05.029 [DOI] [PubMed] [Google Scholar]
- 3.Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer 2008; 8(4):299-308; PMID:18354418; http://dx.doi.org/ 10.1038/nrc2355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Möller P, Herrmann B, Moldenhauer G, Momburg F. Defective expression of MHC class I antigens is frequent in B-cell lymphomas of high-grade malignancy. Int J Cancer 1987; 40(1):32-9; PMID:3298077; http://dx.doi.org/ 10.1002/ijc.2910400107 [DOI] [PubMed] [Google Scholar]
- 5.Bevan MJ. Helping the CD8(+) T-cell response. Nat Rev Immunol 2004; 4(8):595-602; PMID:15286726; http://dx.doi.org/ 10.1038/nri1413 [DOI] [PubMed] [Google Scholar]
- 6.Hannestad K, Kao MS, Eisen HN. Cell-bound myeloma proteins on the surface of myeloma cells: potential targets for the immune system. Proc Natl Acad Sci USA 1972; 69(8):2295-9; PMID:4626403; http://dx.doi.org/ 10.1073/pnas.69.8.2295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kwak LW, Campbell MJ, Zelenetz AD, Levy R. Transfer of specific immunity to B-cell lymphoma with syngeneic bone marrow in mice: a strategy for using autologous marrow as an anti-tumor therapy. Blood 1991; 78(10):2768-72; PMID:1824269 [PubMed] [Google Scholar]
- 8.Bendandi M, Gocke CD, Kobrin CB, Benko FA, Sternas LA, Pennington R, Watson TM, Reynolds CW, Gause BL, Duffey PL et al.. Complete molecular remissions induced by patient-specific vaccination plus granulocyte-monocyte colony-stimulating factor against lymphoma. Nat Med 1999; 5(10):1171-7; PMID:10502821; http://dx.doi.org/ 10.1038/13928 [DOI] [PubMed] [Google Scholar]
- 9.Schuster SJ, Neelapu SS, Gause BL, Janik JE, Muggia FM, Gockerman JP, Winter JN, Flowers CR, Nikcevich DA, Sotomayor EM et al.. Vaccination with patient-specific tumor-derived antigen in first remission improves disease-free survival in follicular lymphoma. J Clin Oncol 2011; 29(20):2787-94; PMID:21632504; http://dx.doi.org/ 10.1200/JCO.2010.33.3005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Geary WA, Frierson HF, Innes DJ, Normansell DE. Quantitative criteria for clonality in the diagnosis of B-cell non-Hodgkin's lymphoma by flow cytometry. Modern Pathol 1993; 6(2):155-61; PMID:8483885 [PubMed] [Google Scholar]
- 11.Liendo C, Danieu L, Al-Katib A, Koziner B. Phenotypic analysis by flow cytometry of surface immunoglobulin light chains and B and T cell antigens in lymph nodes involved with non-Hodgkin's lymphoma. Am J Med 1985; 79(4):445-54; PMID:3931469; http://dx.doi.org/ 10.1016/0002-9343(85)90031-2 [DOI] [PubMed] [Google Scholar]
- 12.Weng J, Cha SC, Matsueda S, Alatrash G, Popescu MS, Yi Q, Molldrem JJ, Wang M, Neelapu SS, Kwak LW. Targeting human B-cell malignancies through Ig light chain-specific cytotoxic T lymphocytes. Clin Cancer Res 2011; 17(18):5945-52; PMID:21813633; http://dx.doi.org/ 10.1158/1078-0432.CCR-11-0970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Topalian SL, Muul LM, Solomon D, Rosenberg SA. Expansion of human tumor infiltrating lymphocytes for use in immunotherapy trials. J Immunol Methods 1987; 102(1):127-41; PMID:3305708; http://dx.doi.org/ 10.1016/S0022-1759(87)80018-2 [DOI] [PubMed] [Google Scholar]
