

Abstract
Approximately 25% of subjects with common variable immunodeficiency (CVID) develop autoimmune disease. We analyzed Tcell subsets, specifically regulatory Tcells along with B cell subsets to determine whether there were changes in regulatory T cells which would correlate with the autoimmune disease clinical phenotype in CVID subjects. We hypothesized that regulatory T cell (CD4+CD25hiCD127lo) suppressive function would be impaired in CVID subjects with autoimmune disease. Using purified, sorted Treg from CVID subjects (n=14) and from healthy controls (HC, n=5) in standard suppression assays, we found the suppressive function of Treg from CVID subjects with autoimmune disease (CVID w/ AI, n=8) to be significantly attenuated compared to CVID subjects with no autoimmune disease (CVID w/o AI, n=6) and to HC (n=5). A number of proteins associated with Treg function were decreased in expression as detected through immunofluorescent antibody via flow cytometry (mean fluorescence intensity (MFI) of FoxP3, Granzyme A, XCL1, pSTAT5, and GITR in Treg was significantly lower (by up to 3 fold) in CVID w/ AI compared to CVID w/o AI and HC. Furthermore, a statistically significant correlation was found between intracellular MFI of FoxP3, Granzyme A, and pSTAT5 in Treg and the degree of Treg dysfunction. These results suggest that attenuation of Treg function is associated with autoimmune disease in CVID subjects and may contribute to autoimmune pathogenesis.
Keywords: Common variable immunodeficiency; T-lymphocytes; Regulatory; Autoimmunity; Immunologic deficiency syndromes; Granzymes; Lymphotactin; Xcl1 protein, human; FOXP3 protein, human; TNFRSF18 protein, human; STAT5 transcription factor
Introduction
Common variable immunodeficiency (CVID) is a primary immunodeficiency characterized by low levels of serum IgG, IgA, and/or IgM, deficient antigen-specific antibody responses, and recurrent infections [1-4]. Approximately 25% of subjects with CVID suffer from autoimmune disease [5,6] which could be the result of immune dysregulation resulting in autoantibody formation to a variety of antigens. The autoimmune diseases vary from autoimmune hemolytic anemia, immune thrombocytopenia purpura, pernicious anemia, vitiligo, psoriasis, vasculitis, rheumatoid arthritis, kerato-conjunctivitis sicca, thyroiditis, to chronic autoimmune neutropenia [6-8]. Much research on B cell immunophenotyping has led to our understanding that CVID w/ AI is associated with decreased numbers of switched memory B cells (CD27+, IgM-, IgD-phenotype) and elevated serum levels of B-cell activating factor of the TNF family (BAFF) and a proliferation-inducing ligand (APRIL); CVID subjects with transmembrane activator and calcium-modulator and cyclophilin ligand interactor (TACI) mutations also have a higher frequency of autoimmunity [8-16]. Recently, the CVID registry, established in 1996, divided 334 patients with CVID into 5 clinical phenotypes and reported that lower levels of CD8+ cells correlated with autoimmunity [17]. However, since CVID is a polygenic disease and since T cells are important to B cell development and function, we focused on Tcell phenotyping. In addition, other laboratories have found that multiple T cell abnormalities ranging from loss of CD4+ naïve cells, disrupted T cell receptor B variable repertoires, to altered cytokine production existing in CVID subjects [3,18-20]. Therefore, we designed our studies to test whether a specific subtype of T cells, the regulatory T cell, was impaired in CVID patients with autoimmune disease.
T regulatory cells (Treg) have been shown to be involved in a number of autoimmune diseases, but little research has studied their role in autoimmune disease in CVID. We have focused on Treg since their repertoire may be established very early on in life, and has implications for the proper suppression of immune dysfunction in childhood and adulthood [21]. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX), which is due to a genetic defect in FoxP3 (forkhead box) leading to Treg deficiency, is associated with severe autoimmune disease [22-24]. Subjects with active systemic lupus erythematosus (SLE) have significantly lower numbers of Treg than subjects with either inactive disease or without autoimmunity [25]. In subjects with multiple sclerosis, Treg function is decreased [26], while Treg numbers are decreased in subjects with active Crohn’s disease compared to those with inactive disease [27].
Recent published work has showed that CVID subjects have lower number of Treg compared to healthy controls [20]. A correlation between splenomegaly and Treg numbers was also shown, but a comparison between Treg and autoimmune disease was not performed in these CVID subjects. Furthermore, the functional characteristics of Treg were not analyzed in these subjects.
Published work with Treg cells has revealed a number of proteins that are necessary for the development and/or function of these cells. FoxP3 is a transcription factor that is associated with Treg development and function [28] and Granzyme A is involved in the granzyme/perforin pathway by which Treg cells kill target cells [29,30]. XCL1 (lymphotactin) is a chemokine that has been previously shown to be directly correlated with Treg function in suppression and cytotoxicity [31] and pSTAT5 (phosphorylated signal transducers and activator of transcription 5) is an intracellular protein critical for in vivo accumulation of functional Treg [32]. GITR (glucocorticoid-induced TNF receptor) and CTLA-4 have been shown to be important for modulating Treg cell function [33-35].
In this study, CVID subjects with and without a diagnosis of autoimmune disease followed at Stanford Hospital and Clinics and Lucille Packard Children’s Hospital were evaluated for Treg function and phenotype. Healthy control subjects were recruited for comparison analysis. Disease controls with X-linked agammaglobulinema (XLA) without autoimmunity were also analyzed.
Materials and methods
Human subjects
The study was approved by the Stanford Administrative Panel on Human Subjects in Medical Research. All subjects signed informed consent forms before participating in the study. Fourteen subjects with CVID were recruited from Stanford Hospital and Clinics and Lucile Packard Children’s Hospital, as well as 5 healthy adult controls. Two subjects with X-linked agammaglobulinemia (XLA) as identified by Bruton’s tyrosine kinase deficiency were enrolled as controls. The study was carried out under the Declaration of Helsinki Guidelines. Blood was drawn at least two times during the course of the study from each subject to determine reproducibility of phenotyping and function of Treg.
Clinical laboratory testing
Stanford Clinical Laboratories performed most laboratory testing stated in Table 1 except for T cell proliferation (antigen and mitogen tests) studies which were performed at FOCUS Diagnostic laboratories (Cypress, California). Regulatory T cell (CD4+CD25hiCD127lo and CD4+CD25neg cell counts were calculated from complete blood count and total CD3+ counts provided by the Stanford Clinical laboratories. Autoantibody testing confirmation was performed for each subject prior to the start of immunoglobulin therapy. Autoantibody screening occurred for each subject (antineutrophil antibody, anti-thyroglobulin antibody, anti-thyroid peroxidase, anti-intrinsic factor, Coombs’ positivity, antinuclear antibody, and rheumatoid factor).
