

ABSTRACT
Myeloid-derived suppressor cells (MDSC) are a heterogeneous population of immature myeloid cells with the capacity to inhibit immunological responses. During cancer progression, MDSC are recruited to the tumor sites and secondary lymphoid organs, leading to the suppression of the antitumor function of NK and T cells. Here, we show that the TLR7/8 agonist resiquimod (R848) has a direct effect on MDSC populations in tumor-bearing mice. Systemic application of R848 led to a rapid reduction in both intratumoral and circulating MDSC. The subpopulation of monocytic MDSC (m-MDSC) was the most affected by R848 treatment with an up to 5-fold decrease in the tumor. We found that TLR7 stimulation in tumor-bearing mice led to a maturation and differentiation of MDSC with upregulation of the surface molecules CD11c, F4/80, MHC-I, and MHC-II. MDSC treated with R848 lost their immunosuppressive function and acquired instead an antigen-presenting phenotype with the capability to induce specific T-cell proliferation. Importantly, we found that MDSC co-injected s.c. with CT26 tumor cells lost their ability to support tumor growth after pretreatment with R848. Our results demonstrate that treatment of tumor-bearing mice with a TLR7/8 agonist acts directly on MDSC to induce their maturation and leads them to acquire a non-suppressive status. Considering the obstacles posed by MDSC for cancer immunotherapy, targeting these cells by a TLR7/8 agonist may improve immune responses against cancer.
KEYWORDS: Immunotherapy, MDSC, myeloid-derived suppressor cells, R848, TLR7
Introduction
The tumor microenvironment crucially contributes to cancer progression by supporting proliferation of tumor cells and at the same time suppressing antitumor immune responses.1 Myeloid-derived suppressor cells (MDSC) form a heterogeneous population of immature myeloid cells within this microenvironment that are instrumental for tumor-associated immune suppression.2 During cancer development, MDSC numbers increase at the tumor site as well as in secondary lymphoid organs. Reports have shown a correlation between MDSC frequency and tumor progression both in mice and in cancer patients.3
MDSC are immature cells that are characterized by the co-expression of the granulocyte marker Gr1 and the myeloid cell marker CD11b.4 Under physiological conditions, Gr1+ CD11b+ cells undergo maturation and differentiate into macrophages, dendritic cells, and granulocytes. In tumor-bearing animals, circulating factors released by the tumor block the differentiation of Gr1+ CD11b+ cells, leading to the accumulation of MDSC in the tumor and lymphoid organs.5 This cell population impairs host immunity by several different mechanisms, including degradation of amino acids essential for T-cell proliferation and activation, secretion of IL-10 which promotes regulatory T-cell expansion, ROS and TGFβ production which inhibit NK-cell functions or production and nitric oxide that targets the T-cell receptor of CD8+ T cells.2 MDSC thus represent a substantial obstacle to successful cancer immunotherapy. Different strategies have been investigated to target MDSC for the treatment of cancer, such as the inhibition of MDSC suppressive functions, or the depletion of MDSC by inducing their apoptosis or by promoting their differentiation into mature cells that are non-suppressive.6
The Toll-like receptor 7 (TLR7) is a sensor for single-stranded viral RNA that is present in the endosomal membrane of specialized immune cells including monocytes, macrophages, and dendritic cells.7 Targeting TLR7 by synthetic agonists such as resiquimod (R848), imiquimod, or 3M-052 as single therapy or as adjuvant induces a potent activation of both the innate and the adaptive immune systems8. In tumor-bearing hosts, TLR7 stimulation leads to an activation of antitumoral immunity that can improve disease outcome in several cancer models.9,10 A recent report has shown that TLR7 stimulation can have beneficial effects on MDSC in vitro by inducing the maturation of these cells;11 however, in vivo data supporting this observation as well as the impact of TLR-driven MDSC maturation on immunotherapy are missing. In the present study, we show that treatment of tumor-bearing mice with the TLR7 agonist R848 drastically decreased MDSC numbers in tumors and in secondary lymphoid organs. Furthermore, MDSC from R848-treated mice showed a block in their inhibitory function and a modification of their phenotype toward a mature antigen-presenting cell (APC) phenotype. Together, these results show that MDSC can be efficiently targeted by TLR7 agonists to promote antitumor immunity.
Results
TLR7-based immunotherapy decreases MDSC numbers in tumor-bearing mice
To investigate the impact of systemic TLR7 stimulation on MDSC numbers and distribution, tumor-bearing mice were treated with the TLR7 agonist R848. Balb/c mice bearing substantially sized subcutaneous CT26 colon carcinoma-derived tumors (average size 60 mm2) received two injections of R848 subcutaneously on the opposite site of the tumor at a 24-h interval. Organs were harvested for flow cytometry analysis of MDSC 18 h after the second injection. It is well established that MDSC can be subdivided into two populations according to their expression of Gr1 and CD11b.12 Polymorphonuclear or granulocytic MDSC (g-MDSC) are defined by Gr1hi and CD11b+ whereas monocytic MDSC (m-MDSC) are characterized by Gr1med and CD11b+ and are considered to be, on a per cell basis, the most immunosuppressive population.13 We found that in mice treated with R848, the number of intratumoral m-MDSC was strongly decreased compared to control-injected mice, even at this early time point after treatment (Fig. 1). As the m-MDSC subpopulation represents the majority of MDSC in CT26 tumors, this decrease also led to a strong reduction in total intratumoral MDSC. An important reduction in m-MDSC was also observed in blood (3-fold decrease) and in spleen (5-fold decrease) of R848-treated mice, indicating that the effect of R848 on MDSC is not limited to the tumor itself but also affects systemic MDSC. Unlike m-MDSC, the g-MDSC population showed a tendency to be increased after R848 treatment, although this was not statistically significant. In the bone marrow we assessed total MDSC rather than MDSC subpopulations, as these were not easily distinguished. Total MDSC numbers were also significantly reduced in the bone marrow of R848-treated mice. We also investigated the effect of R848 on MDSC in orthotopic tumors in the 4T1 breast cancer model. As for CT26 tumors, we observed a decrease in intratumoral m-MDSC, although this was not significant (Fig. S1). These mice presented an important MDSC accumulation in the spleen that was strongly decreased by TLR7 stimulation. Thus, we show that treatment of tumor-bearing mice with a TLR7 ligand leads to a rapid decrease in the number of m-MDSC, both intratumorally and systemically.
