

ABSTRACT
Toll-like receptor (TLR) 7 agonists are effective in topical application for the immunotherapy of skin cancers, but their performance for the systemic treatment of solid tumors is limited by the development of TLR tolerance. In this study, we describe a novel strategy to overcome TLR tolerance and enhance TLR7-dependent antitumor immune responses through reprogramming of TLR signaling pathways. The sensitivity of TLR7 signaling in dendritic cells (DC) was increased by prior stimulation with the dsRNA poly(I:C) that mimics virally induced immune activation. Timing of the stimulations was important, as sequential stimulation with poly(I:C) and the TLR7 agonist R848 interspaced by 24 h induced higher MAPK and NFkB signaling in DC than the simultaneous application of the same ligands. DC activated by sequential poly(I:C)/R848 stimulation efficiently induced Th1 differentiation and primed NK-cell and cytotoxic T-cell responses. We have developed a treatment regimen taking advantage of TLR7 reprogram-ming that cured over 80% of large immunogenic tumors in mice by the action of NK cells and cytotoxic T cells. These results have direct implications for the use of these clinically established ligands in the immunotherapy of cancer.
KEYWORDS: Cancer immunotherapy, dendritic cells, poly(I:C), RIG-I-like receptors agonists, Toll-like receptors
Abbreviations
- BMDC
bone marrow-derived dendritic cell
- CTL
cytotoxic T lymphocyte
- DC
dendritic cell
- dsRNA
double-stranded RNA
- IFN
interferon
- IRF-3
IFN regulatory factor 3
- MDA-5
melanoma differentiation-associated protein 5
- NK cell
natural killer cells
- poly(I:C)
polyinosinic:polycytidylic acid
- PRR
pattern-recognition receptor
- RIG-I
retinoic acid-inducible gene I
- RLR
RIG-I-like receptor
- TLR
Toll-like receptor
- TNF
tumor-necrosis factor
Introduction
Breaking tolerance toward tumor tissue by promoting cytotoxic T-cell and natural killer (NK) cell responses is the main goal of cancer immunotherapy.1 Stimulation of pattern-recognition receptors (PRR) induces maturation and activation of dendritic cells (DC), leading to secretion of pro-inflammatory cytokines such as IL-12 and type-I interferons, which are important for the effector functions of T and NK cells.2,3 In this respect, nucleotide-sensing PRR such as toll-like receptor (TLR)3, TLR7, and RIG-I-like receptors (RLR) are of special interest for cancer therapy due to their potent stimulation of these cytokines.4 Indeed, synthetic agonists for the RNA-sensing TLR7 are now an approved treatment for certain skin cancers and in clinical development for other malignancies.5–7 Ligands targeting the RLR, RIG-I, and MDA-5 are also currently under development for cancer immunotherapy.4,8,9 To transmit intra-cellular signaling, RLR and TLR employ different adaptor molecules; RLR utilize MAVS,10 whereas TLR are coupled to MyD88 with the exception of TLR3, which exclusively signals via the adaptor TRIF.11 Due to the different pathways triggered, cross-talk of receptor families can lead to enhanced immune responses.12 The combination of poly(I:C), a ligand for TLR3 and MDA-5,13,14 and TLR7 ligands can synergistically enhance secretion of IL-12,15 which is a potent inducer of antitumor immune responses in preclinical studies.16
We have recently demonstrated that poly(I:C) exposure, which mimics the immune response induced by viral infection, globally reprograms PRR pathways within 24 h, leading to a sensitization of TLR7 and simultaneously to a block in RLR signaling.17 These findings highlight the importance of the timing and sequence of PRR stimulation for immunotherapeutic strategies. Indeed, we found earlier that repetitive stimulation of the TLR7 pathway led to unresponsiveness to TLR7 ligands and other MyD88-dependent agonists.18 Im-portantly, it was possible to circumvent this tolerance by well-timed applications of TLR7 agonists and thus improve the efficacy of antitumor therapy.18 Whether the reprogramming of PRR signaling pathways seen following viral exposure can affect NK and effector T-cell responses, and whether this phenomenon and the associated enhancement of TLR7 signaling can be harnessed for tumor therapy is currently unknown. Here, we investigated whether TLR7-targeted cancer immunotherapy can be further improved by timing-dependent combination strategies employing poly(I:C) to take advantage of TLR reprogramming and facilitate crosstalk of MyD88-dependent and independent agonists.
Results
Sequential stimulation of MyD88-independent and MyD88-dependent signaling pathways enhances immune activation
To study the impact of timing on combined activation of MyD88-independent and MyD88-dependent PRR, we compared cytokine production induced by the simultaneous application of the ligands poly(I:C) (MDA5/TLR3) and R848 (TLR7) with a sequential stimulation by the two ligands. Freshly isolated murine bone marrow cells, which comprise of a mixture of myeloid cells that are responsive to PRR stimulation, were stimulated twice at an interval of 24 h with poly(I:C), R848, or a sequential combination of the two stimuli. Stimulation with poly(I:C) alone did not result in strong cytokine secretion in these cells, as previously shown17 (Fig. 1A). A single application of the TLR7 ligand R848 led to clear production of the cytokines TNF-α, IL-12p40, IL-12p70, and IL-6, whereas a second stimulation with R848 24 h later resulted in much decreased cytokine levels. We have previously demonstrated that this block in cytokine secretion is due to a state of tolerance that lasts for several days after TLR7 stimulation.18 As expected, the simultaneous application of poly(I:C) and R848 at a single time point led to higher cytokine production in bone marrow cells than stimulation with R848 alone. However, stimulation of bone marrow cells with first poly(I:C) followed by R848 24 h later consistently resulted in even higher production of the cytokines IL-12p40, IL-12p70, and TNF-α (Fig. 1A). Interestingly, this was not the case for IL-6. We observed a similar effect in BMDC: The secretion of these cytokines was increased when BMDC were stimulated sequentially with poly(I:C) and R848, compared to BMDC activated simultaneously with the two ligands or to stimulation with the single agents (Fig. 1A). Of note, sequential stimulation with the inverse sequence, R848 followed by poly(I:C), did not result in changes in cytokine secretion compared to poly(I:C) alone (not shown).
Figure 1.
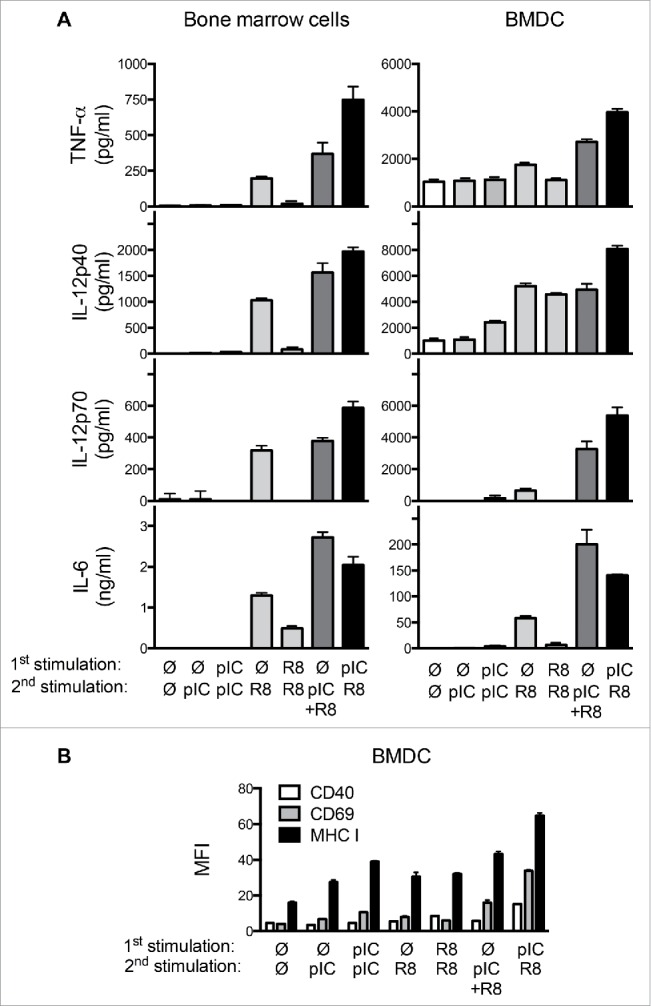
Sequential PRR stimulation with poly(I:C) and R848 enhances activation of immune cells. (A) Cytokine levels in supernatants of bone marrow cells or BMDC stimulated with combinations of poly(I:C) and R848 at a 24 h interval were measured by ELISA 18 h after the second stimulation. Mean + SEM of triplicates are shown. (B) Expression of CD40, CD69, and MHC-I on CD11c+ BMDC stimulated as in (A). Mean fluorescence intensities (MFI) + SEM of quadruples are shown. Data are representative of at least three independent experiments.