Table 1.
Clinical characteristics of CVID patients with and without autoimmune disease
| Pt | Age (yrs) | Sex | CVID main clinical features | Details if T cell dysfunction present | Autoimmune disease | Details on auto antibody testing | Infected at time of study | Associated disease features |
|---|---|---|---|---|---|---|---|---|
| 1 | 41 | F | Low IgG, IgA and IgM, Ab dysfunction and T cell dysfunction | Poor response to tetanus on T cell proliferation studies | Thyroiditis | Antithyroglobulin antibodies present | No | Bronchiectasis |
| 2 | 39 | M | Low IgG, IgA and IgM, Ab dysfunction and T cell dysfunction | Poor response to candida on T cell proliferation studies | Neutropenia, anemia | Bone marrow with increase ingranulocytes and a T-cell infiltrate suggestive of an autoimmune induced neutropenia; responsive to GCSF), direct Coombs positive anemia | No | Chronic thrush, Diarrhea (non infectious etiology to date) |
| 3 | 55 | F | Low IgG, IgA and IgM, Ab dysfunction | Thyroiditis | Antithyroglobulin antibodies present | No | Bronchiectasis | |
| 4 | 16 | M | Low IgG, IgA and IgM, Ab dysfunction | Neutropenia | Antineutrophil antibodies present | No | Bronchiectasis, anemia (negative Coombs however, no autoAb to intrinsic factor or parietal cells performed to rule out pernicious anemia), nephrotic syndrome | |
| 5 | 8 | M | Low IgG, IgA and IgM, Ab dysfunction and T cell dysfunction | Poor response to tetanus and candida on T cell proliferation studies | Anemia, inflammatory bowel disease (diagnosis by colonoscopy) | Direct Coombs positive anemia | No (stool culture, O&P, Giardia, and C. dificile all negative) | |
| 6 | 6 | F | Low IgG, IgA and IgM, Ab dysfunction and T cell dysfunction | Poor response to tetanus and candida on T cell proliferation studies | Anemia, inflammatory bowel disease (no biopsy was performed)* | Direct Coombs positive anemia | No (stool culture, O&P, Giardia, and C. dificile all negative) | |
| 7 | 6 | F | Low IgG, IgA and IgM, Ab dysfunction | Hepatitis, anemia | Direct Coombs positive anemia | No | Anemia also secondary to B- thalassemia trait | |
| 8 | 66 | F | Low IgG, IgA and IgM, Ab dysfunction | Thyroiditis | Antithyroglobulin antibodies present | No | Bronchiectasis | |
| 9 | 21 | M | Low IgG, IgA and IgM, Ab dysfunction | N/A | None detected | No | Bronchiectasis | |
| 10 | 31 | F | Low IgG, IgA and IgM, Ab dysfunction | N/A | None detected | No | Bronchiectasis | |
| 11 | 11 | M | Low IgG, IgA and IgM,, Ab dysfunction | N/A | None detected | No | Asthma, bronchiectasis | |
| 12 | 33 | M | Low IgG, IgA and IgM,, Ab dysfunction | N/A | None detected | No | Bronchiectasis, asthma | |
| 13 | 67 | M | Low IgG, IgA and IgM, Ab dysfunction | N/A | None detected | No | Bronchitis, sinusitis | |
| 14 | 21 | M | Low IgG, IgA and IgM, Ab dysfunction | N/A | None detected | No | Gastritis, asthma, schizoaffective disorder | |
| 15 | 16 | M | Bruton’s XLA | N/A | None detected | No | Right eye blindness due to CMV infection | |
| 16 | 35 | M | Bruton’s XLA | N/A | None detected | No | Left lower leg paralysis due to polio infection in infancy |
Note: No subject underwent splenectomy.
Antibodies
FITC-conjugated anti-CD25 (ACT-1) antibody used in sorting was purchased from DakoCytomation (Chicago, IL). The following antibodies and cognate isotypes were purchased from the indicated sources and used as per manufacturer’s instructions: Live/Dead, CD3, GITR, CD4, CD25, CD45RO, CD45RA, CD28, CD127, CD19, pSTAT5, CTLA4, CD19, CD27, IgM and IgD (BD Biosciences). Granzyme A (PE-conjugated, Caltag), FoxP3 (BioLegend clone 259D-Alexa Fluor 488), and XCL1 (R&D Systems).
Cell purification
CD4+ T cells were first isolated from whole blood with CD4+ Rosette Kit (Stemcell Technologies). CD4+ T cells were incubated with anti-CD127-APC and anti-CD25-PE and anti CD4-FITC antibodies (BD Biosciences) and sorted by flow cytometry using a FACS Aria (BD Biosciences) into CD4+CD25hiCD127lo/− Treg and CD4+CD25- responder T cells.
T-cell suppression assay
Suppression assays were performed in round-bottom 96-well microtiter plates. 3.75 × 103 CD4+CD25neg responder T cells, 3.75 × 103 autologous Treg (except in described mixing studies), 3.75 × 104 allogeneic irradiated CD3-depleted PBMCs were added. All wells were supplemented with anti-CD3 (clone HIT3a at 5.0 μg/ml). T cells were cultured for 7 days at 37 °C in RPMI 1640 medium supplemented with 10% fetal bovine serum. Sixteen hours before the end of the incubation, 1.0 μCi of 3H-thymidine was added to each well. Plates were harvested using a Tomtec cell harvester and 3H-thymidine incorporation determined using a Perkin Elmer scintillation counter. Antigen-presenting cells (APC) consisted of PBMC depleted of T cells using StemSep human CD3+ T cell depletion (StemCell Technologies) followed by 40 Gy of irradiation.
Stimulation for phospho-STAT5 analysis
Treg were stimulated with anti-CD3 and anti-CD28 conditions and then stained with Live/Dead Red viability dye (Invitrogen), and surface markers CD4, CD25, and CD127. Cells were then fixed in the Lyze/Fix PhosFlow buffer®, and then permeabilized in denaturing Permbuffer III® (BD Biosciences). They were then stained with antibodies against phospho-STAT5 according to manufacturer’s instructions.