Figure 1.
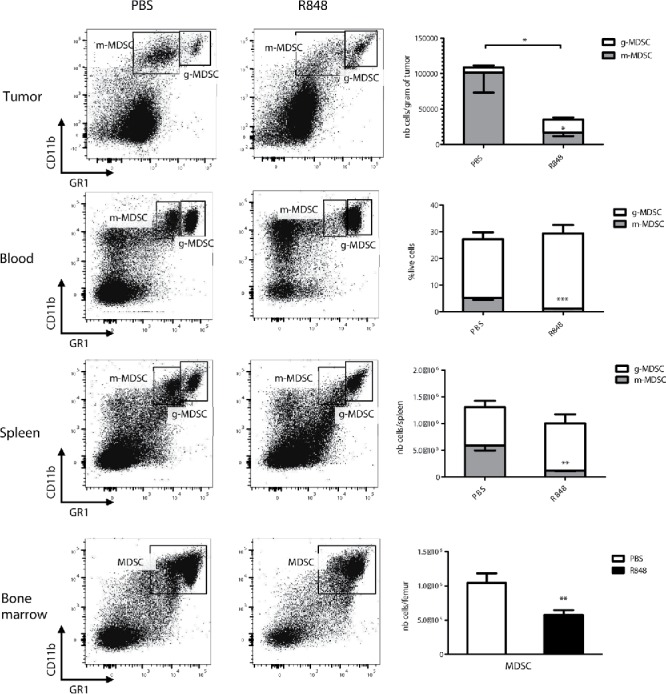
TLR7 stimulation decreases the number of MDSC in CT26 tumor-bearing mice. Flow cytometry analysis of MDSC subpopulations in different organs from CT26 tumor-bearing mice that were injected twice at a 24-h interval with 25 µg of R848 or with PBS and sacrificed 18 h after the last injection. Dot plots are representative of one mouse per group and graphs show the mean of 5 mice/group ± SEM. Data are representative of three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001; Student's t-test.
TLR7-based immunotherapy induces the maturation and differentiation of MDSC in tumor-bearing mice
In order to further characterize the effect of TLR7-based immunotherapy on MDSC, we analyzed the phenotype of splenic MDSC from R848-treated tumor-bearing mice. MDSC are defined by an immature status with low expression of myeloid differentiation markers such as F4/80 or CD11c, as well as low expression of maturation markers such as MHC-I and MHC-II and the costimulatory molecule CD80. We examined the expression levels of these cell-surface markers on splenic MDSC from CT26 tumor-bearing mice 18 h after the last of two R848 injections. We observed a clear upregulation of the differentiation markers F4/80 and CD11c, as well as of the maturation markers MHC-I, MHC-II, and CD80 on MDSC in response to TLR7 stimulation (Fig. 2A and B). Treatment with a TLR7 agonist thus clearly drives the differentiation and maturation of MDSC in vivo.
Figure 2.
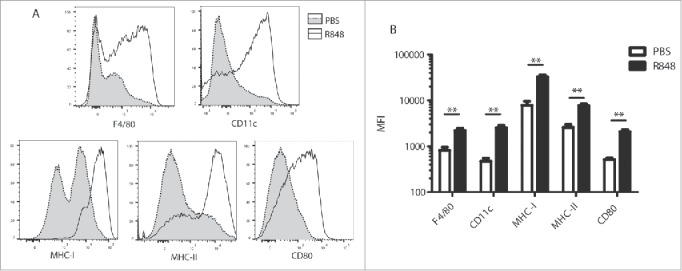
TLR7 stimulation induces the maturation and differentiation of MDSC in tumor-bearing mice. Expression of F4/80, CD80, MHC-I, CD11c, and MHC-II was analyzed by flow cytometry on splenic MDSC from tumor-bearing mice treated with R848 as in Fig. 1. (A) representative histograms of R848-treated (black line) and PBS-treated mice (light shading). (B) Mean fluorescence intensity (MFI) (5 mice/group). Data are representative of three independent experiments, mean ± SEM are shown in (B). **p < 0.01; Student's t-test.