To determine the phenotype of BMDC stimulated with poly(I:C) and R848, we examined surface expression of the activation. Increased expression of CD40, CD69, and MHC-I markers was observed in BMDC treated with combinations of poly(I:C) and R848, with the highest expression after sequential activation (Fig. 1B). In contrast, CD80, CD86, and MHC-II surface expression was not significantly altered (Fig. S1A). Interestingly, although CCL2 chemokine secretion was enhanced by sequential stimulation, other DC migration-associated markers such as CCR7 and CXCR4 were not upregulated by stimulation by PRR agonists (Fig. S1B and C). In summary, we demonstrate that stimulation of a MyD88-independent pathway followed by stimulation of a MyD88-dependent pathway leads to stronger DC activation than two stimulations with a single ligand or simultaneous stimulation with both ligands.
Sequential PRR activation of BMDC enhances intracellular signaling
To investigate the intracellular mechanisms leading to enhanced cytokine secretion following sequential PRR activation, we performed qRT-PCR analysis of BMDC stimulated twice with poly(I:C), R848, or a combination of the two at a 24 h interval. Il-6 transcripts were increased in BMDC treated with the combination of poly(I:C) and R848 independently of the timing of stimulation. Expression of the il-12a transcript, which codes for the p35 subunit of IL-12, was induced by R848 stimulation, but not further increased by prestimulation with poly(I:C) (Fig. 2A). In contrast, il-12b transcripts, that encode the p40 subunit of IL-12, were expressed at significantly higher levels in BMDC treated sequentially with poly(I:C) and R848 than in all other conditions. Indeed, several studies have proposed that regulation of IL-12 production occurs at the level of the p40 subunit (reviewed in19).
Figure 2.
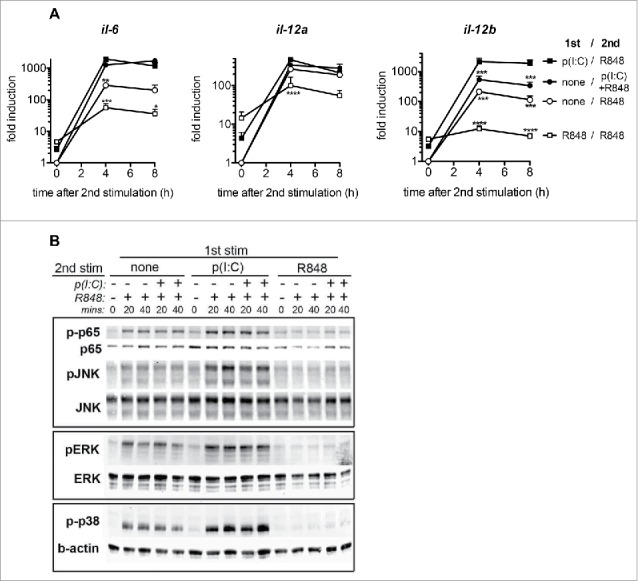
Sequential PRR stimulation of BMDC enhances cytokine mRNA expression and NFκB and MAP kinase signaling. (A) Il-6, Il-12a, and Il-12b mRNA expression 4 and 8 h after the second stimulation in BMDC stimulated as in Fig. 1. Mean + SEM of four to eight independent experiments are shown. (B) Immunoblot analysis of p65 and MAPK in BMDC stimulated with combinations of poly(I:C) and R848, at 0, 20 and 40 min, after the second stimulation. Individual blots are indicated by rectangles and are representative of three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001, and ****p < 0.0001, compared to the sequential PRR stimulation; one-way ANOVA, Dunetts post-test.
The expression of the TLR7 receptor itself was not altered by poly(I:C) stimulation (data not shown). We next examined whether the signaling pathway of TLR7 was affected in BMDC by the timing of poly(I:C) administration. We found that phosphorylation of the NFκB subunit p65 as well as of MAP kinases downstream of TLR7 was greatly increased and prolonged in cells that were treated with poly(I:C) prior to stimulation with R848 (Fig. 2B). Interestingly, simultaneous stimulation with poly(I:C) did not increase the target phosphorylation induced by R848. Furthermore, a first stimulation with R848 24 h prior to the second stimulation with either R848 or with the simultaneous poly(I:C)/R848 combination led to a complete block in TLR7 pathway activation as indicated by the lack of phosphorylated p65 and MAP kinases. Taken together, our results show that the enhanced cytokine secretion induced by sequential stimulation with poly(I:C) and R848 is associated with increased activation of the TLR7 signaling pathway, as shown by the phospho-patterns of intracellular signaling proteins.
Sequential PRR stimulation increases activation of effector T cells and NK cells by DC
An important function of DC is to instruct T cells and NK cells in order to prime effective immune responses.2,3 In a first set of experiments, we assessed the bystander activation exerted by PRR-activated DC on lymphocytes, as this antigen-independent DC function supports TCR signaling and influences the migratory patterns of lymphocytes.20,21 To this end, BMDC were activated by different ligand sequences and co-cultured with splenocytes from naive mice. BMDC that had been stimulated simultaneously by poly(I:C) and R848 induced high expression of the activation marker CD69 on T cells, but BMDC stimulated sequentially with these ligands at a 24 h interval showed the highest capacity to induce CD69 expression on both CD4+ and CD8+ T cells (Fig. 3A). A similar effect was seen when BMDC were co-cultured with purified NK cells: BMDC sequentially activated with poly(I:C) and R848 induced the highest phenotypic activation of NK cells (Fig. 3B). We also examined the effect of BMDC stimulation on the function of effector lymphocytes. We observed that sequentially stimulated BMDC co-cultured with either splenocytes or purified NK cells induced the highest secretion of IFNγ (Fig. 3C), which is an important mediator for the antitumor activity of both NK cells and effector T cells.
Figure 3.

Sequential PRR stimulation of BMDC increases activation of effector T cells and NK cells. (A) Expression of CD69 on CD4+ and CD8+ splenocytes added to sequentially stimulated BMDC 1.5 h after the second stimulation and cocultured for 24 h. Representative histograms and MFI + SEM of quadruples are shown. (B) Expression of CD69 on purified NK cells co-cultured with BMDC as in (A). (C) IFNγ levels in supernatants of splenocytes and purified NK cells co-cultured with BMDC as in (A) and (B). (D) Experimental protocol for the stimulation of BMDC and co-culture with splenocytes or purified NK cells. MFI + SEM of quadruples are shown. Data are representative of two independent experiments.