Flow cytometric analysis
Blood samples from CVID patients were freshly collected, peripheral blood mononuclear cells (PBMCs) were prepared as described previously [36]. PBMCs were stained for 20 min at 4 °C with a mixture of antibodies at optimal concentrations. Acquisition occurred was performed via LSR II (BD Biosciences). Data were analyzed with FlowJo analysis software (Tree Star, Inc. Ashland, OR).
Statistical analysis
Statistical analysis was performed using GraphPad Prism software. Comparisons between two groups were made by using the two-tailed, unpaired Student’s t-test. Comparisons between three groups were made by using 1-way ANOVA. p values less than 0.05 were considered significant (two-tailed). Correlation between two groups were determined to be statistically significant via the Spearman coefficient if the r value was greater than 0.7 or less than −0.7.
Results
Description of subjects
From 2005 to 2008, 14 CVID subjects were enrolled in our study. All these subjects were under longitudinal medical care by a Clinical Immunologist at Stanford Medical Center. Healthy controls (n = 5) and XLA subjects (n = 2) were assessed, determined not to have autoimmune disease by history and chart review and every attempt to sex-match and age-match was done (Table 1). The CVID subjects ranged from 6 years to 67 years of age with a mean age of 32 years of age. 53% were male and 47% were female. Of the 14 CVID subjects, 8 had autoimmune disease (as diagnosed with standard laboratory tests and as per medical history and physical exam findings), while 6 did not. Three subjects had autoimmune thyroiditis, 2 had autoimmune neutropenia, 2 had autoimmune anemia and inflammatory bowel disease (without protein losing enteropathy), and 1 had autoimmune hepatitis. Four of the CVID subjects had T cell proliferation responses which were below those of normal calibrated responses (FOCUS Diagnostic laboratories, standard mitogen and antigen proliferative response assays) (Table 1). None of the subjects had lymphadenopathy, splenomegaly or granulomas as determined by physical exam and/or abdominal ultrasound. All CVID and XLA subjects are currently alive and all currently receive immunoglobulin therapy. None of the patients were on immunosuppressive therapy (i.e. antiproliferatives or steroids) or any drug that could cause hypogammaglobulinemia (i.e. sulfasalazine, phenytoin, gold salts, and antimalarial drugs) at the time of the blood draw.
Since many groups established the importance of classifying CVID subjects by phenotyping of B cell subpopulations [8,17,37] and a recent study showed, after evaluating 105 CVID subjects, that reduced numbers of switched memory B cells (≤ to 0.55% of B cells) were an independent risk factor of autoimmune diseases [16], we performed CD27, IgD and IgM classifications on B cells in the CVID and HC subjects (XLA subjects had no B cells) (Supplementary Fig. 1 for B cell gating, Table 2). In addition, we also enumerated Treg and CD4+ T cells (Table 2).
Table 2.
Cell counts of CVID patients with and without autoimmune disease
| Patient | % Memory/switched B cell phenotype (CD19+CD27+IgM−IgD−) | % CD45RO Treg phenotype | Absolute # CD4+ T cells (×103 per mL) | Absolute number CD4+CD25hi Tregs (×103 per mL) |
|---|---|---|---|---|
| 1 | 0.5 | 80 | 431 | 6 |
| 2 | 0.7 | 70 | 247 | 3 |
| 3 | 1.2 | 64 | 378 | 4 |
| 4 | 0.6 | 73 | 469 | 7 |
| 5 | 1.3 | 60 | 362 | 4 |
| 6 | 0.9 | 56 | 435 | 7 |
| 7 | 1.0 | 64 | 394 | 6 |
| 8 | 0.4 | 89 | 429 | 5 |
| 9 | 3.1 | 32 | 254 | 13 |
| 10 | 2.1 | 36 | 391 | 16 |
| 11 | 3.5 | 30 | 462 | 22 |
| 12 | 2.9 | 35 | 395 | 20 |
| 13 | 3.3 | 30 | 304 | 15 |
| 14 | 2.8 | 26 | 283 | 13 |
| 15 | 0 | 45 | 460 | 19 |
| 16 | 0 | 44 | 327 | 15 |
Decreased number of switched memory B cells in CVID subjects with autoimmune disease
Our data demonstrated that CVID w/ AI subjects had lower levels of switched memory B cells. Differences between males and females were not statistically significant. Treg numbers differed significantly between CVID w/ AI vs CVID w/o AI, HC, XLA groups (p≤0.05) (Table 2). Blood was drawn from subjects at least two times during the course of our study (with time intervals of the second blood draw being 6 months to 12 months from the first blood draw). All results (excluding autoantibody testing which was done before immunoglobulin therapy was initiated) were reproducible in each subject. We did not detect significant variability in Treg function/phenotyping or variability in B cell phenotype numbers over the time period between blood draws in each subject.
Functional suppression by Treg is significantly decreased in CVID subjects with autoimmune disease
Standard Treg function tests for suppression were performed and representative subjects are shown (Fig. 1a). Responder T cells and Treg were mixed at 1:1 ratios. Healthy Control (HC) Treg suppressed the proliferation of activated responder T cells but Treg from CVID subjects with autoimmune disease had impaired capacity to suppress activated T cells. Treg from XLA subjects worked effectively to suppress autologous responder T cell proliferation.
Figure 1.