Direct TLR7 stimulation of MDSC induces their maturation and blocks their suppressive activity
We have previously shown that CpG, a TLR9 ligand, does not act directly on MDSC but only indirectly via cytokines produced by dendritic cells.14 To investigate whether TLR7 stimulation directly affects MDSC, we assessed the impact of R848 stimulation on in vitro differentiated MDSC with respect to their differentiation and maturation. Bone marrow cells were cultured in the presence of GM-CSF and IL-6 as previously described.15 After 4 d, most cells showed double expression of Gr1 and CD11b and strongly suppressed T-cell proliferation. These bone marrow-derived MDSC (BM-MDSC) were stimulated in culture with R848 for 48 h. As seen previously with MDSC following TLR7 treatment in vivo, we observed an increase in differentiation markers (F4/80 and CD11c) and maturation markers (MHC-I and MHC-II) on BM-MDSC upon in vitro R848 stimulation (Fig. 3A). We also performed in vitro R848 stimulation of primary MDSC isolated from the spleen of tumor-bearing mice, and observed a similar strong upregulation of differentiation and maturation markers (Fig. 3B). Thus, TLR7 stimulation directly activates the MDSC population, which has been shown to express TLR7.11
Figure 3.
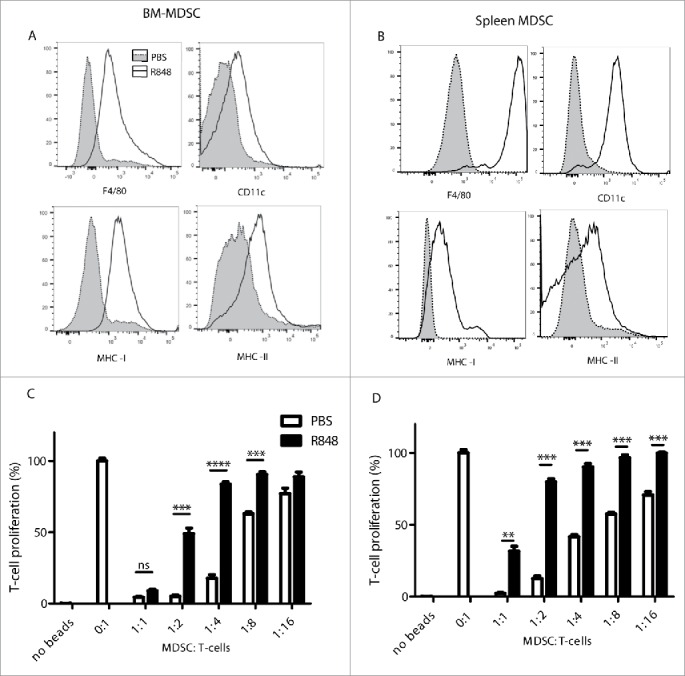
Direct TLR7 stimulation of MDSC induces their maturation and blocks their suppressive activity. (A) BM-MDSC were stimulated with or without R848 (2 µg/mL) for 48 h and surface markers were assessed as in Fig. 2. (B) Splenic MDSC isolated from untreated CT26 tumor-bearing mice were stimulated as in (A). (A, B) representative histograms of R848-stimulated (black line) and PBS-treated MDSC (light shading) are shown. (C) BM-MDSC were stimulated with R848 (2 µg/mL) for 48 h prior to a 3 d co-culture with CFSE-labeled T cells stimulated with anti-CD3/anti-CD28-coated beads. T-cell proliferation was determined by flow cytometry. (D) Splenic MDSC isolated from untreated CT26 tumor-bearing mice were assessed as in (C). Data are representative of three independent experiments. Mean ± SEM are shown. *p < 0.05, **p < 0.01, ***p < 0.001. Student's t-test.
We next examined whether TLR7 stimulation affects the immunosuppressive function of MDSC, which interfere with T-cell activity and inhibit T-cell proliferation.16 BM-MDSC were stimulated with R848 for 48 h and co-cultured with T-cells activated to proliferate with anti-CD3/anti-CD28-coated beads. Control PBS-treated BM-MDSC clearly inhibited T-cell proliferation in a dose-dependent manner (Fig. 3C), whereas R848-stimulated BM-MDSC consistently showed a decreased capacity to inhibit T-cell proliferation. Splenic MDSC isolated from CT26 tumor-bearing mice also suppressed T-cell proliferation, whereas ex vivo stimulation of the splenic MDSC with R848 strongly reduced their immunosuppressive ability (Fig. 3D). Taken together, we demonstrate that R848 acts directly on MDSC, leading to the differentiation and maturation of these cells and to a loss of their suppressive function.
R848-stimulated MDSC acquire the ability to present antigen and to prime T-cell responses
Since TLR7-stimulated MDSC upregulate the expression of maturation markers and of lineage markers associated with APC, we examined whether these MDSC also gained the ability to present antigen and to prime T-cell responses. We performed an antigen-presentation assay with R848-stimulated MDSC exposed to ovalbumin (OVA), and co-cultured these MDSC with OVA-specific CD4+ T cells from OT-II TCR transgenic mice. We then assessed whether these cells induced proliferation of the T cells. Interestingly, R848-stimulated MDSC acquired the ability to induce T-cell proliferation in an antigen-specific manner (Fig. 4). Thus, direct TLR7 stimulation leads to transdifferentiation of MDSC into mature APC.
Figure 4.
