

ABSTRACT
Tumor cells and tumor-infiltrating macrophages produce the chemokine CCL22, which attracts regulatory T cells (Tregs) into the tumor microenvironment, decreasing anticancer immunity. Here, we investigated the possibility of targeting CCL22-expressing cells by activating specific T cells. We analyzed the CCL22 protein signal sequence, identifying a human leukocyte antigen A2- (HLA-A2-) restricted peptide epitope, which we then used to stimulate peripheral blood mononuclear cells (PMBCs) to expand populations of CCL22-specific T cells in vitro. T cells recognizing an epitope derived from the signal-peptide of CCL22 will recognize CCL22-expressing cells even though CCL22 is secreted out of the cell. CCL22-specific T cells recognized and killed CCL22-expressing cancer cells. Furthermore, CCL22-specific T cells lysed acute monocytic leukemia cells in a CCL22 expression-dependent manner. Using the Enzyme-Linked ImmunoSPOT assay, we examined peripheral blood mononuclear cells from HLA-A2+ cancer patients and healthy volunteers for reactivity against the CCL22-derived T-cell epitope. This revealed spontaneous T-cell responses against the CCL22-derived epitope in cancer patients and in healthy donors. Finally, we performed tetramer enrichment/depletion experiments to examine the impact of HLA-A2-restricted CCL22-specific T cells on CCL22 levels among PMBCs. The addition or activation of CCL22-specific T cells decreased the CCL22 level in the microenvironment. Activating CCL22-specific T cells (e.g., by vaccination) may directly target cancer cells and tumor-associated macrophages, thereby modulating Treg recruitment into the tumor environment and augmenting anticancer immunity.
KEYWORDS: Antigen, anti-Tregs, CCL22, T cells, Tregs
Introduction
Solid tumors comprise both cancerous cells and the stroma that provide a supportive framework and enables immune evasion. To create and sustain an immune-permissive environment, tumors attract immune suppressive cells, such as dendritic cell subtypes, myeloid-derived suppressor cells (MDSC), M2 (or tumor-associated) macrophages, and factor forkhead box P3 (Foxp3)-positive regulatory T cells (Tregs), a subpopulation of immunosuppressive T lymphocytes.1 Cancer patients reportedly harbor increased numbers of Tregs, which progressively accumulate in the blood and lymphoid organs, and abundantly infiltrate the tumor tissue itself.2 Intratumoral Tregs suppress CD8+ T-cell responses locally at the tumor site, and high numbers of tumor-infiltrating Tregs correlate with poor prognosis.3
Chemokines are a family of small and structurally related chemotactic cytokines that interact with their chemokine receptors to orchestrate cell migration and homing in the body. CC chemokine receptor 4 (CCR4) is a transmembrane protein that acts as the receptor for two CC chemokine ligands (CCLs): CCL22 (a macrophage-derived chemokine to which CCR4 shows the highest affinity) and CCL17 (a thymus- and activation-regulated chemokine). CCR4 is predominantly expressed by Tregs, and also expressed by Th2 cells and cutaneous lymphocyte antigen-positive skin-homing T cells. Its ligand CCL22 is abundantly expressed in many types of human cancer, including ovarian and prostate tumors, gliomas, and esophageal, gastric, and breast carcinomas.4-8
CCL22 reportedly promotes Treg recruitment to cancer tissue.4,5 Zou and colleagues first described the importance of CCL22 for the specific recruitment of Tregs, which foster immune privilege.4 In their study of patients with ovarian carcinoma, they found that Tregs suppressed tumor-specific T-cell immunity and demonstrated that Treg presence at the tumor site was associated with reduced survival. They further showed that CCL22 mediated the trafficking of Tregs to the tumor. Since CCL22 was mainly produced by tumor cells and tumor-associated macrophages in the primary ovarian tumors, Tregs accumulated in tumors and ascites, and rarely entered the draining lymph nodes in later cancer stages. In a subsequent study, Li and coworkers demonstrated that breast tumor-cell-derived CCL22 is an independent prognostic predictor of survival among breast cancer patients.10 Moreover, it was recently reported that circulating CCL22 levels are related to both glioma risk and survival duration.6
Researchers are presently investigating various potential therapeutic strategies for targeting immune suppression in cancer, particularly the use of monoclonal antibodies to block checkpoint inhibitory pathways (e.g., PD-1/PD-L1). Anticancer immunity could also potentially be increased by blocking Treg migration or function. In mouse models, CCR4 antagonists reportedly prevent CCL22-mediated recruitment of Treg and Th2 cells to the tumor, which is associated with better survival, indicating that targeting CCL22–CCR4 interaction can be an effective cancer treatment approach.9 In the present study, we examined the possibility of using stimulation of CCL22-specific T cells to target the recruitment of regulatory cells to the microenvironment.
Results
CCL22-specific T cells
We screened the amino acid sequence of human CCL22 protein for possible HLA-A2-binding peptide epitopes using the SYFPEITHI epitope prediction algorithm available at www.syfpeithi.de. Interestingly, all high-scoring epitopes were located in the signal peptide portion of the sequence, which is cleaved off before the protein is secreted. One of the high-scoring CCL22-derived peptides was RLQTALLVVL, hereafter referred to as pCCL223–12. To characterize pCCL223–12-specific T cells, we acquired peripheral blood mononuclear cells (PBMCs) from a melanoma patient and stimulated these cells using autologous dendritic cells (DCs) or PBMCs pulsed with the pCCL223–12 peptide. After five stimulations, we performed pCCL223–12-tetramer staining, which revealed small but distinct population of tetramer-positive cells (Fig. 1A, left). The tetramer-positive populations were successfully isolated and expanded using the rapid expansion protocol (Fig. 1A, right). On the expanded culture, we performed intracellular staining for INFγ and TNFα in response to peptide stimulation. Around 30% of CD8+ cells secreted interferon-gamma (IFNγ) and TNFα in response to pCCL223–12 peptide stimulation (Fig. 1B). Only CD8+ T cells secreted INFγ and TNFα in response to the pCCL223–12 peptide, with no response detected from CD4+ T cells. The same results were found with intracellular staining before tetramer isolation and expansion (data not shown).