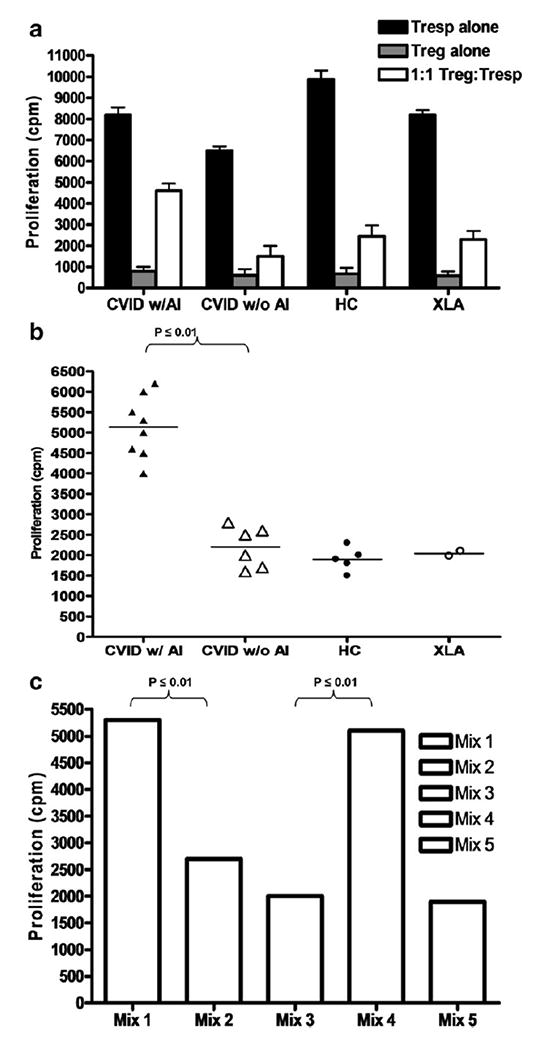
(a) Treg suppression assay in a representative CVID subject with and a representative CVID without autoimmune disease (AI) and representative healthy controls (HC) and representative XLA subject. The x-axis shows responder cells (activated autologous CD4+ effector Tcells) alone in the black bar and natural regulatory T cells alone in the gray bar (shown for confirmation of anergy) which were then mixed at 1:1 ratio as shown in the white bar. The y-axis shows proliferation as measured by uptake of 3H-thymidine (in cpm or counts per minute) The bars represent the standard deviation from the mean. (b) Treg suppression assay results displayed for all subjects (as calculated by autologous Tsuppression assay with Treg:Tresp as a 1:1 ratio) in CVID subjects with and without autoimmune disease compared to healthy controls and XLA subjects. The y-axis shows the numerical quantity of Treg dysfunction as uptake of 3H-thymidine (in cpm or counts per minute). Each dot represents an individual subject. (c) Mixing experiments of Treg and Tresp cells from different patients at 1:1 ratios. Treg derived from CVID w/ AI with Tresp from CVID w/o AI (Mix 1), Treg derived from CVID w/o AI with Tresp from CVID w/ AI (Mix 2), Treg derived from HC with Tresp from CVID w/ AI (Mix 3), Treg derived from CVID w/ AI with Tresp from HC (Mix 4) and Treg derived from XLA with Tresp from HC (Mix 5). A representative pair for each mixing experiment is shown. The y-axis shows the numerical quantity of Treg dysfunction as uptake of 3H-thymidine (in cpm or counts per minute).
To summarize the data with all the subjects tested, we graphed the 3H Thymidine proliferation (counts per minute or cpm) for each autologous Treg:Teff co culture for each subject. CVID subjects with autoimmune disease have a 4 to 6 times greater fold Treg dysfunction compared to healthy controls (p≤0.01) (Fig. 1b). Furthermore, CVID subjects with autoimmune disease have a statistically significant greater fold difference in Treg dysfunction compared to CVID subjects without autoimmune disease (p≤0.01). In contrast, CVID subjects without autoimmune disease have a 1 to 1.5 greater fold Treg dysfunction compared to healthy controls, which is not statistically significant (p>0.05). With limited numbers of subjects, there were no trends for differences in Treg dysfunction in subtypes of autoimmune disease in the CVID subjects.
Next, we tested whether the defect in Treg suppressive function was due to overactivity of responder T cells (Tresp) or impaired Treg in CVID subjects. Therefore, we mixed Treg derived from CVID w/ AI with Tresp from CVID w/o AI in a 1:1 ratio (Mix 1), Treg derived from CVID w/o AI with Tresp from CVID w/ AI (Mix 2), Treg derived from HC with Tresp from CVID w/ AI (Mix 3), Treg derived from CVID w/ AI with Tresp from HC (Mix 4) and Treg derived from XLA with Tresp from HC (Mix 5). A representative pair for each mixing experiment is shown in Figure 1c. The Treg purified from the subjects who had decreased Tcell responses to mitogens and antigens were independently dysfunctional in mixing assays.
From this analysis, the data suggest that it is the Treg, not Tresp, from CVID w/ AI which lead to the attenuation of suppressive function. In addition to Treg numbers being decreased in peripheral blood cells from CVID w/ AI subjects, Treg from these same subjects were dysfunctional compared to HC, CVID, and XLA subjects.
Protein expression associated with Treg function is significantly lower in CVID subjects with autoimmune disease
Because Treg appeared to be dysfunctional in CVID subjects with autoimmune disease, we evaluated the expression of proteins associated with Treg function in these subjects. Specifically, we examined levels of FoxP3, Granzyme A, XCL1 (lymphotactin), pSTAT5, and GITR [28,29,31–33] via flow cytometry analysis of directly conjugated immunofluorescent labeled antibodies. Treg (CD4+CD25hiCD127lo) were gated as shown (Fig. 2) and co stained independently with antibodies for the above markers. The percentage and mean fluorescence intensity (MFI) of FoxP3 in Treg were significantly lower in CVID subjects with autoimmune disease compared to healthy controls (p ≤ 0.001) (Fig. 3a). The percentage and intracellular MFI of Granzyme A in Treg were also significantly lower in CVID subjects with autoimmune disease compared to healthy controls (p ≤ 0.001) (Fig. 3b). However, no statistically significant differences were found between the percentage of Granzyme A-expressing Treg or the MFI of Granzyme A in Treg between CVID subjects with autoimmune disease compared to those without autoimmune disease. The percentage of XCL1-expressing Treg and the MFI of XCL1 in Treg was significantly lower in CVID subjects with autoimmune disease compared to healthy controls (p ≤0.001) (Fig. 3c). The percentage and the MFI of pSTAT5 in Treg was significantly lower in CVID subjects with autoimmune disease compared to healthy controls (p ≤ 0.001 and p ≤ 0.01 respectively) (Fig. 3d). The percentage of pSTAT5-expressing Treg was also significantly decreased in CVID subjects with autoimmune disease compared to those without autoimmune disease (p≤0.05). The percentage and the MFI of GITR in Treg was significantly lower in CVID subjects with autoimmune disease compared to healthy controls (p ≤ 0.001) (Fig. 3e). However, the percentage of GITR-expressing Treg and the MFI of GITR in Treg was not statistically different in CVID subjects with autoimmune disease compared to those without autoimmune disease (p > 0.05) or healthy controls (p > 0.05). We also examined CTLA-4 levels in Treg from CVID subjects with and without autoimmune disease vs. HC and no differences were detected (data not shown).
Figure 2.