Sequential injections of TLR3/MDA5 and TLR7 ligands increase NK cell priming in vivo
To examine whether the sequence of application of poly(I:C) and R848 impacts the priming of NK cells, naive mice were injected twice with poly(I:C), R848, or a combination of the two ligands at a 24 h interval, at a well-established dosage for tumor therapy.18,22 The simultaneous application of poly(I:C) and R848 was highly toxic at the concentrations used and was thus excluded from in vivo experimentation. Although all sequences tested led to upregulation of the activation marker CD69 on splenic NK1.1+ cells, the highest upregulation was clearly observed following the sequential injection of poly(I:C) and R848 (Fig. 4A). Furthermore, we demonstrated that sequential poly(I:C)/R848 treatment of mice led to an increased proportion of IFNγ+ NK cells in the spleen (Fig. 4B). We also observed higher serum levels of IFNγ 4 h and 8 h after the second injection (Fig. 4C). This rapid and potent increase in circulating IFNγ may result from the elevated secretion by NK cells and/or bystander-activated T cells. In summary, we have shown that sequential poly(I:C)/R848 treatment led to a higher immune activation of DC and NK cells than other application sequences.
Figure 4.
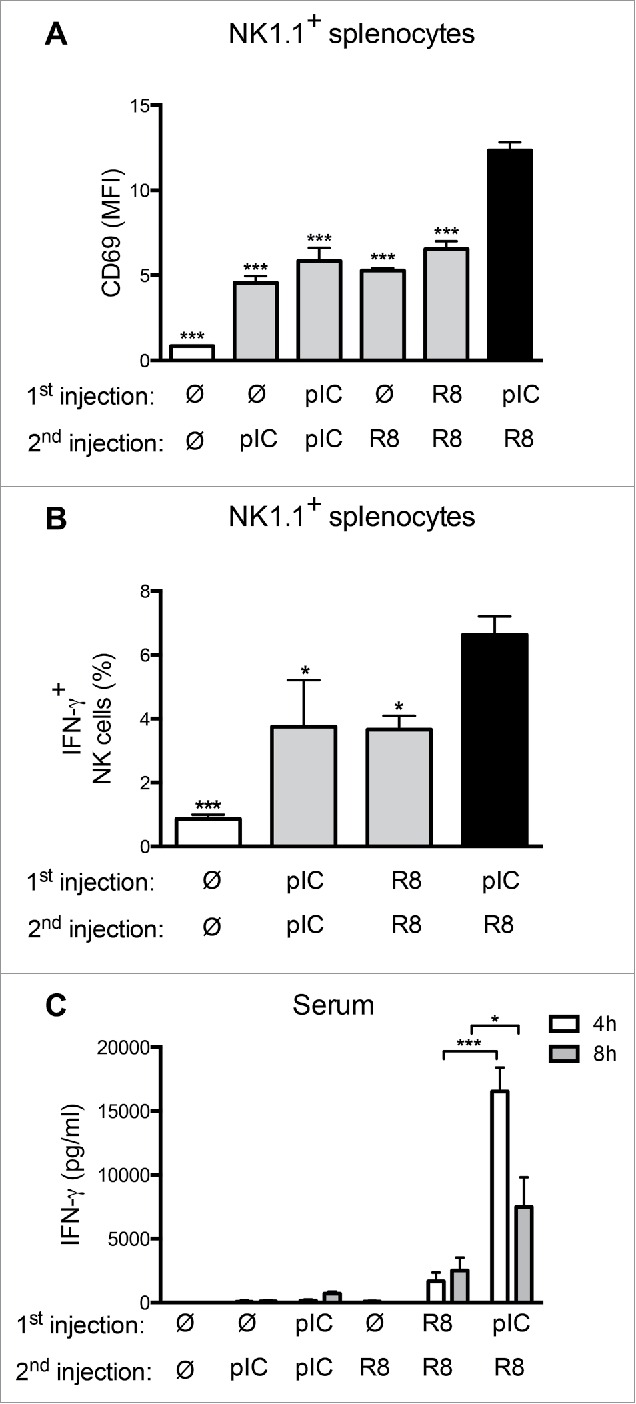
Sequential application of PRR ligands enhances NK-cell activation and IFNγ production in vivo. (A) Expression of CD69 on NK1.1+/CD3− splenic NK cells isolated from mice 4 h after two applications of poly(I:C) (100 µg i.p.), R848 (25 µg s.c.) or a combination of both at a 24 h interval. MFI + SEM of four to five mice/group are shown. (B) Percentage of IFNγ+ splenic NK cells from mice treated as in (A). Mean + SEM of 9 to 10 mice/group are shown. (C) Serum IFNγ levels in mice treated as in (A), 4 or 8 h after the second injection. Mean + SEM of four to five mice are shown. *p < 0.05 and ***p < 0.001 compared to the sequential PRR stimulation; one-way ANOVA, Dunetts post-test.
Sequential PRR activation induces higher cytotoxic T-cell responses and Th1-cell differentiation
We next assessed the efficiency of sequential PRR stimulation for the generation of specific T-cell immunity. To assess the capacity of poly(I:C)/R848-stimulated DC to prime adaptive immune responses, ovalbumin (OVA)-specific CD8+ and CD4+ cells derived from the TCR-transgenic OT-I and OT-II mice, respectively, were co-cultured with PRR-stimulated and OVA-pulsed BMDC. BMDC stimulated with the sequential poly(I:C)/R848 combination elicited the highest conversion toward IFNγ-producing CD8+ T cells and the highest levels of secreted IFNγ (Fig. 5A). We then analyzed the T helper cell polarization induced by BMDC that were activated by sequential PRR stimulation and pulsed with OVA. We observed that these DC efficiently polarized naive OT-II cells into IFNγ+ Th1 cells. Low numbers of IL-17+ cells and no IL-4+ cells were detected (Fig. 5B). The total levels of IFNγ secreted by Th1 cells were highest when DC were activated by sequential poly(I:C)/R848 stimulation.
Figure 5.