Figure 1.

CCL22-specific CD8+ T cells are cytotoxic. (A) Left: pCCL223–12 peptide (RLQTALLVVL) reactive T cells among PMBC of a melanoma patient after in vitro stimulation with the epitope as measured by pCCL223–12-tetramer (PE and APC) staining. Right: tetramer staining of isolated and expanded CCL223–12-specific T cells from the same donor. (B) Intracellular IFNγ and TNFα staining of CD8+ T cells after pCCL223–12 epitope (right) and HIV control peptide (left) stimulation. (C) 51Cr release assay showing lysis of pCCL223–12 and HIV peptide pulsed T2 cells by CCL22-specific T cells. (D) Cytotoxic activity of CCL22-specific T cells against K562 cells lacking HLA-A2 or K562 cells stably transfected with HLA-A2 with or without addition of pCCL223–12 peptide. (E) CCL22-specific T cell cytotoxicity against T2 cells cross presenting the CCL22 signal peptide (MDRLQTALLVVLVLLAVALQAT) or an irrelevant long peptide (CILDSKLEVEALAQLLTFALK), compared to lysis of T2 cells pulsed with pCCL223–12. T2 cells were cultured with CCL22 signal peptide or the irrelevant control peptide overnight before analysis. (F) Peptide titration 51Cr release assay using T2 cells and pCCL223–12-specific T cells. Effector to target ratio of 3:1 was maintained and a 10-fold peptide titrations of 9-mer pCCL223–11 (RLQTALLVV) or the 10-mer pCCL223–12 epitope.
pCCL223–12-specific T cells exhibit HLA-A2-restricted killing
Next, we examined the cytotoxic capability of CCL22-specific T cells. The pCCL223–12-specific T cells were able to lyse pCCL223–12-pulsed T2 cells but did not recognize T2 cells pulsed with a negative control peptide from HIV (Fig. 1C). To confirm the HLA-A2-restricted reactivity of CCL22-specific T cells, we used HLA-A2-transfected K562 cells pulsed with and without pCCL223–12 as targets (Fig. 1D) and found that the T cells only recognized the K562-A2 cells pulsed with pCCL223–12. As an additional control, we examined non-transfected HLA-negative K562 cells, determining that these cells were not recognized by the pCCL223–12-specific T cells even when pulsed with pCCL223–12.
Previous reports show that T2 cells can efficiently cross-present long peptides.10,11 Thus, we further examined whether the pCCL223–12 epitope could be cross-presented from the 22-mer signal peptide sequence of the CCL22 protein (termed the CCL22-signal peptide; MDRLQTALLVVLVLLAVALQAT). Indeed, pCCL223–12-specific T cells lysed the T2 cells that were pulsed with CCL22-signal peptide (Fig. 1E), indicating that T2 cells could cross-present the pCCL223–12 epitope even without TAP expression in these cells. We then examined the T-cell avidity of pCCL223–12-specific T cells toward the pCCL223–11 peptide (RLQTALLVV), which is one amino acid shorter than pCCL223–12 and was predicted by the computer algorithm to bind to HLA-A2 with high affinity. Although the T cells reacted toward both peptides, they showed the highest avidity toward the decamer pCCL223–12 epitope (Fig. 1F).
Cytotoxicity against CCL22-expressing cancer cells
The CCL22-specific cytotoxic T-cell lymphocyte (CTL) culture was tested for reactivity against the leukemia cell lines THP-1, RPMI6666, and SET-2. PCR analysis revealed CCL22 expression in all of these cell lines, although SET-2 showed only a weak band. We found that pCCL223–12-specific cells lysed most of the investigated cancer cell lines, both with and without pre-treatment with IFNγ (Fig. 2A-C). IFNγ reportedly induces CCL22 expression in cancer cell lines12 and increases surface expression of HLA. Chromium release assay revealed that SET-2 cells were not killed (Fig. 2B), which were only weakly positive for CCL22 mRNA on PCR and showed no detectable CCL22 expression as measured by CCL22 ELISA (Fig. 2D). To confirm that the killing of cancer cells by CCL22-specific CTLs was indeed dependent on CCL22 expression, we used siRNA transfection to inhibit CCL22 expression in IFNγ pre-treated THP-1 cells. This transfection rescued these cells from T-cell-mediated lysis (Fig. 2E). Fig. 2F depicts the CCL22 inhibition in THP-1 cells after siRNA transfection as measured by ELISA from the supernatant.
Figure 2.
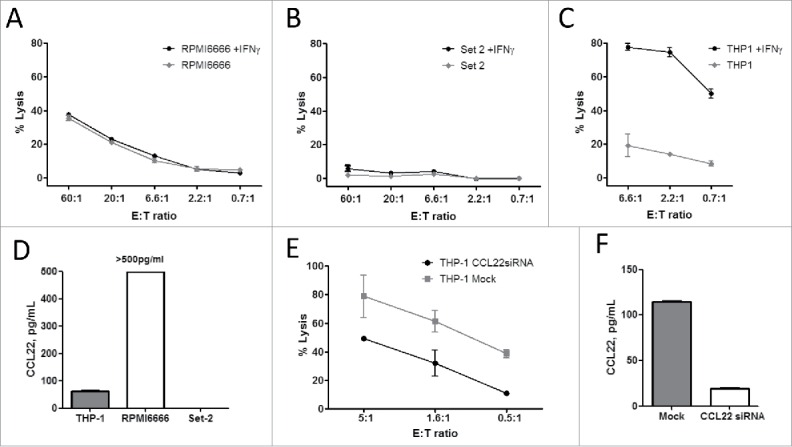
CCL22-reactive T cells are able to recognize and kill CCL22-expressing cancer cell lines. 51Cr release assays of IFNγ non-treated or pretreated cancer cell lines: RPMI6666—Hodgkin's lymphoma (A); Set-2—essential thrombocytemia (B); THP-1—acute monocytic leukemia (C). The same effector CCL22-specific T cell culture was used as effector cells. (D) CCL22 expression in the supernatant of cancer cell lines as measured by CCL22 ELISA. (E) Lysis of IFNγ induced THP-1 cells transfected with CCL22 siRNA transfection or Mock transected by CCL22-specific T cells. Assay performed 48 h after transfection. (F) ELISA analysis of CCL22 expression in the supernatant from siRNA transfected THP-1 cells compared to THP-1 cells transfected with Mock control, 48 h after electroporation.