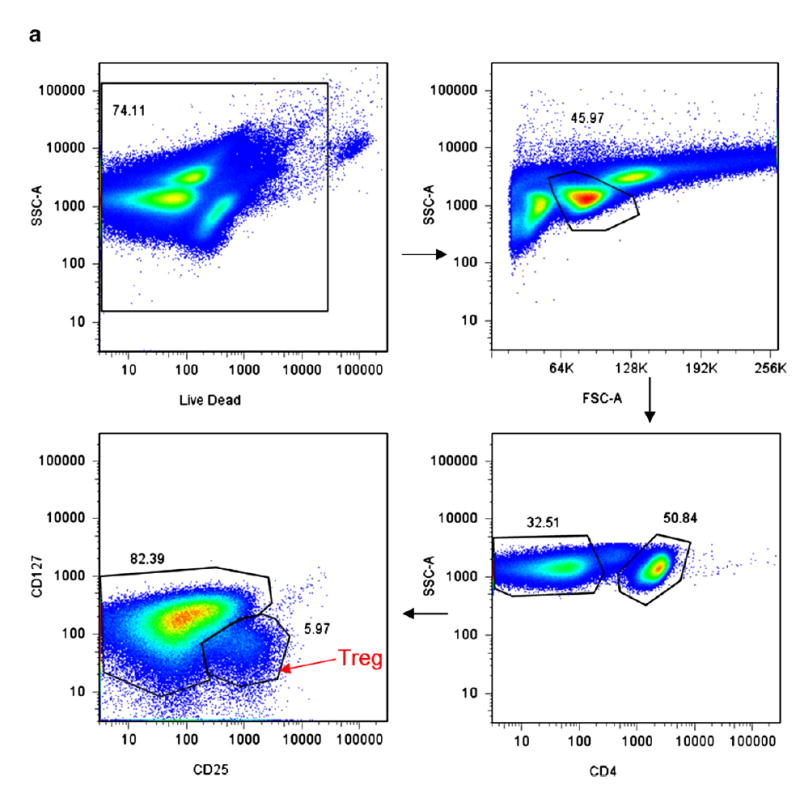
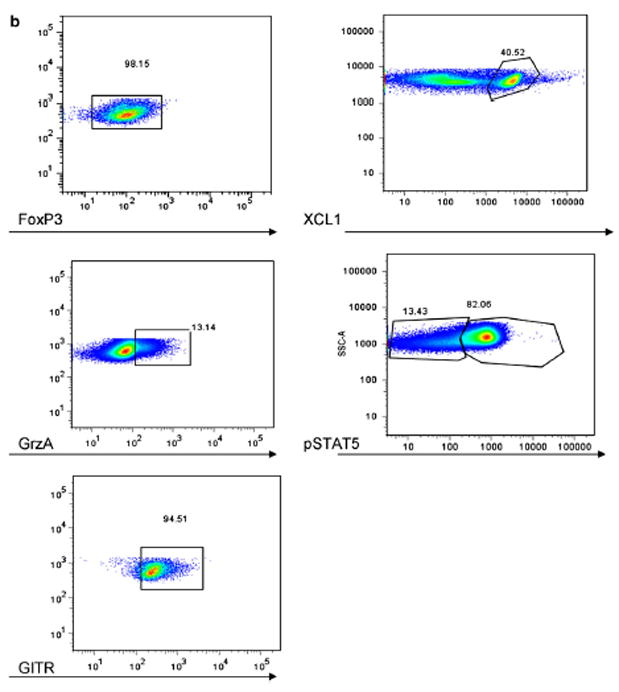
(a) Representative HC sample for Treg gating strategy. PBMC (peripheral blood mononuclear cells) were stained for LiveDead Stain (Invitrogen), CD4+, CD25hi, CD127lo. They were then gated by Live/Dead, then SSC and FSC for the lymphocyte gate, then CD4+, then CD127lo/CD25hi cells. This gating strategy was used to gate for all Treg. Using this Treg gating, additional markers were then assessed as in panel b. (b) Representative HC sample for gating strategies for the additional markers co stained with the Treg population (panel a).
Figure 3.
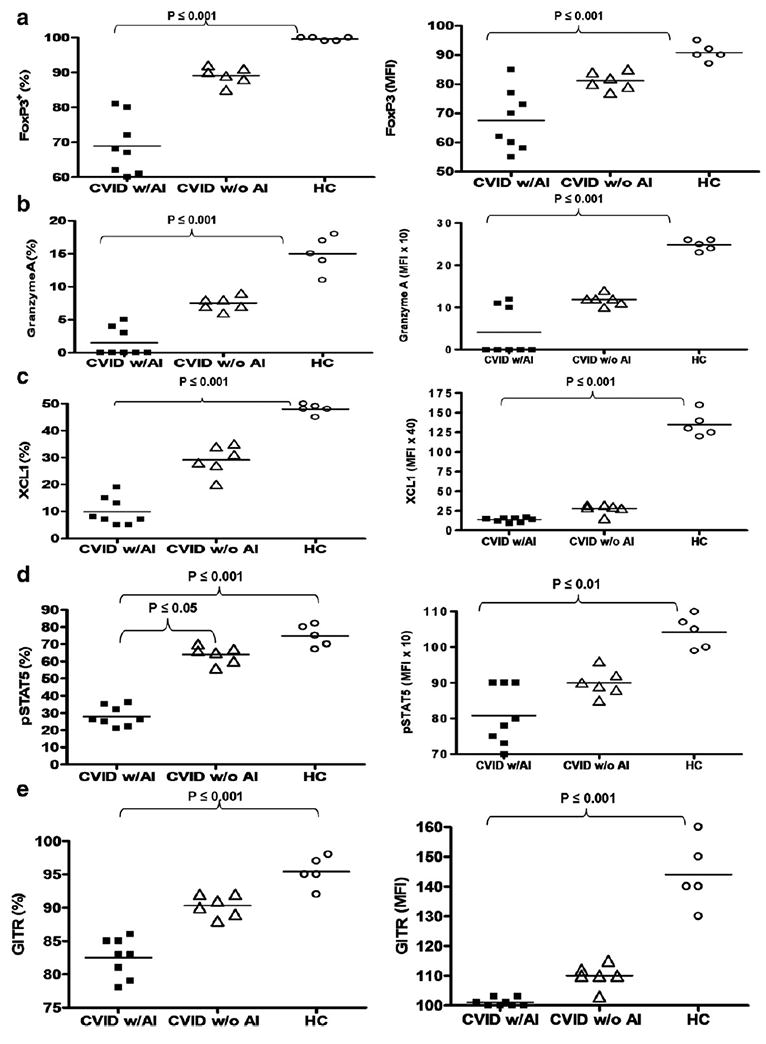
Percentages and MFI (mean fluorescence intensity) of FoxP3 (a), Granzyme A (b), XCL1 (c), pSTAT5 (d), and GITR (e) in Treg of CVID subjects with and without autoimmune disease and healthy controls. Each dot represents an individual subject. The bar represents the mean value and p values ≤ 0.05 were considered significant (two-tailed).