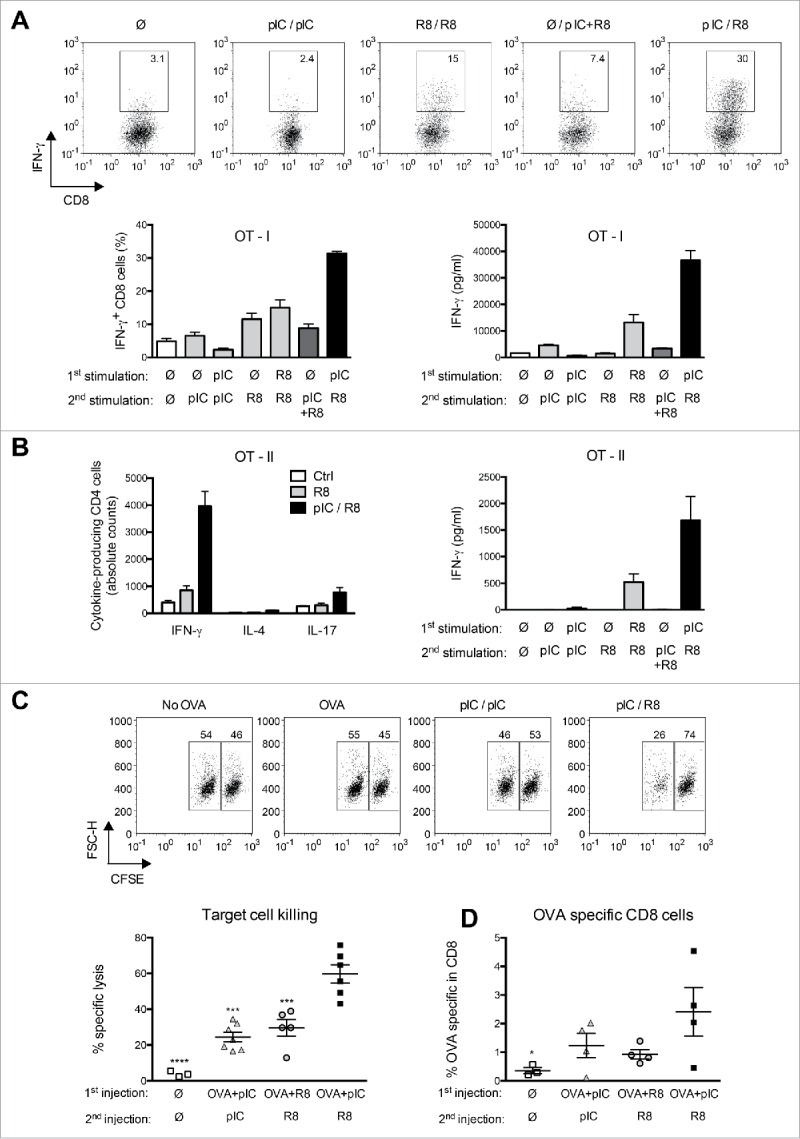
Sequential PRR activation induces higher cytotoxic T-cell responses and Th1 cell differentiation. (A) Percentage of IFNγ+ CD8+ T cells and secreted IFNγ in 3d co-cultures of OT-I cells with antigen-pulsed BMDC stimulated as in Fig. 3A. Representative histograms and mean percentage + SEM of quintuplicates are shown. (B) Total numbers of IFNγ, IL-4, and IL-17-expressing CD4+ T cells and secreted IFNγ in 4-d co-cultures of OT-II T cells with BMDC stimulated as in (A). Mean + SEM of sextuplicates are shown. Data are representative of at least three independent experiments. (C) Specific lysis of target cells in mice immunized s.c. with different combinations of poly(I:C) and R848 together with OVA. Representative histograms show the percentage of antigen-pulsed target (CFSElo) and control (CFSEhigh) cells. Specific lysis for individual mice (n = 3–6) is shown. ***p < 0.001 and ****p < 0.0001 compared to the sequential PRR stimulation; one-way ANOVA, Dunnetts post-test. (D) Frequencies of OVA-specific CD8+ T cells in individual mice (n = 3 to 4) immunized as in (C). *p ≤ 0.05 compared to the sequential PRR stimulation; one-way ANOVA, Dunnetts post-test.
To investigate whether sequential PRR stimulation induces the priming of antigen-specific CTL in vivo, we performed a cytotoxicity assay. Mice were immunized with OVA together with poly(I:C) or R848 followed by a second injection of poly(I:C) or R848 24 h later. One week later, specific killing of OVA peptide-loaded target cells was assessed in vivo. Although all PRR ligand sequences tested led to specific lysis of target cells, sequential PRR activation most efficiently induced OVA-specific killing (Fig. 5C). We further observed increased frequencies of SIINFEKL-reactive CD8+ T cells by pentamer staining of splenocytes from mice that were immunized with a combination of OVA and sequential injections of poly(I:C) and R848 (Fig. 5D). In light of the combined in vivo and in vitro data presented here, we would argue that the enhanced killing of target cells (Fig. 5C) is mediated by a combination of increased frequencies and augmented effector function of specific T cells. In conclusion, sequential application of poly(I:C) and R848 leads to enhanced antigen-specific T-cell responses, which encouraged us to assess the efficacy of sequential PRR stimulation in a mouse model of cancer.
Sequential injections of TLR3/MDA5 and TLR7 ligands are more efficient than single agents for the treatment of established tumors
Mice bearing large established tumors (43 ± 9 mm2) were treated with injections of poly(I:C), R848, or their sequential combination on two consecutive days. The treatment cycle was repeated twice at 5 d intervals to circumvent the occurrence of TLR7 tolerance18. R848 treatment and single injections of poly(I:C) only partly reduced tumor growth (Fig. 6A and B). Repeated poly(I:C) treatment stabilized disease for the duration of therapy, but all tumors resumed growth after the end of treatment (Fig. 6A and B). In contrast, sequential therapy with poly(I:C)/R848 led to a durable antitumor response; complete tumor regression was achieved in 80% of mice (Fig. 6A). The small residual tumors measured after three cycles of sequential therapy consisted only of scar tissue. However, large established B16F10 tumors were far less susceptible to PRR-targeted therapy with only modest effects on tumor growth by sequential therapy and repeated poly(I:C) administration (Fig. S2), suggesting that PRR-targeted therapy is most efficient in tumors readily accessible to immune control. To determine which immune cell subsets were essential for the success of poly(I:C)/R848 therapy in the CT26 model, we depleted CD4+, CD8+, and NK cells in tumor-bearing mice before and during sequential therapy. We observed a complete loss of therapeutic efficacy in mice treated with CD8+-depleting antibodies. Interestingly, NK-cell depletion strongly inhibited therapeutic efficacy during the first week of treatment but not at later time points. CD4+ cell depletion only marginally affected therapy. Thus, the alternating application of MyD88-independent and MyD88-dependent ligands enhances the efficacy of antitumor therapy with PRR agonists. This effect relies mainly on the action of cytotoxic T cells and at early stages on NK cells.
Figure 6.
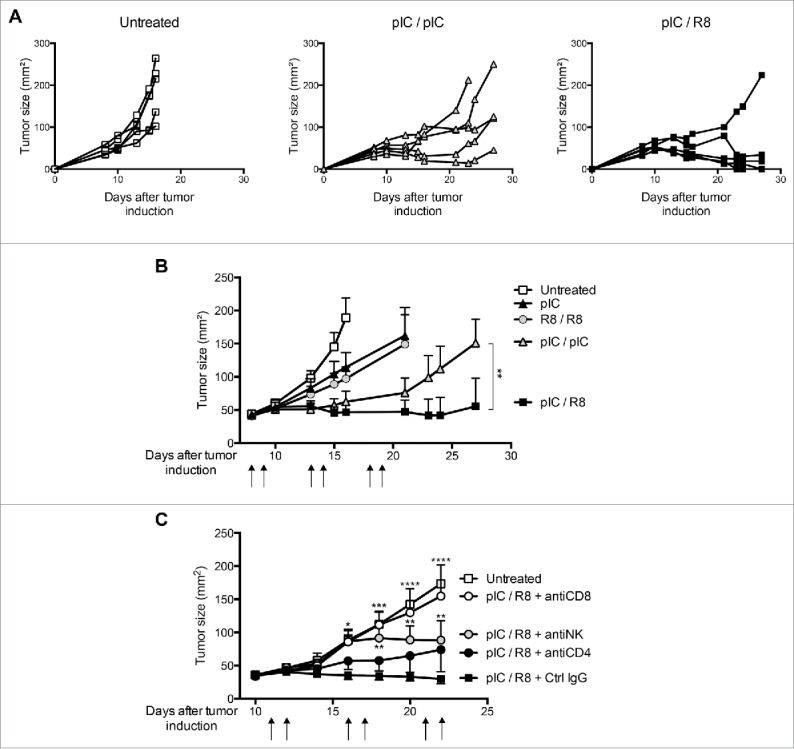
Sequential injections of TLR3/MDA5 and TLR7 ligands are more efficient than single agents for the treatment of established tumors. Growth of CT26 tumors in mice treated with combinations of poly(I:C) and R848 on two consecutive days, repeated twice (arrows). (A) Tumor size is shown for individual mice. (B) Mean + SEM of tumor size (n = 5) are shown. Data are representative of three independent experiments. (C) Tumor size for mice treated as in (A) combined with immune cell depletion. Mean + SEM of tumor size (n = 5) are shown. *p < 0.05; **p < 0.01; ***p < 0.001; and ****p < 0.0001, compared to the sequential PRR stimulation; two-way ANOVA, Bonferroni post-test.