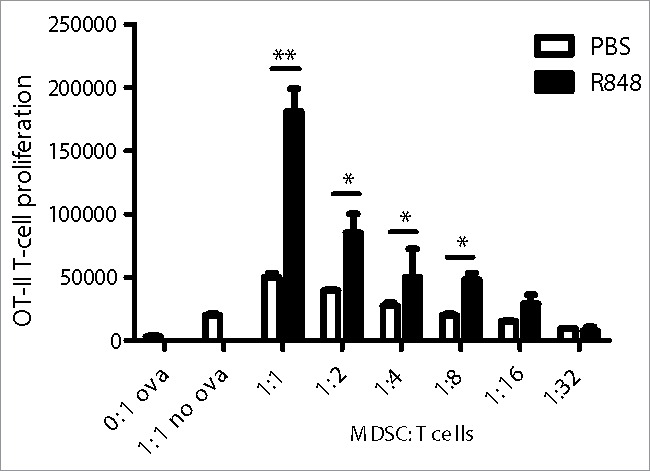
R848-stimulated MDSC acquire the ability to present antigen and to prime T-cell responses. BM-MDSC were stimulated for 48 h with R848 (2 µg/mL) or PBS. Cells were then incubated with OVA for 90 min. After washing, MDSC were co-incubated with OVA-specific, CFSE-labeled CD4+ T-cells for three days. T-cell proliferation was determined by flow cytometry. Data are representative of three independent experiments. Mean ± SEM are shown. **p < 0.01, ***p < 0.001. Student's t-test.
R848 stimulation abolishes the MDSC supporting function on tumor growth
It is well established that the presence of MDSC in the tumor microenvironment supports tumor growth.17 In order to observe the effect of R848 treatment on MDSC for tumor-supporting functions, we co-injected subcutaneously CT26 tumor cells together with BM-MDSC, pretreated with R848 or untreated, and measured tumor growth. As expected, we observed that co-injection of untreated MDSC significantly promoted CT26 tumor growth. In contrast, tumor cells co-injected with R848-treated MDSC showed the same tumor growth rate as tumor cells injected in the absence of MDSC. Thus, treatment of MDSC by R848 abolished their supporting function for tumor growth (Fig. 5).
Figure 5.
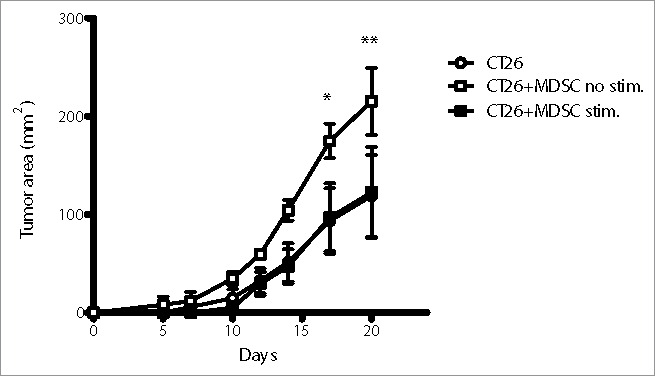
R848-treated MDSC lose their supporting function for promoting tumor growth. Balb/c mice were subcutaneously injected with CT26 alone or co-injected with CT26 and MDSC treated or not with R848 as in Fig. 3. Tumor inoculations were done with 2.5 × 105 CT26 cells (empty circle), or 2.5 × 105 CT26 and 2.5 × 105 BM-MDSC without stimulation (empty square) or 2.5×105 CT26 and 2.5 × 105 BM-MDSC stimulated with R848 (full square). Mean ± SEM of tumor area measured for individual mice (n = 4) are shown. *p < 0.05, **p < 0.01. ANOVA with Bonferroni's post-test.
Discussion
Toll-like receptor agonists are promising therapeutic agents in the context of cancer. The TLR7 agonist imiquimod is indeed already used as standard care for the topical treatment of some primary skin tumors.8,18 We and others have shown that the systemic use of TLR7 agonists also blocks tumor growth in mice, an effect that is at least in part due to the enhancement of the antitumoral function of CD8+ T cells and NK cells as well as to the inhibition of Treg function.10,19 Indeed, repeated treatment of CT26 tumor-bearing mice with the TLR7 ligand R848 inhibits tumor progression (Fig. S2). In the present study, we analyzed the effect of R848 treatment on MDSC populations after a single cycle of two injections, in order to detect early effects of TLR7 stimulation. We show that administration of R848 drastically decreased the number of intratumoral MDSC in CT26 tumor-bearing mice. Although all MDSC have immunosuppressive activity, intratumoral MDSC are more suppressive than peripheral or splenic MDSC, and a reduction in their numbers is thus highly predictive for treatment outcome.20-22 We also show that TLR7 activation significantly decreased the number of MDSC in the bone marrow, which functions as a reservoir for these cells.2 We made similar observations in the 4T1 orthotopic model of breast cancer, suggesting that these results are not limited to a single cancer model. Thus, administration of a TLR7 agonist impacts on numbers of MDSC in the periphery as well as those located within the tumor.
The two subpopulations of MDSC, g-MDSC and m-MDSC, suppress adaptive immunity in tumor-bearing hosts.12 Reports however show that on a per cell ratio basis, m-MDSC are more suppressive than g-MDSC.13 Moreover, m-MDSC have the ability to induce expression of FOXP3 in T cells and consequently to increase the proportion of T regulatory cells in the tumor microenvironment and in secondary lymphoid organs, making them an important therapeutic target to prevent tumor-associated immunosuppression.23 We show that R848 treatment of tumor-bearing mice mainly affected m-MDSC, leading to a strong reduction of this subpopulation in the tumor but also in the spleen and within circulating MDSC. Taken together, our findings show that TLR7 activation decreases the global number of MDSC and shifts the ratio of remaining MDSC toward the less suppressive g-MDSC subpopulation.