- 14.Xiaoling G, Ying L, Jing L, Huifang L, Xia Z, Qingqing F, Ping Z. Induction of anti B-cell malignance CTL response by subfamily-shared peptides derived from variable domain of immunoglobulin heavy chain. Cancer Immunol Immunotherapy 2005; 54(11):1106-14; PMID:15889252; http://dx.doi.org/10835683 10.1007/s00262-005-0696-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Trojan A, Schultze JL, Witzens M, Vonderheide RH, Ladetto M, Donovan JW, Gribben JG. Immunoglobulin framework-derived peptides function as cytotoxic T-cell epitopes commonly expressed in B-cell malignancies. Nat Med 2000; 6(6):667-72; PMID:10835683; http://dx.doi.org/ 10.1038/76243 [DOI] [PubMed] [Google Scholar]
- 16.Southwood S, Sidney J, Kondo A, del Guercio MF, Appella E, Hoffman S, Kubo RT, Chesnut RW, Grey HM, Sette A. Several common HLA-DR types share largely overlapping peptide binding repertoires. J Immunol 1998; 160(7):3363-73; PMID:9531296 [PubMed] [Google Scholar]
- 17.Lampen MH, van Hall T. Strategies to counteract MHC-I defects in tumors. Curr Opin Immunol 2011; 23(2):293-8; PMID:21295956; http://dx.doi.org/ 10.1016/j.coi.2010.12.005 [DOI] [PubMed] [Google Scholar]
- 18.Wang RF. The role of MHC class II-restricted tumor antigens and CD4+ T cells in antitumor immunity. Trends Immunol 2001; 22(5):269-76; PMID:11323286; http://dx.doi.org/ 10.1016/S1471-4906(01)01896-8 [DOI] [PubMed] [Google Scholar]
- 19.Bluestone JA, Mackay CR, O'Shea JJ, Stockinger B. The functional plasticity of T cell subsets. Nat Rev Immunol 2009; 9(11):811-6; PMID:19809471; http://dx.doi.org/ 10.1038/nri2654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science 2003; 299(5609):1057-61; PMID:12522256; http://dx.doi.org/ 10.1126/science.1079490 [DOI] [PubMed] [Google Scholar]
- 21.Nurieva RI, Chung Y, Hwang D, Yang XO, Kang HS, Ma L, Wang YH, Watowich SS, Jetten AM, Tian Q et al.. Generation of T follicular helper cells is mediated by interleukin-21 but independent of T Helper 1, 2, or 17 cell lineages. Immunity 2008; 29(1):138-49; PMID:18599325; http://dx.doi.org/ 10.1016/j.immuni.2008.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eyerich S, Eyerich K, Pennino D, Carbone T, Nasorri F, Pallotta S, Cianfarani F, Odorisio T, Traidl-Hoffmann C, Behrendt H et al.. Th22 cells represent a distinct human T cell subset involved in epidermal immunity and remodeling. J Clin Invest 2009; 119(12):3573-85; PMID:19920355; http://dx.doi.org/ 10.1172/JCI40202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol 2005; 6(11):1123-32; PMID:16200070; http://dx.doi.org/ 10.1038/ni1254 [DOI] [PubMed] [Google Scholar]
- 24.Purwar R, Schlapbach C, Xiao S, Kang HS, Elyaman W, Jiang X, Jetten AM, Khoury SJ, Fuhlbrigge RC, Kuchroo VK et al.. Robust tumor immunity to melanoma mediated by interleukin-9-producing T cells. Nat Med 2012; 18(8):1248-53; PMID:22772464; http://dx.doi.org/ 10.1038/nm.2856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Muranski P, Boni A, Antony PA, Cassard L, Irvine KR, Kaiser A, Paulos CM, Palmer DC, Touloukian CE, Ptak K et al.. Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood 2008; 112(2):362-73; PMID:18354038; http://dx.doi.org/ 10.1182/blood-2007-11-120998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu Y, Hong S, Li H, Park J, Hong B, Wang L, Zheng Y, Liu Z, Xu J, He J et al.. Th9 cells promote antitumor immune responses in vivo. J Clin Invest 2012; 122(11):4160-71; PMID:23064366; http://dx.doi.org/ 10.1172/JCI65459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Annunziato F, Romagnani S. The transient nature of the Th17 phenotype. Eur J Immunol 2010; 40(12):3312-6; PMID:21110314; http://dx.doi.org/ 10.1002/eji.201041145 [DOI] [PubMed] [Google Scholar]