Discussion
In spite of their efficacy for generating both innate and adaptive responses, there are to date few RLR and TLR agonists that are clinically approved or under clinical evaluation for cancer immunotherapy.4,7 In this study, we show how two of these established agents can be combined to enhance effector immune responses in order to improve anticancer therapy. By targeting different receptor pathways with the right timing and sequence, we have achieved higher activation of innate and adaptive immunity, resulting in over 80% cure rate of immunogenic murine tumors.
The simultaneous activation of multiple PRR pathways has been employed to facilitate IL-12 secretion and enhance CD4+ Th1-cell polarization,15,23 and has been assessed for antitumor activity.24–26 Here, we show that the sequential triggering first of a MyD88-independent pathway, and then of a MyD88-dependent pathway, leads to even higher Th1 responses than synergy relying on simultaneous stimulation, but with a different mechanism: We have demonstrated earlier that the viral activation of RLR reprograms the TLR7 receptor pathway and renders this pathway more sensitive to subsequent stimuli in a type I IFN-dependent manner.17 A similar IFN-dependent reprogramming phenomenon has been described for NOD receptors following viral infection.27 This contrasts with the synergy achieved by simultaneous activation of PRR pathways, which seems to be independent of type-I interferon.15,27,28 We have shown that the IFN-dependent TLR7 sensitization was mimicked by stimulation with poly(I:C) and was maximal after 8 to 24 h,17 whereas synergistic poly(I:C)/TLR7 stimulation showed maximal efficiency when ligands were given 4 h apart.15,28 We thus propose that PRR synergy and PRR reprogramming are independent and complementary phenomena to reinforce anti-viral responses by triggering Th1-type immunity.
The sequential stimulation with poly(I:C) and R848 proved to be much more effective for the treatment of model CT26 tumors than repeated stimulation with the single ligands. In our tumor model, the simultaneous injection of poly(I:C) and R848 at the doses used for sequential stimulation was highly toxic. A careful dose reduction of these ligands may allow safe simultaneous application,26 but dose reduction may also impede the antitumor effect. In our model, applying poly(I:C) only once every 5 d instead of twice rendered this monotherapy nearly ineffective, indicating that dose reduction to limit toxicity can also limit therapeutic success. Applying lower doses of poly(I:C) more frequently may induce a state of receptor tolerance and thus also limit treatment efficacy. Poly(I:C) has however shown dose-limiting toxicity in animal models as well as in clinical trials, and the reduction of poly(I:C) doses is therefore advised for patient safety.22,29-32 The sequential application of poly(I:C)/R848 may therefore allow better therapeutic efficacy with lower doses of poly(I:C) by sensitizing the TLR7 receptor pathway.
In addition to the enhanced activation of immune effector cells and reduced toxicity, the sequential combination of poly(I:C) and TLR7 agonists may have further advantages for immunotherapy. In lung cancer, TLR7 stimulation was shown to exert net tumor-promoting capacities such as enhanced tumor growth and resistance to chemotherapy, due to TLR expression by tumor cells.33,34 It is possible that the pro-apoptotic and autophagic effects of poly(I:C)8,35–37 may counterbalance this property of TLR7 agonists, thus resulting in a net antitumoral effect mediated by immune cells.
The antitumoral functions of TLR7 ligands rely on their ability to activate NK cells and cytotoxic T-cell responses.38–41 Furthermore, TLR ligands interfere with the migration of regulatory T cells into tumors42 and block the suppressive function of myeloid-derived suppressor cells.43 Poly(I:C) is also known to induce protective T-cell and NK-cell responses against tumors. We have demonstrated here that CD8+ T cells and, in the early stage of tumor growth, NK cells were decisive factors for the therapeutic success of sequential poly(I:C)/R848 therapy. It is well-known that NK cells, like T cells, require accessory cells to develop their effector functions; in particular, the effect of either poly(I:C) or R848 on NK-cell activation is largely dependent on DC support.3 Indeed, we have shown that sequential stimulation of DC enhanced both NK-cell and cytotoxic T-cell function more than stimulation with single ligands or simultaneous activation.
The ablation of CD8+ cells abolished the therapeutic effect of sequential poly(I:C)/R848 therapy in CT26 tumors, indicating a dominant role for cytotoxic T cells in this model. In contrast, large B16F10 tumors, classically considered poorly immunogenic due to low MHC-I expression,44 were only mildly affected by sequential therapy or monotherapy with poly(I:C) or R848. This suggests that for tumors with low mutational loads that lack neoantigens, or for tumors that are poorly T cell-inflamed, PRR-targeted therapy may need to be combined with other approaches targeting the T-cell repertoire, such as vaccination or checkpoint blockade45. We have shown here that sequential therapy in combination with a model antigen can efficiently prime antigen-reactive cytotoxic T cells de novo, highlighting the applicability of a cancer vaccine enhanced by sequential PRR stimulation.
In conclusion, we demonstrate for the first time that the sequential combination of two clinically established PRR ligands for non-overlapping signaling pathways can be safely administered to tumor-bearing mice leading to remarkable cure rates in immunogenic tumors. These findings provide a seminal contribution to the development of PRR-targeted anticancer therapies, underscoring the importance of well-timed stimulation of different receptor families for maximal efficacy and safety.
Materials and methods
Mice and reagents
Wild-type C57BL/6J and BALB/cJ mice were purchased from Janvier Labs (Le Genest-St-Isle, France). TCR transgenic OT-I and OT-II mice were from Charles River (Sulzfeld, Germany). All experiments were performed on a C57BL/6 background unless indicated otherwise. Mice were maintained under specific pathogen-free conditions and used at 6−14 weeks of age. All procedures were approved by the local regulatory agency. Poly(I:C) (low molecular weight), EndoFit Ovalbumin and OVA257–264 (SIINFEKL) peptide were from Invivogen (Toulouse, France). Resiquimod (R848) was obtained from Enzo Life Sciences (New York, NY).
Generation of bone-marrow derived DC (BMDC) and lymphocyte isolation
Myeloid BMDC were generated as described previously.17 Microbead-based kits were used to isolate NK cells (STEMCELL Technologies; British Columbia, Canada) or CD8+ OT-I and CD4+ OT-II T cells (Miltenyi Biotec; Bergisch Gladbach, Germany) from mouse spleens. Purity of isolated cells was routinely 85−95%.
Immune cell activation in culture
0.5–10 × 105 bone marrow cells or BMDC were stimulated for 24 h with 200 μg/mL poly(I:C) or 0.1 μg/mL R848 in RPMI complete medium containing 10% fetal calf medium, 2 mmol/L L-glutamine, 100 μg/mL streptomycin and 1 IU/mL penicillin. After washing, BMDC were restimulated with poly(I:C) or R848 for additional 16−18 h and analyzed by flow cytometry or ELISA. The concentrations of PRR ligands were established earlier17,18 and did not lead to signs of toxicity in myeloid cells. For co-culture experiments, BMDC were activated as above, but for the second stimulation, ligands were added in Opti-MEM (Life Technologies, Invitrogen) with or without OVA (1 μg/mL) for 1.5 h. Medium was replaced to avoid PRR ligand-mediated toxicity on lymphocytes and 2 × 105 splenocytes or 1 × 105 NK, CD8+ OT-I or CD4+ OT-II cells were added and co-cultured for the indicated time. For IFNγ staining, OT-I and OT-II cells were re-stimulated after 3−4 d with ionomycin (1μg/mL, Serva Electrophoresis, Mannheim, Germany) and PMA (30 ng/mL, Invitrogen) in the presence of brefeldin-A (1 μg/mL, Amresco, Solon, OH) for 3 h.