MDSC are a heterogeneous population of immature cells from the myeloid lineage.24 They are characterized by an absence of maturation and differentiation markers due to a block in differentiation exerted by tumor-derived factors.12 As these cells are known to impair tumor immunity, different strategies have been applied to target MDSC in cancer. Several approaches are under investigation to inhibit these cells pharmacologically, including the induction of MDSC apoptosis (by gemcitabine, sunitinib, or 5-fluorouracil)25-27, the inhibition of MDSC function (with sildenafil and cyclooxygenase 2 inhibitors)28, 29 and the promotion of maturation of MDSC into non-suppressive cells (all-trans retinoic acid, vitamin D).30 Here, we have shown that TLR7 stimulation leads to the upregulation of maturation markers on MDSC in vitro, a finding that is in accordance with previous reports.11,31 In contrast to what we have shown with the TLR9 agonist CpG that matures MDSC indirectly through the action of cytokines produced by dendritic cell14, we show here that the TLR7 agonist R848 directly targeted MDSC. Importantly, we show that following treatment of mice with R848, splenic MDSC rapidly upregulate differentiation and maturation markers, demonstrating that TLR7 activation also promotes MDSC maturation in vivo.
One of the key characteristics of MDSC is their suppressive activity on T-cell function, which is closely linked to their immature phenotype.2 Depending on their environment, MDSC are able to differentiate into fully mature APC such as dendritic cells or macrophages, or into immunosuppressive phenotypes such as tumor-associated macrophages (TAM). Indeed, exposure of m-MDSC to hypoxic stress drives the maturation of m-MDSC into TAM via the action of the transcription factor HIF1-α.22 In the absence of HIF1-α expression, m-MDSC differentiate into DC after hypoxic stress.22 In addition, hypoxia selectively reduces STAT3 activity in MDSC, thus, favoring differentiation to TAM.32 In the present study, we observed that TLR7 activation directs the differentiation of MDSC toward a mature APC phenotype and decreases MDSC-mediated T-cell suppression, indicating that differentiation into TAM is prevented. Furthermore, we show that TLR7 stimulation of MDSC abolishes their tumor-promoting effect. Altogether, we demonstrate that TLR7-activated MDSC lose their capacity for immunosuppression and acquire a phenotype that is closer to differentiated APC such as macrophages or dendritic cells than to TAM. The detailed molecular mechanisms leading to the altered MDSC phenotype after TLR7 stimulation remain to be elucidated. Since STAT3 activation is crucial for the immunosuppressive function of MDSC, activation of the TLR7/NFB pathway may interact with signaling via the JAK-STAT pathway and may thus explain the effect of TLR7 stimulation on MDSC differentiation.33,34
Our findings show that both peripheral and intratumoral MDSC can be efficiently targeted by TLR7 agonists. We propose that TLR7 ligands could be successfully combined with other anticancer therapies to inhibit unwanted MDSC activity. For example, it was recently shown that neutralization of the chemoattractant CCL2 prevented metastasis by retaining tumor-promoting myeloid cells in the bone marrow.35 However, interruption of the anti-CCL2 treatment led to a generalized efflux of these myeloid cells and consequently increased metastasis. A combination therapy with an anti-CCL2 agent and a TLR7 agonist may be a beneficial strategy to rapidly mature myeloid progenitor cells and prevent MDSC-associated immunosuppression upon cessation of the anti-CCL2 treatment. MDSC targeting by TLR7 agonists could thus open new strategies for combination immunotherapy.
Materials and methods
Mice
Female BALB/c and C57BL/6 mice were purchased from Janvier (Le Genest-Saint-Isle, France). TCR transgenic OT-II mice were obtained from Charles River (Sulzfeld, Germany). Mice were housed under specific pathogen-free conditions at the animal facility of the University of Fribourg and used at 6–12 weeks of age for in vivo experiments. All animal procedures were conducted in strict compliance with the Swiss federal legislation for animal experimentation.
Cell culture and tumor inoculation
The CT26 tumor cell line was cultured in complete medium: RPMI 1640, 1% (v/v) sodium pyruvate, 50 U/mL penicillin, 50 µg/mL streptomycin, 5 × 10−5 M 2-mercaptoethanol (all from PAA Laboratories) and 10% (v/v) fetal calf serum (Life Technologies, Grand Island, NY). CT26 tumor inoculation was performed with 2.5 ×105 cells injected in the flank of 6–8-week-old mice. Tumor area was calculated by the product of the perpendicular diameters of individual tumors and expressed in square millimeter. 4T1 primary tumors were initiated by orthotopic injection of 4T1 (ATCC; CRL-2539) mammary carcinoma cells into the fourth mammary fat pad of female BALB/c mice as described.36
Primary cell isolation
Bone marrow cells were flushed from tibia and femur and passed through a 40 µm cell strainer. Splenocytes were obtained by cutting the spleen into small pieces and passing through a 40 µm cell strainer. Blood was taken by cardiac puncture directly after mice were euthanized. Single cell suspensions from tumor tissue were obtained by cutting tumors in small pieces followed by incubation with “Tumor Dissociation Kit” enzymes (Miltenyi Biotec) for 45 min at 37°C under agitation. Tumor lysates were then washed once with PBS and passed through 100 µm and 40 µm cell strainers. MDSC isolation was performed from single cell suspensions of tumors or splenocytes using the Myeloid-Derived Suppressor Cell Isolation Kit (Miltenyi Biotec) according to the manufacturer's protocol.