Spontaneous T-cell responses against CCL22
We next acquired PBMCs from 13 cancer patients and 10 healthy individuals and stimulated these cells with the pCCL223–12 peptide for 2 weeks in the presence of low-dose IL-2. We used the IFNγ Enzyme-Linked ImmunoSPOT (ELISPOT) assay to analyze the reactivity toward the pCCL223–12 peptide. Spontaneous specific T-cell reactivity against pCCL223–12 was detected in a number of melanoma patients and healthy donors (Fig. 3A). Notably, the overall responses appear to be similar in healthy donors and cancer patients (Fig. 3B).
Figure 3.
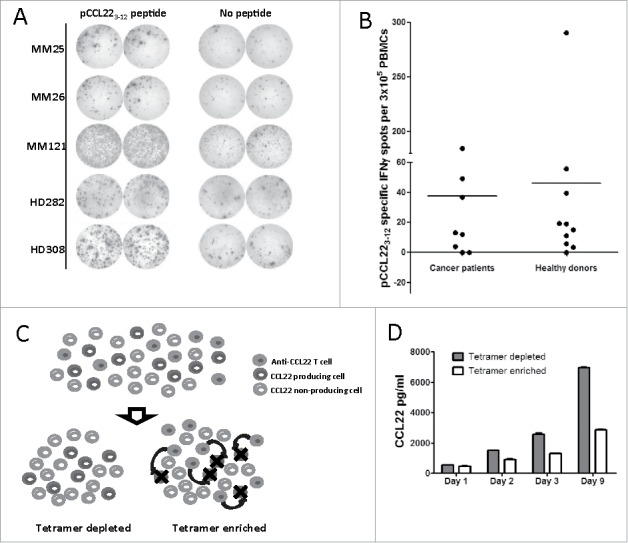
CCL223–12-reactive T cells are present in healthy donors and cancer patients. (A) IFNγ ELISPOT examples showing T-cell responses against the pCCL223–12 epiotpe in three melanoma patients (MM) and two healthy donors (HD). (B) pCCL223–12 peptide-specific IFNγ ELISPOT responses in cancer patients and healthy donors. The average number of pCCL223–12-specific spots (after subtraction of spots in wells without added peptide) was calculated per 3 × 105 PBMC for each donor. (C) Experimental setup of pCCL223–12-specific T-cell depletion/enrichment in PBMCs. PBMCs from a healthy donor were stimulated twice with pCCL223–12 peptide before the pCCL223–12-specific T cells (anti-CCL22 T cells) were isolated using pCCL223–12-PE-tetramer in combination with anti-PE magnetic beads. The remaining depleted PBMCs were divided into two cultures before pCCL223–12-tetramer-isolated cells were added into one of these, resulting in a tetramer-enriched culture. CCL22-specific T cells in a tetramer-enriched culture target CCL22-producing T cells (as indicated by black arrows). (D) ELISA analysis of CCL22 in the supernatants from pCCL223–12-tetramer-enriched compared to tetramer-depleted PBMC cultures over time.
T-cell-mediated decrease in CCL22 levels in the microenvironment
Donor PBMCs that showed a pCCL223–12 response in ELISPOT were then stimulated twice with pCCL223–12 peptide in the presence of IL2. Next, the culture was depleted of pCCL223–12-reactive T cells using HLA-A2/pCCL223–12-tetramer and magnetic beads. This T-cell-depleted culture was divided into two portions, and the HLA-A2/pCCL223–12-tetramer-isolated T cells were added back to one of the portions (Fig. 3C). We then monitored the CCL22 concentration in the supernatants of both cultures. Notably, CCL22 levels were lower in the culture with added pCCL223–12-specific T cells after only 1 d, and this difference increased over the culturing time. After 9 d of culture, the CCL22 concentration was over two times higher in the tetramer-depleted culture compared to that in the tetramer-enriched culture (Fig. 3D).
pCCL223–12 stimulation decreased the CCL22 levels in the microenvironment
To mimic a setting in which cancer patients are vaccinated with CCL22-derived peptides, we stimulated PMBCs with the pCCL223–12 peptide epitope and IL-2 in vitro. We then investigated whether this activation of pCCL223–12-specific T cells affected the overall CCL22 concentration among the PMBCs. First, donor PBMCs that showed a pCCL223–12 response in ELISPOT were stimulated using the pCCL223–12 peptide, and we measured the CCL22 concentration in the supernatant over 1 week (Fig. 4A). CCL22 expression was lower in the cultures stimulated with pCCL223–12 peptide compared to cultures stimulated with an HIV control peptide. This difference was apparent after 2 d of culture and reached significance after 1 week (p = 0.01).
Figure 4.