Flow cytometry and ELISA
For flow cytometry, cells were stained with anti-mouse CD3 (17A2), CD8a (53-6.7), CD4+ (RM4-5 CD8+ (53-6.7), ), CD19 (6D5), NK1.1 (eBR2a), CD11b (M1/70), CD11c (N418), CD69 (H1.2F3), IFNγ (XMG1.2), IL-4 (11B11), and IL-17A (TC11-18H10.1) antibodies from BioLegend (San Diego, CA) and anti-mouse CD40 (1C10) and MHC Class I H-2Kb antibodies (AF6-88.5.5.3) from Miltenyi Biotec. For intracellular cytokine staining, cells were additionally treated with BD Cytofix/Cytoperm (Becton Dickinson, San Diego, CA) and specificity of staining was routinely confirmed with antibody isotype controls. Analyses were performed on an MACSQuant Flow Cytometer (Miltenyi Biotec) using FlowJo software (FLOWJO, Ashland, OR). Cytokine levels in cell culture supernatants or murine serum were quantified by ELISA (BD Biosciences and BioLegend, San Diego, CA).
Serum cytokines and in vivo NK-cell activation
Mice were injected with combinations of 200 μg poly(I:C) i.p. and 25 µg R848 s.c. at a 24 h interval. Serum cytokines and splenic NK-cell activation were measured at the indicated times. To assess IFNγ secretion by NK cells, splenocytes were isolated 4 h after the last injection and incubated for additional 4 h in RPMI complete medium containing 1 μg/mL of brefeldin-A.
Immunoblot analyses and quantitative real-time PCR
BMDC (2 × 106) were stimulated twice with combinations of PRR agonists at a 24 h interval. Cells were lysed at different time points, and Western blots and qRT-PCR were performed as described previously.17 Following primary and labeled secondary antibodies were used: anti-β-actin (8H10D10), anti-phospho-p38 (D3F9), anti-p38 (D13E1), anti-phospho-JNK (G9), anti-JNK (56G8), anti-phospho-p65 (93H1), anti-p65 (L8F6 or D14E12), anti-phospho-ERK1/2 (D13.14.4E), anti-ERK1/2 (3A7), and goat anti-rabbit IgG (H+L) DyLight™ 680 Conjugate (Cell Signaling Technologies, Danvers, MA), IRDye® 800CW goat-anti-mouse antibody (LI-COR, Inc., Lincoln, NE). Following primers were used (all from Eurogentec, Seraing, Belgium): il-12a: 5′-ttctsgacaagggcatgctg-3′ (forward), 5′-gcagagtctcgccattatga-3′ (reverse); il-12b: 5′-accctgaccatcactgtcaa-3′ (forward), 5′-gtggagcagcagatgtgagt-3′ (reverse); il-6: 5′-tgcaagagacttccatccag-3′ (forward), 5′-tgaagtctcctctccggact-3′ (reverse); Gapdh: 5′-caaagtggagattgttgcca-3′ (forward), 5′-gccttgactgtgccgttgaa-3′ (reverse).
In vivo cytotoxicity assay
Mice were immunized s.c. with OVA (50 μg/mouse) admixed with either poly(I:C) (50 μg/mouse) or R848 (25 μg/mouse). Twenty-four h later a second injection of poly(I:C) or R848 s.c. was administered at the same site. One week later, splenocytes from untreated mice were labeled with 0.5 μM or 5 μM CFSE (BioLegend). CFSElow-labeled cells were loaded with 1 μg/mL OVA257–264 (SIINFEKL) peptide and coinjected with CFSEhigh cells at a 1:1 ratio. After 18 h, splenocytes were analyzed by flow cytometry. Specific lysis was calculated using the following formula: specific killing (% ) = 1 − [ (percentage of CFSElow/CFSEhigh cells in immunized animal)/( (percentage of CFSElow/CFSEhigh cells in control animal)] × 100. For the detection of ovalbumin-specific cytotoxic T cells, mice were immunized as for the in vivo cytotoxicity assay. One week later, the draining lymph nodes were isolated and nodal cells were analyzed for the presence of CD8+/CD19- ovalbumin-reactive T cells with the H2Kb-SIINFEKL pentamer according to the manufacturer's protocol (ProImmune, Oxford, UK).
Tumor induction
BALB/c and C57BL/6 mice were inoculated respectively with 2.5 × 105 CT26 or 2.5 × 105 B16F10 tumor cells s.c. (ATCC, LGC Standards, Wesel, Germany) and treatment was started with poly(I:C) (200 µg i.p.) or R848 (10 μg s.c., contralaterally to the tumor) when tumors reached an average size of 35−45 mm2. Tumor size is expressed as the product of the perpendicular diameters (mm2). Mice were killed when they reached one of the criteria set by the local regulatory agency and the last measured value was included in calculation of the mean for remaining time points. For immune cell depletion, mice were injected i.p. with 250 μg rat anti-mouse CD8a, CD4+ or control IgG2b (clones 2.43, GK1.5 and LTF-2; BioXcell, West Lebanon, NH), or 50 μg anti-asialo GM1 Ab (eBioscience, San Diego, CA) at days 10, 13, 17, and 21 after tumor induction. Target cell depletion was confirmed by flow cytometry.
Statistics
Statistical significance was evaluated using a two-tailed Student's test and ANOVA with Dunnetts or Bonferroni post-tests for multiple statistical comparisons as appropriate. pvalues < 0.05 were considered as significant. Analyses were performed with GraphPad Prism 6 software (GraphPad Softwares Inc., CA, USA).
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work is part of the medical doctoral thesis of L.C.R. at the Ludwig-Maximilian University Munich, Munich, Germany, and of the Ph.D. theses of M.T. and T.S. at the University of Fribourg, Fribourg, Switzerland. We thank Jerôme Widmer for excellent technical assistance in animal experimentation.
Funding
This work was supported by the Swiss Cancer League Grant KLS-2910-02-2012 (to C.B. and C.H.), Swiss National Science Foundation Grants 31003A_138284, 310030-156372, ProDoc PDFMP3_137079 and National Center of Competence in Research Bio-Inspired Materials (to C.B.).
Author contributions
C.H. and C.B. conceived and guided the study. C.H., M.T., I.M., L.C.R. A.O., M.P., L.S., T.S. and T.H. performed and analyzed experiments. C.H., M.T. and C.B. interpreted the results and wrote the manuscript.