To test whether lower expression of GITR, XCL1, Granzyme A, and pSTAT5 was associated with lower expression of FoxP3 in CVID w/ AI subjects, we found that the decreases in MFI of XCL1 and of pSTAT5 correlated with decreases in MFI of FoxP3 but that decreases in MFI of Granzyme A and of GITR were independent of FoxP3 MFI expression levels (representative correlation studies of pSTAT5 and Granzyme A with FoxP3 are shown in Figures 4a and 4b, respectively).
Figure 4.
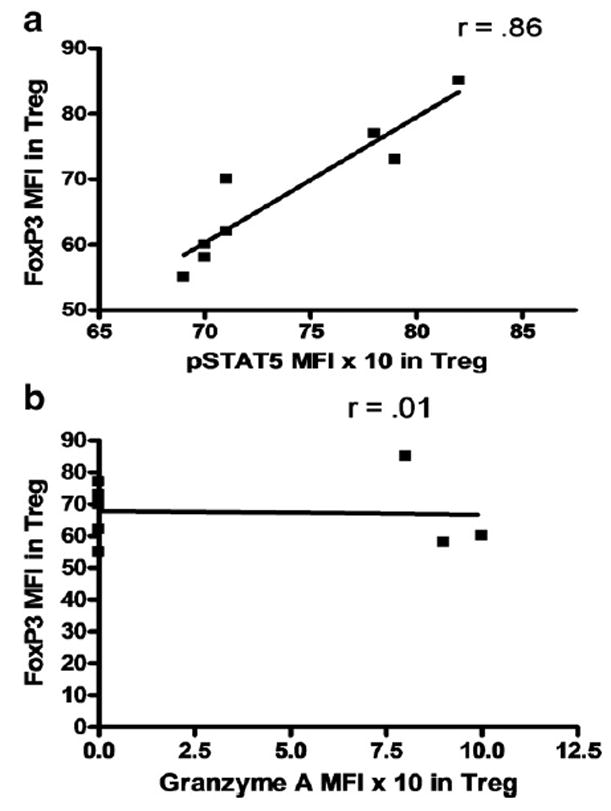
Correlation studies of MFI of Foxp3 and pSTAT5 MFI (a) and of Foxp3 and Granzyme A MFI (b) in Treg from CVID w/ AI subjects. Each dot represents an individual subject. r value>0.7 was considered significant (two-tailed).
We also evaluated if pSTAT5 decreases correlated with CD25 expression changes on the surface of Treg since we have previously found that pSTAT5b homozygote deficiency leads to CD25 decreases [32]. Over the time course of the pSTAT5 stimulation experiment, we did not detect any changes in CD25 expression on Treg (data not shown).
Correlation between protein expression associated with Treg function and amount of Treg dysfunction in CVID subjects with autoimmune disease
We analyzed if Treg dysfunction correlated with the expression of proteins involved in Treg function in CVID subjects with autoimmune disease. We found statistically significant correlations between the percentage of FoxP3, Granzyme A, and pSTAT5 expression on Treg and Treg dysfunction (Figs. 5a,b,d). However, we did not find a statistically significant correlation between the percentage of XCL1 and GITR expression in Treg and Treg dysfunction in CVID subjects with autoimmune disease (Figs. 5c, e).
Figure 5.
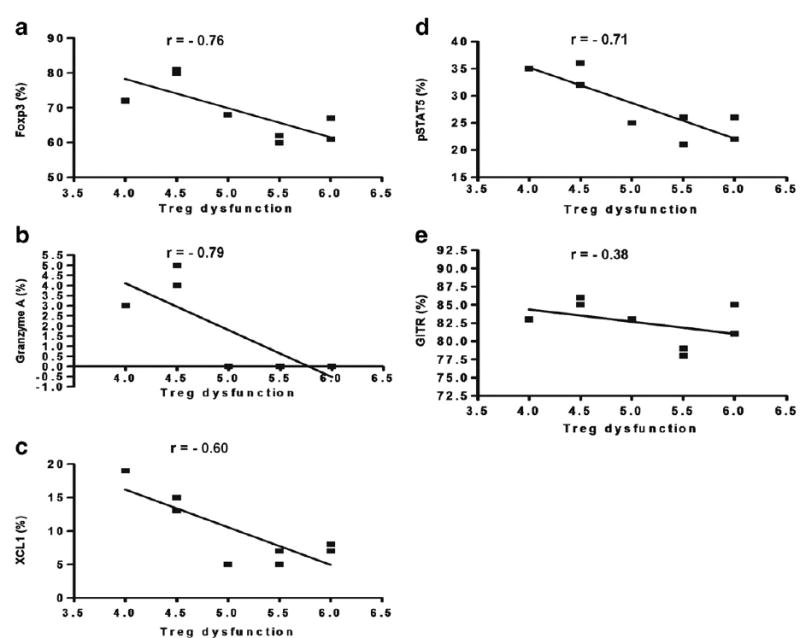
Correlation studies of % positive for FoxP3 (a), Granzyme A (b), XCL1 (c), pSTAT5 (d), and GITR (e) in Treg of CVID w/ AI subjects. Each dot represents an individual subject. r value<−0.7 was considered significant (two-tailed).
CD45RO expression in Treg is significantly increased in CVID subjects with autoimmune disease
Memory T cells are often distinguished by the CD45RO marker, and Treg activity is reported to involve both the memory and naïve T cells from the CD4+CD25hiCD127lo fraction of PBMC in healthy controls [30,38]. Treg (CD4+CD25hiCD127lo) were gated as shown (Fig. 2a) and co stained with antibodies for CD45RO and RA (Figs. 6a–c). We discovered that the percentage of memory CD45RO+-expressing Treg was significantly increased in CVID subjects with autoimmune disease compared to CVID subjects without autoimmune disease (p≤0.001) (Fig. 7a), even with the younger subjects in whom a higher amount of CD45RA+ cells would be expected. The percentage of CD45RA+-expressing Treg is significantly lower in CVID subjects with autoimmune disease compared to CVID subjects without autoimmune disease (p≤0.001) (Fig. 7b). Since we had examined percentages of switched memory B cell percentages (Table 2), we then tested whether there was a correlation with percentages of CD45RO+ Treg. Our data demonstrates that increases in CD45RO+ Treg are associated with decreases in switched memory B cells (Fig. 8).