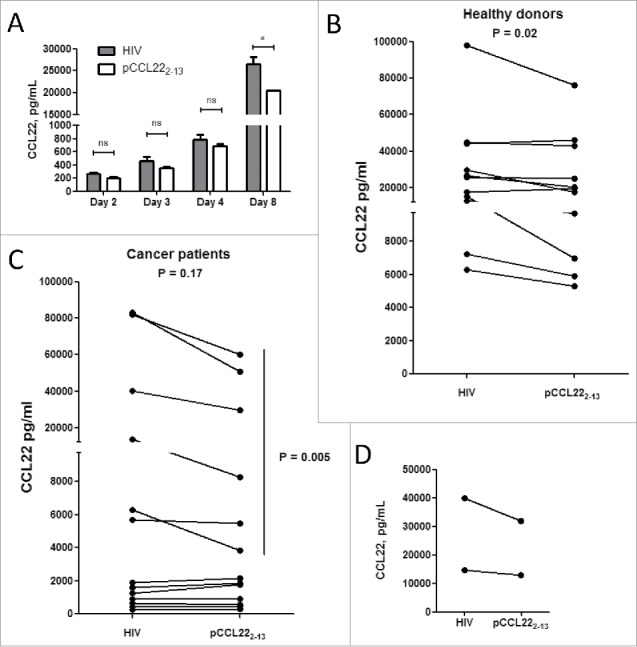
Activation of CCL22-specific T cells decreases CCL22 levels in the microenvironment. (A) CCL22 levels in supernatants from PBMC isolated from a healthy donor after stimulation with either pCCL223–12 or HIV peptide as measured by CCL22 ELISA (*p = 0.01, ns—not significant, t-test). Experiments were performed in triplicates for each peptide. (B) Changes in CCL22 levels in supernatants from PBMC isolated from 11 healthy donor PBMCs after stimulation with pCCL223–12 peptide compared to HIV control peptide as measured by CCL22 ELISA on day 7. (C) Changes in CCL22 levels in in supernatants from PBMC isolated from 13 cancer patients after stimulation with pCCL223–12 peptide compared to HIV control peptide as measured by CCL22 ELISA on day 7. Experiments were performed in triplicates or duplicates for each peptide. (D) CCL22 levels in supernatants from cells isolated from ovarian cancer ascites isolated from two ovarian cancer patients after stimulation with either pCCL223–12 or HIV peptide as measured by CCL22 ELISA.
We subsequently used pCCL223–12 peptide or an HIV control peptide to stimulate PBMCs from 11 healthy donors and 13 cancer patients, and then measured the CCL22 concentration in the supernatants one week after stimulation. In PBMCs from healthy donors, the CCL22 concentration significantly decreased following stimulation with pCCL223–12 peptide (p = 0.02) (Fig. 4B). On the other hand, in PBMCs from cancer patients, the overall decrease in CCL22 concentration after stimulation with pCCL223–12 did not reach significance (p = 0.17) (Fig. 4C). When PBMCs from cancer patients were stratified according to low CCL22 expression (≤ 2,000 pg/mL) and high CCL22 expression (≥ 5,000 pg/mL) (Fig. 4C), the high-expression group showed a significant decrease in CCL22 concentration after pCCL223–12 stimulation (p = 0.005).
Ovarian ascetic fluid reportedly contains a mixture of cancer cells and immune-infiltrating cells, along with high levels of CCL22.4 To examine whether pCCL223–12-specific T cells may influence CCL22 concentration directly in the tumor microenvironment, we collected ascetic fluid from five patients with HLA-A2-positive epithelial ovarian cancer and isolated the ascites cells. The ascites cells from two of these patients showed low viability and, thus, we could only analyze cells from three patients. The ascites cells from one of these patients did not include any T lymphocytes. The ascites cells from the remaining two ovarian cancer patients were stimulated with pCCL223–12 peptide, which led to a decrease in the overall CCL22 levels in the supernatants at 1 week after stimulation (Fig. 4D).
pCCL223–12 stimulation influenced the cytokine milieu
We further examined the PBMC supernatants from 11 cancer patients and 10 healthy donors with regard to changes in cytokine levels after 1 week of stimulation with pCCL223–12 peptide compared to with an HIV control peptide. The PBMCs from cancer patients that were stimulated with CCL22 peptide showed a significant increase in IL-6 level (p = 0.02). A similar increase was observed in cultures of PBMCs from healthy donors, although this change did not reach significance (p = 0.06) (Fig. 5A). We also observed a tendency of decreasing TNFα levels in cultures of PBMCs from healthy donors (7 out of 10) and cancer patients (7 out of 11); however, these changes did not reach significance (p = 0.7 and p = 0.16, respectively) (Fig. 5B). We further examined the concentrations of IL-1β, IL-10, and IFNγ in the culture supernatants. We detected no unambiguous differences in these cytokines between cultures stimulated with pCCL223–12 peptide versus control peptide. After stimulation, IL-10 was almost undetectable in the supernatants, and IL-1β was induced after stimulation with pCCL223–12 in only one cancer patient and two healthy donors (Fig. S1).
Figure 5.
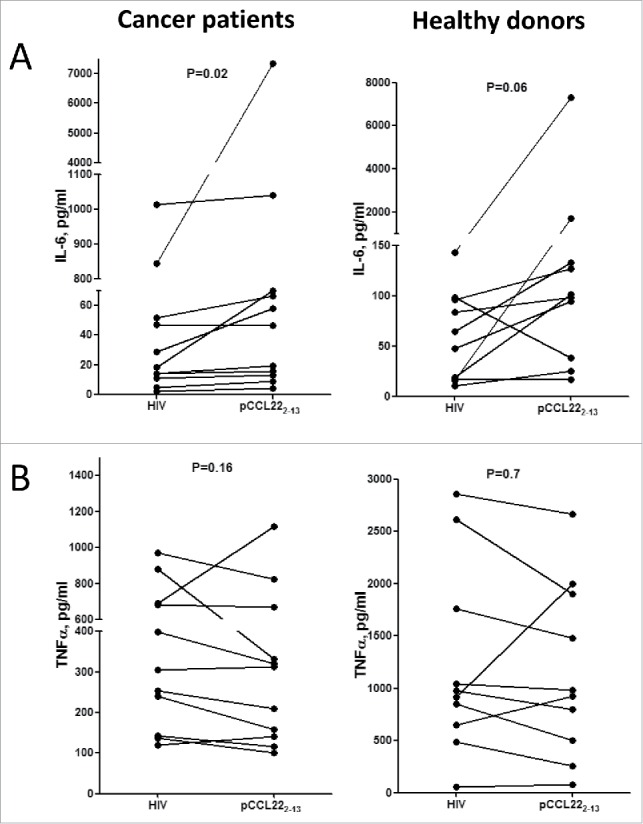
Stimulation of CCL22-specific T cells affects the PBMC cytokine profile. (A) Overall changes in IL-6 expression in supernatants from PBMC isolated from 11 cancer patients (left) or 10 healthy donors (right) after pCCL223–12 stimulation compared to HIV control peptide stimulation (p = 0.02 and p = 0.06, respectively, paired t-test). (B) Overall changes in TNFα expression in supernatants from PBMC isolated from 11 cancer patients (left) or 10 healthy donors (right) after pCCL223–12 stimulation compared to HIV control peptide stimulation (p = 0.16 and p = 0.7, respectively, paired t-test).