ORCID
Christian Hotz http://orcid.org/0000-0001-6148-9821
Ines Mottas http://orcid.org/0000-0002-9027-7577
Lorenzo Spagnuolo http://orcid.org/0000-0002-9450-0879
Thibaud Spinetti http://orcid.org/0000-0001-8057-3746
Carole Bourquin http://orcid.org/0000-0003-3862-4583
References
- 1.Makkouk A, Weiner GJ. Cancer immunotherapy and breaking immune tolerance: new approaches to an old challenge. Cancer Res 2015; 75:5-10; PMID:25524899; http://dx.doi.org/ 10.1158/0008-5472.CAN-14-2538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Steinman RM, Hemmi H. Dendritic cells: translating innate to adaptive immunity. Curr Top Microbiol Immunol 2006; 311:17-58; PMID:17048704; http://dx.doi.org/ 10.1007/3-540-32636-7_2 [DOI] [PubMed] [Google Scholar]
- 3.Lucas M, Schachterle W, Oberle K, Aichele P, Diefenbach A. Dendritic cells prime natural killer cells by trans-presenting interleukin 15. Immunity 2007; 26:503-17; PMID:17398124; http://dx.doi.org/ 10.1016/j.immuni.2007.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Junt T, Barchet W. Translating nucleic acid-sensing pathways into therapies. Nat Rev Immunol 2015; 15:529-44; PMID:26292638; http://dx.doi.org/ 10.1038/nri3875 [DOI] [PubMed] [Google Scholar]
- 5.Peris K, Campione E, Micantonio T, Marulli GC, Fargnoli MC, Chimenti S. Imiquimod treatment of superficial and nodular basal cell carcinoma: 12-week open-label trial. Dermatol Surg Off Publ Am Soc Dermatol Surg Al 2005; 31:318-23; PMID:15841634 [DOI] [PubMed] [Google Scholar]
- 6.Sterry W, Ruzicka T, Herrera E, Takwale A, Bichel J, Andres K, Ding L, Thissen MRTM. Imiquimod 5% cream for the treatment of superficial and nodular basal cell carcinoma: randomized studies comparing low-frequency dosing with and without occlusion. Br J Dermatol 2002; 147:1227-36; PMID:12452875; http://dx.doi.org/ 10.1046/j.1365-2133.2002.05069.x [DOI] [PubMed] [Google Scholar]
- 7.Galluzzi L, Vacchelli E, Eggermont A, Fridman WH, Galon J, Sautes-Fridman C, Tartour E, Zitvogel L, Kroemer G. Trial watch. Oncoimmunology 2012; 1:699-716; PMID:22934262; http://dx.doi.org/ 10.4161/onci.20696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Duewell P, Beller E, Kirchleitner SV, Adunka T, Bourhis H, Siveke J, Mayr D, Kobold S, Endres S, Schnurr M. Targeted activation of melanoma differentiation-associated protein 5 (MDA5) for immunotherapy of pancreatic carcinoma. Oncoimmunology 2015; 4:e1029698; PMID:26504669; http://dx.doi.org/ 10.1080/2162402X.2015.1029698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Poeck H, Besch R, Maihoefer C, Renn M, Tormo D, Morskaya SS, Kirschnek S, Gaffal E, Landsberg J, Hellmuth J et al.. 5′-Triphosphate-siRNA: turning gene silencing and Rig-I activation against melanoma. Nat Med 2008; 14:1256-63; PMID:18978796; http://dx.doi.org/ 10.1038/nm.1887 [DOI] [PubMed] [Google Scholar]
- 10.Loo Y-M, Gale M. Immune signaling by RIG-I-like receptors. Immunity 2011; 34:680-92; PMID:21616437; http://dx.doi.org/ 10.1016/j.immuni.2011.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blasius AL, Beutler B. Intracellular toll-like receptors. Immunity 2010; 32:305-15; PMID:20346772; http://dx.doi.org/ 10.1016/j.immuni.2010.03.012 [DOI] [PubMed] [Google Scholar]
- 12.Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011; 34:637-50; PMID:21616434; http://dx.doi.org/ 10.1016/j.immuni.2011.05.006 [DOI] [PubMed] [Google Scholar]
- 13.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 2001; 413:732-8; PMID:11607032; http://dx.doi.org/ 10.1038/35099560 [DOI] [PubMed] [Google Scholar]
- 14.Gitlin L, Benoit L, Song C, Cella M, Gilfillan S, Holtzman MJ, Colonna M. Melanoma differentiation-associated gene 5 (MDA5) is involved in the innate immune response to Paramyxoviridae infection in vivo. PLoS Pathog 2010; 6:e1000734; PMID:20107606; http://dx.doi.org/ 10.1371/journal.ppat.1000734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Napolitani G, Rinaldi A, Bertoni F, Sallusto F, Lanzavecchia A. Selected Toll-like receptor agonist combinations synergistically trigger a T helper type 1–polarizing program in dendritic cells. Nat Immunol 2005; 6:769-76; PMID:15995707; http://dx.doi.org/ 10.1038/ni1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tugues S, Burkhard SH, Ohs I, Vrohlings M, Nussbaum K, Vom Berg J, Kulig P, Becher B. New insights into IL-12-mediated tumor suppression. Cell Death Differ 2015; 22:237-46; PMID:25190142; http://dx.doi.org/ 10.1038/cdd.2014.134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hotz C, Roetzer LC, Huber T, Sailer A, Oberson A, Treinies M, Heidegger S, Herbst T, Endres S, Bourquin C. TLR and RLR signaling are reprogrammed in opposite directions after detection of viral infection. J Immunol Baltim Md 1950 2015; 195:4387-95; PMID:26392465; http://dx.doi.org/ 10.4049/jimmunol.1500079 [DOI] [PubMed] [Google Scholar]
- 18.Bourquin C, Hotz C, Noerenberg D, Voelkl A, Heidegger S, Roetzer LC, Storch B, Sandholzer N, Wurzenberger C, Anz D et al.. Systemic cancer therapy with a small molecule agonist of toll-like receptor 7 can be improved by circumventing TLR tolerance. Cancer Res 2011; 71:5123-33; PMID:21697281; http://dx.doi.org/ 10.1158/0008-5472.CAN-10-3903 [DOI] [PubMed] [Google Scholar]
- 19.Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol 2003; 3:133-46; PMID:12563297; http://dx.doi.org/ 10.1038/nri1001 [DOI] [PubMed] [Google Scholar]
- 20.Gagnon J, Ramanathan S, Leblanc C, Cloutier A, McDonald PP, Ilangumaran S. IL-6, in synergy with IL-7 or IL-15, stimulates TCR-independent proliferation and functional differentiation of CD8+ T lymphocytes. J Immunol Baltim Md 1950 2008; 180:7958-68; PMID:18523259; http://dx.doi.org/ 10.4049/jimmunol.180.12.7958 [DOI] [PubMed] [Google Scholar]
- 21.Heidegger S, Kirchner S-K, Stephan N, Bohn B, Suhartha N, Hotz C, Anz D, Sandholzer N, Stecher B, Rüssmann H et al.. TLR activation excludes circulating naive CD8+ T cells from gut-associated lymphoid organs in mice. J Immunol Baltim Md 1950 2013; 190:5313-20; PMID:23589622; http://dx.doi.org/ 10.4049/jimmunol.1202280 [DOI] [PubMed] [Google Scholar]
- 22.Hartmann D, Adams JS, Meeker AK, Schneider MA, Lenz BF, Talmadge JE. Dissociation of therapeutic and toxic effects of polyinosinic-polycytidylic acid admixed with poly-L-lysine and solubilized with carboxymethyl cellulose in tumor-bearing mice. Cancer Res 1986; 46:1331-8; PMID:3484679 [PubMed] [Google Scholar]
- 23.Zobywalski A, Javorovic M, Frankenberger B, Pohla H, Kremmer E, Bigalke I, Schendel DJ. Generation of clinical grade dendritic cells with capacity to produce biologically active IL-12p70. J Transl Med 2007; 5:18; PMID:17430585; http://dx.doi.org/ 10.1186/1479-5876-5-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aranda F, Llopiz D, Díaz-Valdés N, Riezu-Boj JI, Bezunartea J, Ruiz M, Martínez M, Durantez M, Mansilla C, Prieto J et al.. Adjuvant combination and antigen targeting as a strategy to induce polyfunctional and high-avidity T-cell responses against poorly immunogenic tumors. Cancer Res 2011; 71:3214-24; PMID:21402711; http://dx.doi.org/ 10.1158/0008-5472.CAN-10-3259 [DOI] [PubMed] [Google Scholar]