Preparation of bone marrow-derived MDSC (BM-MDSC)
BM-MDSC were obtained from primary bone marrow cells as previously described.15 After red blood cell lysis (BD Pharm Lyse, BD Biosciences), cells were cultured in complete medium supplemented with 40 ng/mL GM-CSF and 40 ng/mL IL-6 (both from PeproTech, Rocky Hill, NJ) and harvested at day 4. The percentage of Gr1+ CD11b+ cells was over 90%.
In vitro stimulation experiments
Stimulation of the cells was carried out in complete medium with agonists at the concentrations described in the figure legends. For proliferation assays, T cells were purified from the spleen of naive mice by negative selection with the Pan T-cell isolation kit (Miltenyi Biotec, Bergisch Gladbach, Germany). T cells were stained with CFSE (Biolegend, San Diego, CA) according to the manufacturer's protocol and incubated in 96-well plates at 8×105 T cells/well with different ratios of MDSC. Then, cells were stimulated for 3 d with Dynabead mouse T-cell activator anti-CD3/anti-CD28 (Thermo Scientific, Waltham, MA). To measure T-cell proliferation, CFSE dilution was assessed by flow cytometry.37 For antigen presentation assays, MDSC were stimulated with R848 (2 µg/mL) for 48 h and incubated for 1.5 h with OVA protein (1 µg/mL) (Invivogen, Toulouse, France). 8×105 OVA-specific T cells from OT-II mice labeled with CFSE (5 µM) were added to the culture medium. After 3 d, cell proliferation was monitored by flow cytometry.
Flow cytometry
Cells were incubated with Fc-blocking antibody (BioLegend, San Diego, CA) for 15 min and stained with fluorescently labeled antibodies. Anti-mouse CD11b (M1/70), CD11c (N418), CD3 (17A2), CD4+ (RM4–5), Gr1 (RB6-8C5), Ly6c (HK1.4), Ly6g (1A8), F4/80 (BM8), MHC-I H-2Kd (SF1-1.1.1), MHC-II (M5/114.15.2), and CD80 (16-10A1) were purchased from BioLegend (San Diego, CA). All cell acquisitions were recorded using the MACSQuant system from Miltenyi Biotec. Data were analyzed using FlowJo v10.0.8 Software (Tree Stat, Inc., Ashland, USA).
Statistical analysis
Data were analyzed by either unpaired Student's t-test or by ANOVA as appropriate followed by Bonferroni post-tests. Results are shown in column graphs as the mean±standard error of the mean (SEM). The number of asterisks in the figures indicates the level of statistical significance as follows: *p < 0.05; **p < 0.01; ***p < 0.001. All data were analyzed using GraphPad Prism 5.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Anne Oberson for excellent technical assistance and Jérôme Widmer for help with animal experimentation.
Funding
This work was supported by the Swiss Cancer League Grant KLS-2910-02-2012 (to C.B. and C.H.), Swiss National Science Foundation Grants 31003A_138284, 310030-156372, ProDoc PDFMP3_137079 and National Center of Competence in Research Bio-Inspired Materials (to C.B.), Swiss National Science Foundation Grants 31003A_135738 and Medic Foundation (to C.R). This work is part of the Ph.D. theses of T.S., L.S. and M.T. at the University of Fribourg, Fribourg, Switzerland.
ORCID
Thibaud Spinetti http://orcid.org/0000-0001-8057-3746
Lorenzo Spagnuolo http://orcid.org/0000-0002-9450-0879
Inès Mottas http://orcid.org/0000-0002-9027-7577
Christian Hotz http://orcid.org/0000-0001-6148-9821
Carole Bourquin http://orcid.org/0000-0003-3862-4583
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646-74; PMID:21376230; http://dx.doi.org/ 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 2.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol 2012; 12:253-68; PMID:22437938; http://dx.doi.org/ 10.1038/nri3175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Idorn M, Kollgaard T, Kongsted P, Sengelov L, Thor Straten P. Correlation between frequencies of blood monocytic myeloid-derived suppressor cells, regulatory T cells and negative prognostic markers in patients with castration-resistant metastatic prostate cancer. Cancer Immunol Immunother 2014; 63:1177-87; PMID:25085000; http://dx.doi.org/ 10.1007/s00262-014-1591-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gabrilovich DI, Bronte V, Chen SH, Colombo MP, Ochoa A, Ostrand-Rosenberg S, Schreiber H. The terminology issue for myeloid-derived suppressor cells. Cancer Res 2007; 67:425; PMID:17210725; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-3037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sica A, Bronte V. Altered macrophage differentiation and immune dysfunction in tumor development. J Clin Invest 2007; 117:1155-66; PMID:17476345; http://dx.doi.org/ 10.1172/JCI31422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Najjar YG, Finke JH. Clinical perspectives on targeting of myeloid derived suppressor cells in the treatment of cancer. Front Oncol 2013; 3:49; PMID:23508517; http://dx.doi.org/ 10.3389/fonc.2013.00049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hemmi H, Kaisho T, Takeuchi O, Sato S, Sanjo H, Hoshino K, Horiuchi T, Tomizawa H, Takeda K, Akira S. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat Immunol 2002; 3:196-200; PMID:11812998; http://dx.doi.org/ 10.1038/ni758 [DOI] [PubMed] [Google Scholar]