Discussion
CCL22 secretion by tumor cells, as well as by tumor-associated macrophages, attracts and recruits Tregs to the tumor microenvironment, resulting in suppression of anticancer immunity.4,13 Solid tumor production of CCL22 reportedly causes Treg accumulation in many cancers, including ovarian, prostate, esophageal, gastric, and breast carcinomas, and glioblastomas.5,8 On the other hand, tumors lacking CCL22 expression are not infiltrated by Tregs, regardless of the productions of other CCR4-binding chemokines (e.g., CCL17), suggesting that Treg recruitment to the tumor environment occurs via the CCL22:CCR4 axis.5
Tumor-infiltrating Tregs inhibit antitumor immunity and are associated with poor prognosis in several types of human cancer.4,5 Thus, many cancer treatment strategies involve Treg depletion or modulation. Cyclophosphamide and anti-CD25 antibodies have been examined to target Tregs.14 Likewise, clinical trials have investigated the use of a CD25-directed diphtheria toxin (Ontak) to eliminate Tregs in patients suffering from renal cell carcinoma or melanoma.15 In our present study, we suggest the use of activated T cells to target CCL22-expressing cells as a novel method of decreasing Tregs in tumors and thereby increasing anticancer immunity. CCL22 epitopes could easily be added to cancer vaccines as a means of strengthening anticancer immune responses in patients.
Our present findings demonstrated that it was possible for specific T cells to target CCL22-expressing cells. Scrutiny of the CCL22 signal sequence for possible HLA-A2-binding peptides revealed that the most probable epitope sequence was derived from the CCL22 signal peptide. A signal sequence is the N-terminal extension of a newly synthesized secretory or membrane protein, which is usually 16–30 amino acid residues in length. The CCL22 signal peptide is cleaved off by signal peptidases prior to CCL22 secretion from the cell. Hence, T cells that recognize an epitope derived from the CCL22 signal peptide will identify CCL22-expressing cells even though CCL22 has been secreted out of the cell. Several HLA-restricted epitopes derived from protein signal sequences have previously been described.10,16,17
We demonstrated that it was possible for T cells to recognize an HLA-restricted CCL22-derived peptide epitope, and we were thus able to expand CCL22-specific T cells by re-stimulation of PBMCs with a CCL22 peptide in vitro. The results of our chromium release cytotoxicity assays further demonstrated specific recognition and lysis of CCL22-expressing leukemia cells. Moreover, CCL22 knockdown by siRNA transfection rescued cells from being killed by CCL22-specific T cells. These findings suggest that in CCL22-expressing cells, the signal peptide is degraded and the epitope is subsequently processed and presented on the cell surface restricted to HLA-A2 molecules. We also found that the CCL22 signal peptide could be taken up and cross-presented on the surface of non-professional antigen-presenting cells.
We used the ELISPOT assay to examine PBMCs from HLA-A2+ cancer patients and healthy individuals for reactivity against the CCL22-derived T-cell epitope and found that T cells spontaneously reacted to the CCL22-derived peptide. Tetramer enrichment/depletion experiments revealed that the addition of HLA-A2-restricted CCL22-specific T cells to PBMCs decreased the CCL22 level in the microenvironment. We further determined that activation of CCL22-specific T cells via stimulation with the peptide epitope significantly decreased CCL22 levels among PMBCs from both healthy donors and cancer patients with high CCL22 production. Such activation also led to a CCL22 decrease in supernatants of ascites-derived cells isolated from ovarian cancer patients. These findings suggest that CCL22-specific T cells may be used to target CCL22-expressing cells and to thereby suppress CCL22-mediated Treg migration into the tumor microenvironment.
Interestingly, activating CCL22-specific T cells by peptide stimulation also resulted in increased release of IL-6 into the PMBC supernatants, suggesting that CCL22-specific T cells may influence the overall pro-inflammatory microenvironment. ELISPOT results showed spontaneous CCL22-specific T-cell responses in both cancer patients and healthy donors, which was somewhat surprising since CCL22 is abundantly expressed in normal immune cells. It is thought that self-reactive T cells harboring T-cell receptors with high affinity to a target/HLA complex undergo clonal deletion to maintain self-tolerance. Thus, under healthy conditions, T cells should be tolerized to an inflammation-induced protein like CCL22. However, Yu and colleagues recently reported that clonal deletion prunes the T-cell repertoire without fully eliminating self-reactive T cell clones.18 Thus, self-reactive T cells that survive thymic selection (including CCL22-specific T cells) may actively participate in immune regulation. We recently reported the frequent detection of self-reactive pro-inflammatory T cells that recognize regulatory immune cells, which apparently have a function that counteracts the immune-suppressive feedback signals in the immune system. For example, we previously described self-reactive T cells that specifically recognize indoleamine 2,3-dioxygenase (IDO), tryptophan 2,3-dioxygenase (TDO), programmed death-ligand 1 (PD-L1), and Foxp3 (reviewed in refs.19,20). We recently proposed that these cells should collectively be termed anti-regulatory T cells (anti-Tregs).20 CCL22 and other proteins, including Foxp3, PD-L1, and IDO, are commonly expressed in normal immune cells under different physiological conditions, including inflammation and stress. Anti-Tregs could contribute to immune homeostasis, potentially serving to counteract the function of suppressive regulatory immune cells. Essentially, anti-Treg cells may “support” effector T cells by directly eliminating regulatory cells or by locally secreting pro-inflammatory cytokines into the microenvironment. Anti-Treg activation can strongly influence immunity via both direct and indirect mechanisms.20 Our presently described data suggest that CCL22-specific T cells can be defined as anti-Tregs.
In conclusion, our present results showed that CD8+ T cells could recognize an HLA-restricted CCL22 peptide epitope. These T cells recognized and lysed various cancer cell lines in a manner dependent on CCL22 expression and were naturally present in cancer patients and healthy individuals. Activation of CCL22-specific T cells may directly influence the CCL22 concentration in the microenvironment.