- 25.Shojaei H, Oberg H-H, Juricke M, Marischen L, Kunz M, Mundhenke C, Gieseler F, Kabelitz D, Wesch D. Toll-like receptors 3 and 7 agonists enhance tumor cell lysis by human gammadelta T cells. Cancer Res 2009; 69:8710-7; PMID:19887600; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-1602 [DOI] [PubMed] [Google Scholar]
- 26.Whitmore MM, DeVeer MJ, Edling A, Oates RK, Simons B, Lindner D, Williams BRG. Synergistic activation of innate immunity by double-stranded RNA and CpG DNA promotes enhanced antitumor activity. Cancer Res 2004; 64:5850-60; PMID:15313929; http://dx.doi.org/ 10.1158/0008-5472.CAN-04-0063 [DOI] [PubMed] [Google Scholar]
- 27.Kim Y-G, Park J-H, Reimer T, Baker DP, Kawai T, Kumar H, Akira S, Wobus C, Núñez G. Viral infection augments Nod1/2 signaling to potentiate lethality associated with secondary bacterial infections. Cell Host Microbe 2011; 9:496-507; PMID:21669398; http://dx.doi.org/ 10.1016/j.chom.2011.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krummen M, Balkow S, Shen L, Heinz S, Loquai C, Probst H-C, Grabbe S. Release of IL-12 by dendritic cells activated by TLR ligation is dependent on MyD88 signaling, whereas TRIF signaling is indispensable for TLR synergy. J Leukoc Biol 2010; 88:189-99; PMID:20360404; http://dx.doi.org/ 10.1189/jlb.0408228 [DOI] [PubMed] [Google Scholar]
- 29.Hartmann D, Schneider MA, Lenz BF, Talmadge JE. Toxicity of polyinosinic-polycytidylic acid admixed with poly-L-lysine and solubilized with carboxymethylcellulose in mice. Pathol Immunopathol Res 1987; 6:37-50; PMID:3447151; http://dx.doi.org/ 10.1159/000157040 [DOI] [PubMed] [Google Scholar]
- 30.Krown SE, Kerr D, Stewart WE, Field AK, Oettgen HF. Phase I trials of poly(I,C) complexes in advanced cancer. J Biol Response Mod 1985; 4:640-9; PMID:2418162 [PubMed] [Google Scholar]
- 31.Lampkin BC, Levine AS, Levy H, Krivit W, Hammond D. Phase II trial of poly(I,C)-LC, an interferon inducer, in the treatment of children with acute leukemia and neuroblastoma: a report from the Children's Cancer Study Group. J Biol Response Mod 1985; 4:531-7; PMID:2416884 [PubMed] [Google Scholar]
- 32.Stevenson HC, Abrams PG, Schoenberger CS, Smalley RB, Herberman RB, Foon KA. A phase I evaluation of poly(I,C)-LC in cancer patients. J Biol Response Mod 1985; 4:650-5; PMID:2418163 [PubMed] [Google Scholar]
- 33.Chatterjee S, Crozet L, Damotte D, Iribarren K, Schramm C, Alifano M, Lupo A, Cherfils-Vicini J, Goc J, Katsahian S et al.. TLR7 promotes tumor progression, chemotherapy resistance, and poor clinical outcomes in non-small cell lung cancer. Cancer Res 2014; 74:5008-18; PMID:25074614; http://dx.doi.org/ 10.1158/0008-5472.CAN-13-2698 [DOI] [PubMed] [Google Scholar]
- 34.Cherfils-Vicini J, Platonova S, Gillard M, Laurans L, Validire P, Caliandro R, Magdeleinat P, Mami-Chouaib F, Dieu-Nosjean M-C, Fridman W-H et al.. Triggering of TLR7 and TLR8 expressed by human lung cancer cells induces cell survival and chemoresistance. J Clin Invest 2010; 120:1285-97; PMID:20237413; http://dx.doi.org/ 10.1172/JCI36551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Salaun B, Coste I, Rissoan M-C, Lebecque SJ, Renno T. TLR3 can directly trigger apoptosis in human cancer cells. J Immunol Baltim Md 1950 2006; 176:4894-901; PMID:16585585; http://dx.doi.org/ 10.4049/jimmunol.176.8.4894 [DOI] [PubMed] [Google Scholar]
- 36.Tormo D, Checińska A, Alonso-Curbelo D, Pérez-Guijarro E, Cañón E, Riveiro-Falkenbach E, Calvo TG, Larribere L, Megías D, Mulero F et al.. Targeted activation of innate immunity for therapeutic induction of autophagy and apoptosis in melanoma cells. Cancer Cell 2009; 16:103-14; PMID:19647221; http://dx.doi.org/ 10.1016/j.ccr.2009.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao X, Ai M, Guo Y, Zhou X, Wang L, Li X, Yao C. Poly I:C-induced tumor cell apoptosis mediated by pattern-recognition receptors. Cancer Biother Radiopharm 2012; 27:530-4; PMID:23062195; http://dx.doi.org/ 10.1089/cbr.2012.1226 [DOI] [PubMed] [Google Scholar]
- 38.Bourquin C, Schmidt L, Lanz A-L, Storch B, Wurzenberger C, Anz D, Sandholzer N, Mocikat R, Berger M, Poeck H et al.. Immunostimulatory RNA oligonucleotides induce an effective antitumoral NK cell response through the TLR7. J Immunol Baltim Md 1950 2009; 183:6078-86; PMID:19890064; http://dx.doi.org/ 10.4049/jimmunol.0901594 [DOI] [PubMed] [Google Scholar]
- 39.Bourquin C, Wurzenberger C, Heidegger S, Fuchs S, Anz D, Weigel S, Sandholzer N, Winter G, Coester C, Endres S. Delivery of immunostimulatory RNA oligonucleotides by gelatin nanoparticles triggers an efficient antitumoral response. J Immunother Hagerstown Md 1997 2010; 33:935-44; PMID:20948443; http://dx.doi.org/ 10.1097/CJI.0b013e3181f5dfa7 [DOI] [PubMed] [Google Scholar]
- 40.Hart OM, Athie-Morales V, O'Connor GM, Gardiner CM. TLR7/8-mediated activation of human NK cells results in accessory cell-dependent IFN-gamma production. J Immunol Baltim Md 1950 2005; 175:1636-42; PMID:16034103; http://dx.doi.org/ 10.4049/jimmunol.175.3.1636 [DOI] [PubMed] [Google Scholar]
- 41.Wang D, Precopio M, Lan T, Yu D, Tang JX, Kandimalla ER, Agrawal S. Antitumor activity and immune response induction of a dual agonist of Toll-like receptors 7 and 8. Mol Cancer Ther 2010; 9:1788-97; PMID:20515950; http://dx.doi.org/ 10.1158/1535-7163.MCT-09-1198 [DOI] [PubMed] [Google Scholar]
- 42.Anz D, Koelzer VH, Moder S, Thaler R, Schwerd T, Lahl K, Sparwasser T, Besch R, Poeck H, Hornung V et al.. Immunostimulatory RNA blocks suppression by regulatory T cells. J Immunol Baltim Md 1950 2010; 184:939-46; PMID:19966212; http://dx.doi.org/ 10.4049/jimmunol.0901245 [DOI] [PubMed] [Google Scholar]
- 43.Zoglmeier C, Bauer H, Nörenberg D, Wedekind G, Bittner P, Sandholzer N, Rapp M, Anz D, Endres S, Bourquin C. CpG blocks immunosuppression by myeloid-derived suppressor cells in tumor-bearing mice. Clin Cancer Res Off J Am Assoc Cancer Res 2011; 17:1765-75; PMID:21233400; http://dx.doi.org/ 10.1158/1078-0432.CCR-10-2672 [DOI] [PubMed] [Google Scholar]
- 44.Overwijk WW, Restifo NP. B16 as a mouse model for human melanoma. Curr Protoc Immunol Ed John E Coligan Al 2001; Chapter 20:Unit 20.1; PMID: 18432774; http://dx.doi.org/ 10.1002/0471142735.im2001s39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science 2015; 348:69-74; PMID:25838375; http://dx.doi.org/ 10.1126/science.aaa4971 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.