Figure 6.
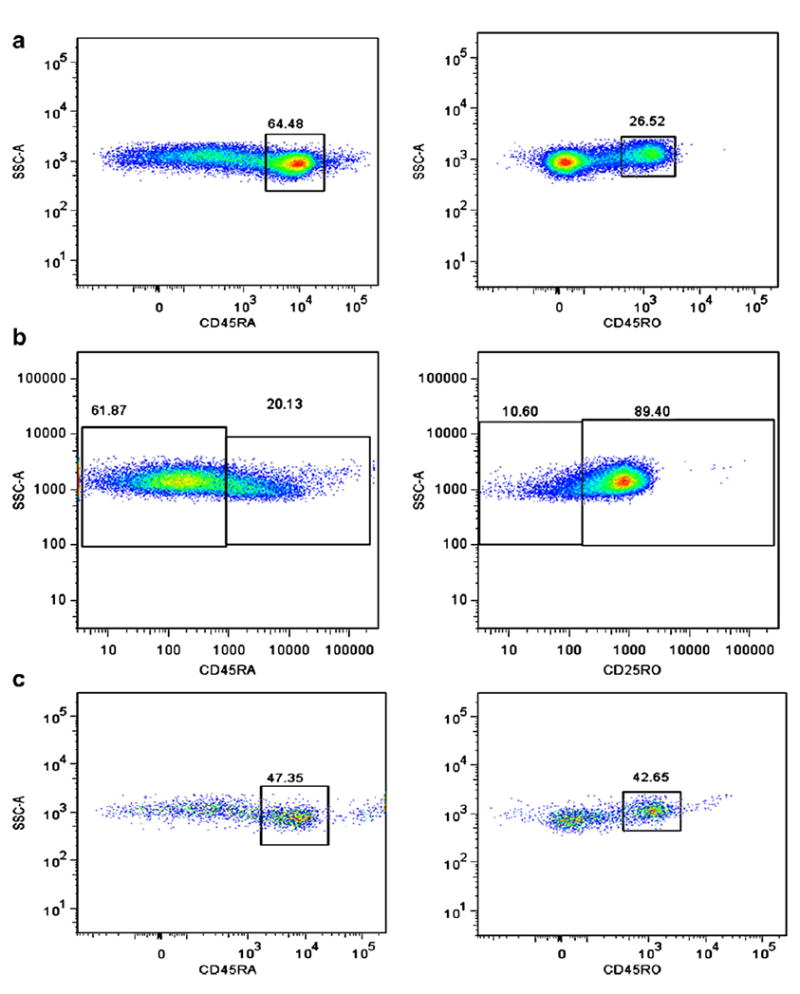
Gating strategy shown for CD45RA+ and CD45RO+ co stains of Treg of representative CVID w/o AI (a), CVID w/ AI (b) and HC (c).
Figure 7.
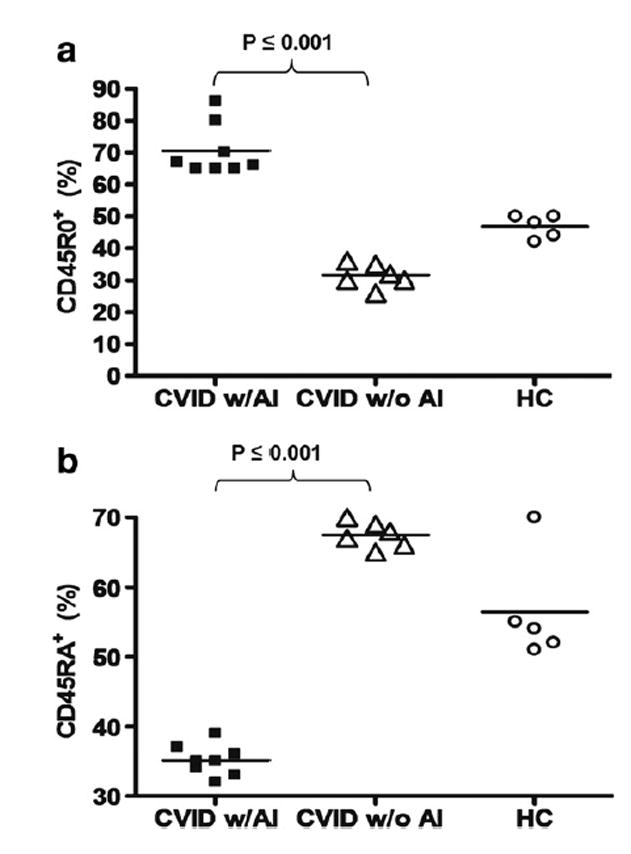
Percentage of CD45RO+ (a) and CD45RA+ (b) expressing Treg of CVID subjects with autoimmune disease, CVID without autoimmune disease, and healthy controls. Each dot represents an individual subject. The bar represents the mean value and p values ≤ 0.05 were considered significant (two-tailed).
Figure 8.
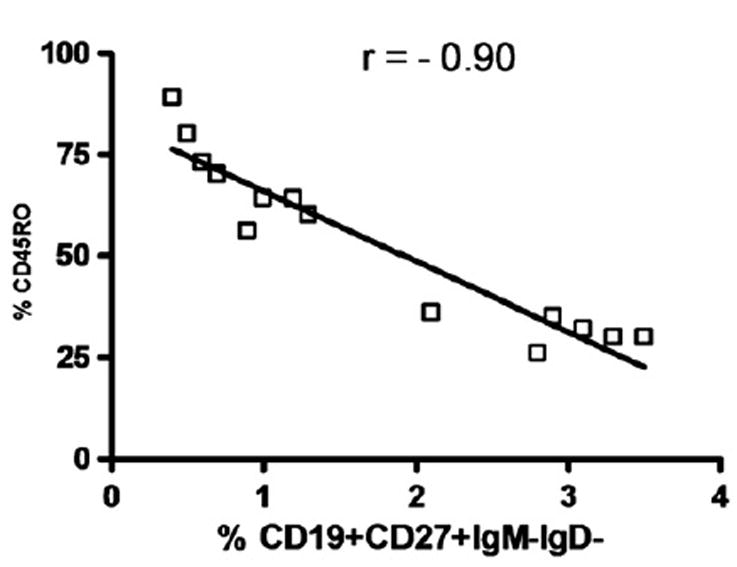
Correlation studies of % CD45RO+ Treg and % CD19+CD27+IgM-IgD- switched memory B cells in all CVID subjects. Each dot represents an individual subject. r value<−0.7 was considered significant (two-tailed).