- 8.Kobold S, Wiedemann G, Rothenfusser S, Endres S. Modes of action of TLR7 agonists in cancer therapy. Immunother 2014; 6:1085-95; PMID:25428647; http://dx.doi.org/ 10.2217/imt.14.75 [DOI] [PubMed] [Google Scholar]
- 9.Sidky YA, Borden EC, Weeks CE, Reiter MJ, Hatcher JF, Bryan GT. Inhibition of murine tumor growth by an interferon-inducing imidazoquinolinamine. Cancer Res 1992; 52:3528-33; PMID:1377595 [PubMed] [Google Scholar]
- 10.Bourquin C, Schmidt L, Lanz AL, Storch B, Wurzenberger C, Anz D, Sandholzer N, Mocikat R, Berger M, Poeck H et al.. Immunostimulatory RNA oligonucleotides induce an effective antitumoral NK cell response through the TLR7. J Immunol 2009; 183:6078-86; PMID:19890064; http://dx.doi.org/ 10.4049/jimmunol.0901594 [DOI] [PubMed] [Google Scholar]
- 11.Lee M, Park CS, Lee YR, Im SA, Song S, Lee CK. Resiquimod, a TLR7/8 agonist, promotes differentiation of myeloid-derived suppressor cells into macrophages and dendritic cells. Arch Pharm Res 2014; 37:1234-40; PMID:24748512; http://dx.doi.org/ 10.1007/s12272-014-0379-4 [DOI] [PubMed] [Google Scholar]
- 12.Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol 2008; 181:5791-802; PMID:18832739; http://dx.doi.org/ 10.4049/jimmunol.181.8.5791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dolcetti L, Peranzoni E, Ugel S, Marigo I, Fernandez Gomez A, Mesa C, Geilich M, Winkels G, Traggiai E, Casati A, et al.. Hierarchy of immunosuppressive strength among myeloid-derived suppressor cell subsets is determined by GM-CSF. Eur J Immunol 2010; 40:22-35; PMID:19941314; http://dx.doi.org/ 10.1002/eji.200939903 [DOI] [PubMed] [Google Scholar]
- 14.Zoglmeier C, Bauer H, Norenberg D, Wedekind G, Bittner P, Sandholzer N, Rapp M, Anz D, Endres S, Bourquin C. CpG blocks immunosuppression by myeloid-derived suppressor cells in tumor-bearing mice. Clin Cancer Res 2011; 17:1765-75; PMID:21233400; http://dx.doi.org/ 10.1158/1078-0432.CCR-10-2672 [DOI] [PubMed] [Google Scholar]
- 15.Marigo I, Bosio E, Solito S, Mesa C, Fernandez A, Dolcetti L, Ugel S, Sonda N, Bicciato S, Falisi E et al.. Tumor-induced tolerance and immune suppression depend on the C/EBPbeta transcription factor. Immunity 2010; 32:790-802; PMID:20605485; http://dx.doi.org/18272812 10.1016/j.immuni.2010.05.010 [DOI] [PubMed] [Google Scholar]
- 16.Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, De Baetselier P, Van Ginderachter JA. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood 2008; 111:4233-44; PMID:18272812; http://dx.doi.org/ 10.1182/blood-2007-07-099226 [DOI] [PubMed] [Google Scholar]
- 17.Qu P, Yan C, Du H. Matrix metalloproteinase 12 overexpression in myeloid lineage cells plays a key role in modulating myelopoiesis, immune suppression, and lung tumorigenesis. Blood 2011; 117:4476-89; PMID:21378275; http://dx.doi.org/ 10.1182/blood-2010-07-298380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peris K, Campione E, Micantonio T, Clare Marulli G, Concetta Fargnoli M, Chimenti S. Imiquimod treatment of superficial and nodular basal cell carcinoma: 12-week open-label trial. Dermatol Surg 2006; 31:318-23; PMID:15841634; http://dx.doi.org/19966212 10.1111/j.1524-4725.2005.31081 [DOI] [PubMed] [Google Scholar]
- 19.Anz D, Koelzer VH, Moder S, Thaler R, Schwerd T, Lahl K, Sparwasser T, Besch R, Poeck H, Hornung V et al.. Immunostimulatory RNA blocks suppression by regulatory T cells. J Immunol 2010; 184:939-46; PMID:19966212; http://dx.doi.org/ 10.4049/jimmunol.0901245 [DOI] [PubMed] [Google Scholar]
- 20.Jia W, Jackson-Cook C, Graf MR. Tumor-infiltrating, myeloid-derived suppressor cells inhibit T cell activity by nitric oxide production in an intracranial rat glioma + vaccination model. J Neuroimmunol 2010; 223:20-30; PMID:20452681; http://dx.doi.org/ 10.1016/j.jneuroim.2010.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maenhout SK, Van Lint S, Emeagi PU, Thielemans K, Aerts JL. Enhanced suppressive capacity of tumor-infiltrating myeloid-derived suppressor cells compared with their peripheral counterparts. Int J Cancer 2014; 134:1077-90; PMID:23983191; http://dx.doi.org/ 10.1002/ijc.28449 [DOI] [PubMed] [Google Scholar]