Methods
Patient material
PBMCs were collected from healthy individuals and cancer patients (melanoma, renal cell carcinoma, and breast cancer patients). Blood samples were drawn a minimum of 4 weeks after termination of any kind of anticancer therapy. PBMC were isolated using Lymphoprep™ separation, HLA-typed, and frozen in FCS with 10% dimethyl sulfoxide (DMSO). Fresh ovarian ascites was filtered with a 70-µm filter and the cells were isolated by centrifugation (1500 RPMI-1640, 5 min). If the ascites contained a high abundance of erythrocytes, they were removed by adding lysis buffer (Ortho-Mune Lysing Solution) to the cells with incubation of 3–5 min. The lysis buffer was quickly washed away and the cells cryopreserved in human serum with 10% DMSO at −140° until use. The protocol was approved by the Scientific Ethics Committee for The Capital Region of Denmark and conducted in accordance with the provisions of the Declaration of Helsinki. Written informed consent from the patients was obtained before study entry.
Cancer cell lines
Cancer cells were cultured in RPMI-1640 (Life technologies) with 10% or 20% FCS. Cell cultures were split 2 to 3 times a week, depending on the observed cell density. Last validation of cancer cell lines: the cancer cell lines were validated upon acquisition from the commercial supplier. The cell lines used were T2, THP-1, RPMI6666 cells (all available at the American Type Culture Collection (ATCC)), and Set-2 cells (available a Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH (DCMZ)).
ELISPOT assay
PBMCs were placed in the bottom of ELISPOT plate (nitrocellulose bottomed 96-well plates by MultiScreen MAIP N45; Millipore) pre-coated with IFNγ capture Ab (Mabtech) and the peptides were added at 5 µg/mL. PBMCs from each patient were set up in duplicates or triplicates for peptide and control stimulations. Cells were incubated in ELISPOT plates in the presence of an antigen for 14–16 h after which they are washed off and secondary biotinylated Ab (Mabtech) was added. After 2 h incubation unbound secondary antibody was washed off and streptavidin conjugated alkaline phosphatase (AP) (Mabtech) was added for 1 h. Next, unbound conjugated enzyme is washed off and the assay is developed by adding BCIP/NBT substrate (Mabtech). Developed ELISPOT plates were analyzed on CTL ImmunoSpot S6 Ultimate-V analyzer using Immunospot software v5.1. Responses were calculated as the difference between average numbers of spots in wells stimulated with pCCL223–12 peptide and control wells.
Peptides
HLA-A2-restricted peptides were predicted using an online epitope prediction database SYFPEITHI available on the web (www.syfpeithi.de). A 10 amino acid long HLA-A2-restricted peptide epitope derived from the signal sequence of CCL22 (here referred to as pCCL223–12, RLQTALLVVL) was synthesized by TAG Copenhagen, Copenhagen, Denmark. Other peptides: pCCL223–11 peptide: RLQTALLVV, HIV peptide HIV-1 pol476-484: ILKEPVHGV, CCL22 signal peptide: MDRLQTALLVVLVLLAVALQAT as well as a long irrelevant random control peptide: CILDSKLEVEALAQLLTFALK. Peptides were dissolved in either DMSO or sterile water.
Establishment of antigen-specific T-cell cultures
CCL22-specific T cell culture was established by stimulation of cancer patient PBMC with irradiated pCCL223–12 peptide-loaded autologous DC or PBMCs. The following day IL-7 and IL-12 (PeproTech, London, UK) were added. Stimulation of the cultures was carried out every 8 d with CCL223–12 peptide-loaded irradiated autologous DC followed by pCCL223–12 peptide-loaded irradiated autologous PBMC. The day after peptide stimulation IL-2 (PeproTech, London, UK) was added. Four DC stimulations and one PBMC stimulation were made in total.
Generation of dendritic cells
DCs were generated from monocytes isolated from PBMCs by adherence on culture dishes at 37°C for 1 to 2 h in RPMI-1640. Adherent monocytes were cultured in RPMI-1640 supplemented with 10% FCS in the presence of IL-4 (250 U/mL) and GM-CSF (1,000 U/mL) for 6 d. DCs were matured by addition of IL-β (1,000 U/mL), IL-6 (1,000 U/mL) TNF-α (1,000 U/mL), and PGE2 (1 μg/mL).
Cytotoxicity assay
Conventional 51Cr-release assays for CTL-mediated cytotoxicity were carried out as described elsewhere.21 HLA-A2-positive cells were used as targets: T2-cells (ATCC), HLA-A2+ Hodgkin's lymphoma cell line (RPMI6666), monocytic leukemia cells (THP-1), and essential thrombocythemia (Set-2) with or without IFNγ (100 U/mL) addition for 2 d prior to performing cytotox assay.
For peptide titration cytotoxicity assay, T2 cells were used as target cells and a constant effector to target ratio of 3:1 was used for all peptide concentrations. 10-fold serial peptide dilutions ranging from 10−2 mM to 10−9 mM of pCCL223–12 and pCCL223–11 were made.
siRNA-mediated CCL22 silencing
A set of three Stealth siRNA duplexes for targeted silencing of CCL22 (HSS109578, HSS184551, HSS184552) were obtained from Invitrogen (Invitrogen, Paisley, UK). siRNA dublex sequences: HSS109578 sense 5′-CCUGGGUGAAGAUGAUUCUCAAUAA-3′ and antisense 5′-UUAUUGAGAAUCAUCUUCACCCAGG-3′; HSS184551 sense 5′-CCCUGCGCGUGGUGAAACACUUCUA-3′ and antisense 5′-UAGAAGUGUUUCACCACGCGCAGGG-3′; HSS184552 sense 5′-GCCAACAUGGAAGACAGCGUCUGCU-3′ and antisense 5′-AGCAGACGCUGUCUUCCAUGUUGGC-3′. For CCL22 silencing experiments, THP-1 cells were transfected with CCL22 siRNA using electroporation parameters as previously described.22
Flow cytometric analysis
Flow cytometry analysis was performed on a FACSCanto™ II (BD Biosciences, San Jose CA, USA), and cell sorting was performed on FACSAria™ (BD Biosciences, San Jose CA, USA).