- 28.Smith CM, Wilson NS, Waithman J, Villadangos JA, Carbone FR, Heath WR, Belz GT. Cognate CD4+ T cell licensing of dendritic cells in CD8+ T cell immunity. Nat Immunol 2004; 5(11):1143-8; PMID:15475958; http://dx.doi.org/ 10.1038/ni1129 [DOI] [PubMed] [Google Scholar]
- 29.Bevan MJ. Helping the CD8+ T-cell response. Nat Rev Immunol 2004; 4(8):595-602; PMID:15286726; http://dx.doi.org/ 10.1038/nri1413 [DOI] [PubMed] [Google Scholar]
- 30.Zhang B, Karrison T, Rowley DA, Schreiber H. IFN-gamma- and TNF-dependent bystander eradication of antigen-loss variants in established mouse cancers. J Clin Invest 2008; 118(4):1398-404; PMID:18317595; http://dx.doi.org/ 10.1172/JCI33522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garrido F, Cabrera T, Aptsiauri N. “Hard” and “soft” lesions underlying the HLA class I alterations in cancer cells: implications for immunotherapy. Int J Cancer 2010; 127(2):249-56; PMID:20178101; http://dx.doi.org/ 10.1002/ijc.25270 [DOI] [PubMed] [Google Scholar]
- 32.Qin Z, Blankenstein T. CD4+ T cell-mediated tumor rejection involves inhibition of angiogenesis that is dependent on IFNγ receptor expression by nonhematopoietic cells. Immunity 2000; 12(6):677-86; PMID:10894167; http://dx.doi.org/ 10.1016/S1074-7613(00)80218-6 [DOI] [PubMed] [Google Scholar]
- 33.Lundin KU, Screpanti V, Omholt H, Hofgaard PO, Yagita H, Grandien A, Bogen B. CD4+ T cells kill Id+ B-lymphoma cells: FasLigand-Fas interaction is dominant in vitro but is redundant in vivo. Cancer Immunol Immunother 2004; 53(12):1135-45; PMID:15696611; http://dx.doi.org/23376950 10.1007/s00262-004-0538-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Braumuller H, Wieder T, Brenner E, Aszmann S, Hahn M, Alkhaled M, Schilbach K, Essmann F, Kneilling M, Griessinger C et al.. T-helper-1-cell cytokines drive cancer into senescence. Nature 2013; 494(7437):361-5; PMID:23376950; http://dx.doi.org/ 10.1038/nature11824 [DOI] [PubMed] [Google Scholar]
- 35.Merlo A, Turrini R, Bobisse S, Zamarchi R, Alaggio R, Dolcetti R, Mautner J, Zanovello P, Amadori A, Rosato A. Virus-specific cytotoxic CD4+ T Cells for the treatment of EBV-related tumors. J Immunol 2010; 184(10):5895-902; PMID:20385879; http://dx.doi.org/ 10.4049/jimmunol.0902850 [DOI] [PubMed] [Google Scholar]
- 36.Marshall NB, Swain SL. Cytotoxic CD4 T cells in antiviral immunity. J Biomed Biotechnol 2011; 2011:954602; PMID:22174559; http://dx.doi.org/ 10.1155/2011/954602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wakabayashi O, Yamazaki K, Oizumi S, Hommura F, Kinoshita I, Ogura S, Dosaka-Akita H, Nishimura M. CD4+ T cells in cancer stroma, not CD8+ T cells in cancer cell nests, are associated with favorable prognosis in human non-small cell lung cancers. Cancer Sci 2003; 94(11):1003-9; PMID:14611679; http://dx.doi.org/ 10.1111/j.1349-7006.2003.tb01392.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Perez-Diez A, Joncker NT, Choi K, Chan WFN, Anderson CC, Lantz O, Matzinger P. CD4 cells can be more efficient at tumor rejection than CD8 cells. Blood 2007; 109(12):5346-54; PMID:17327412; http://dx.doi.org/ 10.1182/blood-2006-10-051318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hunder NN, Wallen H, Cao J, Hendricks DW, Reilly JZ, Rodmyre R, Jungbluth A, Gnjatic S, Thompson JA, Yee C. Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. N Eng J Med 2008; 358(25):2698-703; PMID:18565862; http://dx.doi.org/ 10.1056/NEJMoa0800251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zwaveling S, Mota SCF, Nouta J, Johnson M, Lipford GB, Offringa R, van der Burg SH, Melief CJ. Established human papillomavirus type 16-expressing tumors are effectively eradicated following vaccination with long peptides. J Immunol 2002; 169(1):350-8; PMID:12077264; http://dx.doi.org/ 10.4049/jimmunol.169.1.350 [DOI] [PubMed] [Google Scholar]
- 41.Bijker MS, van den Eeden SJF, Franken KL, Melief CJM, Offringa R, van der Burg SH. CD8+ CTL priming by exact peptide epitopes in incomplete Freund's adjuvant induces a vanishing CTL response, whereas long peptides induce sustained CTL reactivity. J Immunol 2007; 179(8):5033-40; PMID:17911588; http://dx.doi.org/ 10.4049/jimmunol.179.8.5033 [DOI] [PubMed] [Google Scholar]
- 42.Melief CJM, van der Burg SH. Immunotherapy of established (pre)malignant disease by synthetic long peptide vaccines. Nat Rev Cancer 2008; 8(5):351-60; PMID:18418403; http://dx.doi.org/ 10.1038/nrc2373 [DOI] [PubMed] [Google Scholar]
- 43.Sewell AK. Why must T cells be cross-reactive? Nat Rev Immunol 2012; 12(9):669-77; PMID:22918468; http://dx.doi.org/ 10.1038/nri3279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guo Y, Niiya H, Azuma T, Uchida N, Yakushijin Y, Sakai I, Hato T, Takahashi M, Senju S, Nishimura Y et al.. Direct recognition and lysis of leukemia cells by WT1-specific CD4+ T lymphocytes in an HLA class II-restricted manner. Blood 2005; 106(4):1415-8; PMID:15845894; http://dx.doi.org/ 10.1182/blood-2005-01-0413 [DOI] [PubMed] [Google Scholar]
- 45.Quezada SA, Simpson TR, Peggs KS, Merghoub T, Vider J, Fan X, Blasberg R, Yagita H, Muranski P, Antony PA et al.. Tumor-reactive CD4+ T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J Exp Med 2010; 207(3):637-50; PMID:20156971; http://dx.doi.org/ 10.1084/jem.20091918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lyadova IV, Oberdorf S, Kapina MA, Apt AS, Swain SL, Sayles PC. CD4 T cells producing IFN-gamma in the lungs of mice challenged with mycobacteria express a CD27-negative phenotype. Clin Exp Immunol 2004; 138(1):21-9; PMID:15373901; http://dx.doi.org/ 10.1111/j.1365-2249.2004.02573.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mucida D, Husain MM, Muroi S, van Wijk F, Shinnakasu R, Naoe Y, Reis BS, Huang Y, Lambolez F, Docherty M et al.. Transcriptional reprogramming of mature CD4(+) helper T cells generates distinct MHC class II-restricted cytotoxic T lymphocytes. Nat Immunol 2013; 14(3):281-9; PMID:23334788; http://dx.doi.org/ 10.1038/ni.2523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Taniuchi I. Transcriptional regulation in helper versus cytotoxic-lineage decision. Curr Opin Immunol 2009; 21(2):127-32; PMID:19361971; http://dx.doi.org/ 10.1016/j.coi.2009.03.012 [DOI] [PubMed] [Google Scholar]
- 49.Kim H-J, Cantor H. CD4 T-cell subsets and tumor immunity: the helpful and the not-so-helpful. Cancer Immunol Res 2014; 2(2):91-8; PMID:24778273; http://dx.doi.org/ 10.1158/2326-6066.CIR-13-0216 [DOI] [PubMed] [Google Scholar]
- 50.Appay V. The physiological role of cytotoxic CD4(+) T-cells: the holy grail? Clin Exp Immunol 2004; 138(1):10-3; PMID:15373899; http://dx.doi.org/ 10.1111/j.1365-2249.2004.02605.x [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.