- 22.Corzo CA, Condamine T, Lu L, Cotter MJ, Youn JI, Cheng P, Cho HI, Celis E, Quiceno DG, Padhya T et al.. HIF-1alpha regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med 2010; 207:2439-53; PMID:20876310; http://dx.doi.org/ 10.1084/jem.20100587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Serafini P, Mgebroff S, Noonan K, Borrello I. Myeloid-derived suppressor cells promote cross-tolerance in B-cell lymphoma by expanding regulatory T cells. Cancer Res 2008; 68:5439-49; PMID:18593947; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-6621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gallina G, Dolcetti L, Serafini P, De Santo C, Marigo I, Colombo MP, Basso G, Brombacher F, Borrello I, Zanovello P et al.. Tumors induce a subset of inflammatory monocytes with immunosuppressive activity on CD8+ T cells. J Clin Invest 2006; 116:2777-90; PMID:17016559; http://dx.doi.org/ 10.1172/JCI28828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vincent J, Mignot G, Chalmin F, Ladoire S, Bruchard M, Chevriaux A, Martin F, Apetoh L, Rebe C, Ghiringhelli F. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res 2010; 70:3052-61; PMID:20388795; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-3690 [DOI] [PubMed] [Google Scholar]
- 26.Le HK, Graham L, Cha E, Morales JK, Manjili MH, Bear HD. Gemcitabine directly inhibits myeloid derived suppressor cells in BALB/c mice bearing 4T1 mammary carcinoma and augments expansion of T cells from tumor-bearing mice. Int Immunopharmacol 2009; 9:900-9; PMID:19336265; http://dx.doi.org/ 10.1016/j.intimp.2009.03.015 [DOI] [PubMed] [Google Scholar]
- 27.Ko JS, Rayman P, Ireland J, Swaidani S, Li G, Bunting KD, Rini B, Finke JH, Cohen PA. Direct and differential suppression of myeloid-derived suppressor cell subsets by sunitinib is compartmentally constrained. Cancer Res 2010; 70:3526-36; PMID:20406969; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-3278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Serafini P, Meckel K, Kelso M, Noonan K, Califano J, Koch W, Dolcetti L, Bronte V, Borrello I. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J Exp Med 2006; 203:2691-702; PMID:17101732; http://dx.doi.org/ 10.1084/jem.20061104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fujita M, Kohanbash G, Fellows-Mayle W, Hamilton RL, Komohara Y, Decker SA, Ohlfest JR, Okada H. COX-2 blockade suppresses gliomagenesis by inhibiting myeloid-derived suppressor cells. Cancer Res 2011; 71:2664-74; PMID:21324923; http://dx.doi.org/ 10.1158/0008-5472.CAN-10-3055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nefedova Y, Fishman M, Sherman S, Wang X, Beg AA, Gabrilovich DI. Mechanism of all-trans retinoic acid effect on tumor-associated myeloid-derived suppressor cells. Cancer Res 2007; 67:11021-8; PMID:18006848; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-2593 [DOI] [PubMed] [Google Scholar]
- 31.Wang J, Shirota Y, Bayik D, Shirota H, Tross D, Gulley JL, Wood LV, Berzofsky JA, Klinman DM. Effect of TLR agonists on the differentiation and function of human monocytic myeloid-derived suppressor cells. J Immunol 2015; 194:4215-21; PMID:25825448; http://dx.doi.org/ 10.4049/jimmunol.1402004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kumar V, Cheng P, Condamine T, Mony S, Languino LR, McCaffrey JC, Hockstein N, Guarino M, Masters G, Penman E et al.. CD45 phosphatase inhibits STAT3 transcription factor activity in myeloid cells and promotes tumor-associated macrophage differentiation. Immunity 2016; 44:303-15; PMID:26885857; http://dx.doi.org/ 10.1016/j.immuni.2016.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol 2004; 25:280-8; PMID:15145317; http://dx.doi.org/ 10.1016/j.it.2004.03.008 [DOI] [PubMed] [Google Scholar]
- 34.Larangé A, Antonios D, Pallardy M, Kerdine-Römer S. TLR7 and TLR8 agonists trigger different signaling pathways for human dendritic cell maturation. J Leukoc Biol 2009; 85:673-83; PMID:19164127; http://dx.doi.org/25337873 10.1189/jlb.0808504 [DOI] [PubMed] [Google Scholar]
- 35.Bonapace L, Coissieux MM, Wyckoff J, Mertz KD, Varga Z, Junt T, Bentires-Alj M. Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature 2014; 515:130-3; PMID:25337873; http://dx.doi.org/ 10.1038/nature13862 [DOI] [PubMed] [Google Scholar]
- 36.Kuonen F, Laurent J, Secondini C, Lorusso G, Stehle JC, Rausch T, Hull EF-vt, Bieler G, Alghisi GC, Schwendener R et al.. Inhibition of the kit ligand/c-kit axis attenuates metastasis in a mouse model mimicking local breast cancer relapse after radiotherapy. Clin Cancer Res 2012; 18:4365-74; PMID:22711708; http://dx.doi.org/8176234 10.1158/1078-0432.CCR-11-3028 [DOI] [PubMed] [Google Scholar]
- 37.Lyons AB, Parish CR. Determination of lymphocyte division by flow cytometry. J Immunol Meth 1994; 171:131-7; PMID:8176234; http://dx.doi.org/ 10.1016/0022-1759(94)90236-4 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.