Intracellular staining of CCL22-specific T cell cultures was performed after the cells were stimulated with HIV or pCCL223–12 peptides for 5 h (BD GolgiPlug™ was added after the first hour). The cells were then stained for surface markers, and then washed and permeabilized by using Fixation/Permeabilization and Permeabilization Buffer (eBioscience), according to manufacturer's instructions. Antibodies used: IFNγ- PE-Cy7, TNFα- APC, CD4-PerCP, and CD8-FITC (all from BD Biosciences). Dead cells were stained using LIVE/DEAD® Fixable Near-IR Dead Cell Stain Kit according to manufacturer's instructions.
For tetramer staining and sorting, PE- and APC-coupled HLA-A2 multimers with HIV or pCCL223–12 peptides were used in addition to the CD4-PerCP and CD8-FITC (BD Biosciences). HLA-A2 multimers were produced in house by using a previously described MHC peptide exchange technology.23
Analysis of CCL22 expression
Cell culture supernatants from PBMC, cancer cell lines, and ascites cell cultures as well as siRNA-transfected THP-1 cells were analyzed using Human CCL22/MDC DuoSet ELISA kit (R&D Systems) according to manufacturer's instructions.
Peptide stimulation of PBMCs and ascites cells
PBMCs from healthy donors or cancer patients were thawed and rested for 4 h in X-VIVO 15™ (Lonza) before being set up into 24-well plates with 2 × 106 cells/well in X-VIVO medium with 5% human serum. A total of 20 µg/mL of pCCL223–12 peptide in DMSO or HIV peptide in sterile water were added to each well. Appropriate amount of DMSO was added to HIV control wells to control for the solvent of pCCL223–12 peptide. The following day, IL-2 was added to a final concentration of 120 U/mL (360 U for ascites cells stimulation). Supernatant samples were collected after 2 or 7 d of culture. Cells were stimulated twice before being tested in ELISPOT assay.
Cytokine expression LUMINEX
Cell culture supernatants from PBMCs or ascites cells stimulated with CCL22-3 or HIV peptide were analyzed for IFNγ, TNF-α, IL-6, IL-10, and IL-1β using Bio-Plex Pro™ Human Chemokine assays from Bio-Rad. Samples were acquired on the Bio Plex 200 system and analyzed using Bio-Plex Manager™ v6.
Statistical analysis
T-test was used for the statistical analysis of ELISA samples from cancer patients and healthy donors when comparing pCCL223–12 stimulation with HIV control. The same analysis was used to compare IFNγ, TNF-α, IL-6, and IL-10 expression after pCCL223–12 stimulation with HIV control. Wilcoxon-signed rank test was used for statistical analysis of IL-10 expression due to multiple zero values.
Supplementary Material
Disclosure of potential conflicts of interest
The author declares no competing financial interests. However, it should be noted that Mads Hald Andersen is an author of a filed patent application based on the use of CCL22 for vaccination. The rights of the patent application have been transferred to Copenhagen University Hospital, Herlev in accordance with Danish Law of Public Inventions at Public Research Institutions.
Acknowledgments
We would like to thank Merete Jonassen and, Tina Seremet for excellent technical assistance. We thank Per thor Straten for scientific discussions. Supported by the Danish Cancer Society, the Lundbeck foundation, Toyota Foundation, the Danish Council for Independent Research, and Herlev Hospital. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Iellem A, Mariani M, Lang R, Recalde H, Panina-Bordignon P, Sinigaglia F, D'Ambrosio D. Unique chemotactic response profile and specific expression of chemokine receptors CCR4 and CCR8 by CD4(+)CD25(+) regulatory T cells. J Exp Med 2001; 194:847-53; PMID:11560999; http://dx.doi.org/ 10.1084/jem.194.6.847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Colombo MP, Piconese S. Regulatory-T-cell inhibition vs. depletion: the right choice in cancer immunotherapy. Nat Rev Cancer 2007; 7:880-7; PMID:17957190; http://dx.doi.org/ 10.1038/nrc2250 [DOI] [PubMed] [Google Scholar]
- 3.Yu P, Lee Y, Liu W, Krausz T, Chong A, Schreiber H, Fu YX. Intratumor depletion of CD4+ cells unmasks tumor immunogenicity leading to the rejection of late-stage tumors. J Exp Med 2005; 201:779-91; PMID:15753211; http://dx.doi.org/ 10.1084/jem.20041684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M et al.. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med 2004; 10:942-9; PMID:15322536; http://dx.doi.org/ 10.1038/nm1093 [DOI] [PubMed] [Google Scholar]
- 5.Gobert M, Treilleux I, Bendriss-Vermare N, Bachelot T, Goddard-Leon S, Arfi V, Biota C, Doffin AC, Durand I, Olive D et al.. Regulatory T cells recruited through CCL22/CCR4 are selectively activated in lymphoid infiltrates surrounding primary breast tumors and lead to an adverse clinical outcome. Cancer Res 2009; 69:2000-9; PMID:19244125; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-2360 [DOI] [PubMed] [Google Scholar]
- 6.Zhou M, Bracci PM, McCoy LS, Hsuang G, Wiemels JL, Rice T, Zheng S, Kelsey KT, Wrensch MR, Wiencke JK. Serum macrophage-derived chemokine/CCL22 levels are associated with glioma risk, CD4 T cell lymphopenia and survival time. Int J Cancer 2015; 137:826-36; PMID:25604093; http://dx.doi.org/ 10.1002/ijc.29441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Menetrier-Caux C, Faget J, Biota C, Gobert M, Blay JY, Caux C. Innate immune recognition of breast tumor cells mediates CCL22 secretion favoring Treg recruitment within tumor environment. Oncoimmunology 2012; 1:759-61; PMID:22934274; http://dx.doi.org/ 10.4161/onci.19680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cao L, Hu X, Zhang J, Huang G, Zhang Y. The role of the CCL22-CCR4 axis in the metastasis of gastric cancer cells into omental milky spots. J Transl Med 2014; 12:267; PMID:25245466; http://dx.doi.org/ 10.1186/s12967-014-0267-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pere H, Montier Y, Bayry J, Quintin-Colonna F, Merillon N, Dransart E, Badoual C, Gey A, Ravel P, Marcheteau E et al.. A CCR4 antagonist combined with vaccines induces antigen-specific CD8+ T cells and tumor immunity against self antigens. Blood 2011; 118:4853-62; PMID:21908423; http://dx.doi.org/ 10.1182/blood-2011-01-329656 [DOI] [PubMed] [Google Scholar]
- 10.Munir S, Andersen GH, Met O, Donia M, Frosig TM, Larsen SK, Klausen TW, Svane IM, Andersen MH. HLA-restricted cytotoxic T cells that are specific for the immune checkpoint ligand PD-L1 occur with high frequency in cancer patients. Cancer Res 2013; 73:1674-776; PMID:23328583; http://dx.doi.org/12538675 10.1158/0008-5472.CAN-12-3507 [DOI] [PubMed] [Google Scholar]
- 11.Gnjatic S, Atanackovic D, Matsuo M, Jager E, Lee SY, Valmori D, Chen YT, Ritter G, Knuth A, Old LJ. Cross-presentation of HLA class I epitopes from exogenous NY-ESO-1 polypeptides by nonprofessional APCs. J Immunol 2003; 170:1191-6; PMID:12538675; http://dx.doi.org/ 10.4049/jimmunol.170.3.1191 [DOI] [PubMed] [Google Scholar]
- 12.Faget J, Biota C, Bachelot T, Gobert M, Treilleux I, Goutagny N, Durand I, Léon-Goddard S, Blay JY, Caux C et al.. Early detection of tumor cells by innate immune cells leads to T(reg) recruitment through CCL22 production by tumor cells. Cancer Res 2011; 71:6143-52; PMID:21852386; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-0573 [DOI] [PubMed] [Google Scholar]
- 13.Ishida T, Ishii T, Inagaki A, Yano H, Komatsu H, Iida S, Inagaki H, Ueda R. Specific recruitment of CC chemokine receptor 4-positive regulatory T cells in Hodgkin lymphoma fosters immune privilege. Cancer Res 2006; 66:5716-22; PMID:16740709; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-0261 [DOI] [PubMed] [Google Scholar]
- 14.Walter S, Weinschenk T, Stenzl A, Zdrojowy R, Pluzanska A, Szczylik C, Staehler M, Brugger W, Dietrich PY, Mendrzyk R et al.. Multipeptide immune response to cancer vaccine IMA901 after single-dose cyclophosphamide associates with longer patient survival. Nat Med 2012; 18:1254-61; PMID:22842478; http://dx.doi.org/ 10.1038/nm.2883 [DOI] [PubMed] [Google Scholar]
- 15.Dannull J, Su Z, Rizzieri D, Yang BK, Coleman D, Yancey D, Zhang A, Dahm P, Chao N, Gilboa E et al.. Enhancement of vaccine-mediated antitumor immunity in cancer patients after depletion of regulatory T cells. J Clin Invest 2005; 115:3623-33; PMID:16308572; http://dx.doi.org/ 10.1172/JCI25947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Henderson RA, Michel H, Sakaguchi K, Shabanowitz J, Appella E, Hunt DF, Engelhard VH. HLA-A2.1-associated peptides from a mutant cell line: a second pathway of antigen presentation. Science 1992; 255:1264-6; PMID:1546329; http://dx.doi.org/ 10.1126/science.1546329 [DOI] [PubMed] [Google Scholar]
- 17.Wolfel C, Drexler I, Van Pel A, Thres T, Leister N, Herr W, Sutter G, Huber C, Wölfel T. Transporter (TAP)- and proteasome-independent presentation of a melanoma-associated tyrosinase epitope. Int J Cancer 2000; 88:432-8; PMID:11054673; http://dx.doi.org/ 10.1002/1097-0215(20001101)88:3%3c432::AID-IJC16%3e3.0.CO;2-9 [DOI] [PubMed] [Google Scholar]
- 18.Yu W, Jiang N, Ebert PJ, Kidd BA, Muller S, Lund PJ, Juang J, Adachi K, Tse T, Birnbaum ME et al.. Clonal deletion prunes but does not eliminate self-specific alphabeta CD8(+) T lymphocytes. Immunity 2015; 42:929-41; PMID:25992863; http://dx.doi.org/24691076 10.1016/j.immuni.2015.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andersen MH. The targeting of immunosuppressive mechanisms in hematological malignancies. Leukemia 2014; 28:1784-92; PMID:24691076; http://dx.doi.org/ 10.1038/leu.2014.108 [DOI] [PubMed] [Google Scholar]
- 20.Andersen MH. Immune regulation by self-recognition: Novel possibilities for anticancer immunotherapy. J Natl Cancer Inst 2015; 107:154; PMID:26063792; http://dx.doi.org/10490979 10.1093/jnci/djv154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Andersen MH, Bonfill JE, Neisig A, Arsequell G, Sondergaard I, Valencia G, Neefjes J, Zeuthen J, Elliott T, Haurum JS. Phosphorylated peptides can be transported by TAP molecules, presented by class I MHC molecules, and recognized by phosphopeptide-specific CTL. J Immunol 1999; 163:3812-8; PMID:10490979 [PubMed] [Google Scholar]
- 22.Met O, Balslev E, Flyger H, Svane IM. High immunogenic potential of p53 mRNA-transfected dendritic cells in patients with primary breast cancer. Breast Cancer Res Treat 2011; 125:395-406; PMID:20336365; http://dx.doi.org/ 10.1007/s10549-010-0844-9 [DOI] [PubMed] [Google Scholar]
- 23.Toebes M, Coccoris M, Bins A, Rodenko B, Gomez R, Nieuwkoop NJ, van de Kasteele W, Rimmelzwaan GF, Haanen JB, Ovaa H et al.. Design and use of conditional MHC class I ligands. Nat Med 2006; 12:246-51; PMID:16462803; http://dx.doi.org/ 10.1038/nm1360 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.