Discussion
We have shown that CD4+CD25hiCD127lo Treg in CVID subjects with autoimmune disease are lower in number in the peripheral blood and have a significantly reduced ability to suppress proliferation of autologous CD4+ effector cells and allogenic CD4+ effector cells compared to subjects with CVID without autoimmune disease, healthy controls and XLA. This inability to suppress potentially autoreactive T cells suggests that dysfunctional Treg may play a role in autoimmune disease in CVID. The main primary immunodeficiencies that have been described involving reduced or absent Treg are IPEX syndrome and CVID.
The cellular studies of the immune system in CVID subjects to date have shown numerical and functional defects in B, T, natural killer cells, macrophages and monocytes [39–44]. T cell functional abnormalities are common (20% of CVID subjects) [4,45] with defects in cytokine production, T helper function, and T cell signaling [19,46–52]. Our series of subjects, however, is the first in the literature to show that Treg are decreased in number and function in CVID subjects with autoimmune disease.
To investigate the underlying mechanism, we looked at the expression of some of the key proteins involved in the function of Treg, including FoxP3, Granzyme A, XCL1 (lymphotactin), pSTAT5, and GITR. CVID subjects with autoimmune disease show statistically significant decreases in expression of all these proteins compared to healthy controls. Correlation between protein expression and Treg dysfunction was statistically significant for FoxP3, Granzyme A, and pSTAT5. This suggests that these may be the key proteins in Treg-mediated autoimmunity in CVID subjects. This is further supported by the evidence that mutations in FoxP3 cause lethal autoimmunity in the scurfy mouse and in humans suffering from IPEX syndrome. Granzyme A also is a key protein by which Treg likely kill target cells, including potentially autoreactive Tcells, and pSTAT5 is an intracellular protein critical for in vivo accumulation of functional Treg. In our studies, lower levels of FoxP3 expression correlated with pSTAT5 and XCL1 expression levels, but not with Granzyme A or GITR levels. This would suggest that Granzyme A and GITR are separately affected in the overall Treg defect mechanisms associated with CVID with autoimmune diseases.
We examined switched memory B cell percentages based on classifications in the literature [8,16,17] and tested whether there was a correlation with CD45RO+ Treg since we found that increases in CD45RO+ Treg in CVID with AI. Our data demonstrates that increases in CD45RO+ Treg are associated with decreases in switched memory B cells (p≤0.001) (Table 2 and Fig. 8). CD45RO+ Treg have been found to be less suppressive and produce less IL-10 compared to CD45RA+ Treg [53]. It is of interest to note that ICOS-deficient subjects produce low levels of IL-10 and this could be associated with defective B cell memory due to impairment of germinal center formation [54]. Therefore increased CD45RO+ Treg could lead to the decreased switched memory B cell phenotype in CVID subjects with autoimmunity. Further studies are needed to address this hypothesis.
To perform rigorous statistics, we performed ANOVA analysis with intergroup comparisons of CVID subjects with autoimmune disease, CVID subjects without autoimmune disease, XLA, and healthy controls, rather than Student’s t-test analysis on CVID subjects with autoimmune disease vs. CVID subjects without autoimmune disease. There were many trends for Treg phenotypic differences in CVID subjects with autoimmune disease and CVID subjects without autoimmune disease. It is possible that specific genetic causes of CVID will lead to differential outcomes of Treg function and phenotype. Stratifying the subject population for each particular autoimmune disease may also be important; we will continue to enroll larger subject numbers for this analysis working with cooperative networks. This will also enable our laboratory to study Treg enumeration, phenotype, and function in subjects with other clinical categories in CVID subjects: splenomegaly, lymphoproliferative disorders, and granulomas [8,16,17]. Some of the CVID subjects without autoimmune disease may develop autoimmune disease in the future and, as such, we plan on following all our subjects with CVID longitudinally.
In conclusion, we show for the first time that Treg from CVID subjects with autoimmune disease are decreased in the peripheral blood and have a significantly decreased ability to suppress CD4+ effector cells. Treg dysfunction may play an important role in autoimmunity in CVID. Furthermore, the examination of FoxP3, Granzyme A, pSTAT5, and CD45RO may be important in phenotyping Treg in CVID subjects with autoimmune disease.
Supplementary Material
Acknowledgments
We would like to thank Dr. David Lewis for the critical review of the manuscript. We appreciate the subjects and their families for their time and their blood samples. This study was funded by the Stanford Children’s Health Research Program and the Immunity, Transplantation and Infectious Disease Institute at Stanford University School of Medicine.
Footnotes
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.clim.2008.12.006.
References
- 1.Chapel H, Geha R, Rosen F. Primary immunodeficiency diseases: an update. Clin Exp Immunol. 2003;132:9–15. doi: 10.1046/j.1365-2249.2003.02110.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Conley ME, Notarangelo LD, Etzioni A. Diagnostic criteria for primary immunodeficiencies. Representing PAGID (Pan-American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies) Clin Immunol. 1999;93:190–197. doi: 10.1006/clim.1999.4799. [DOI] [PubMed] [Google Scholar]
- 3.Cunningham-Rundles C. Common variable immunodeficiency. Curr Allergy Asthma Rep. 2001;1:421–429. doi: 10.1007/s11882-001-0027-1. [DOI] [PubMed] [Google Scholar]
- 4.Cunningham-Rundles C, Bodian C. Common variable immunodeficiency: clinical and immunological features of 248 patients. Clin Immunol. 1999;92:34–48. doi: 10.1006/clim.1999.4725. [DOI] [PubMed] [Google Scholar]
- 5.Knight AK, Cunningham-Rundles C. Inflammatory and autoimmune complications of common variable immune deficiency. Autoimmun Rev. 2006;5:156–159. doi: 10.1016/j.autrev.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 6.Park MA, Li JT, Hagan JB, Maddox DE, Abraham RS. Common variable immunodeficiency: a new look at an old disease. Lancet. 2008;372:489–502. doi: 10.1016/S0140-6736(08)61199-X. [DOI] [PubMed] [Google Scholar]
- 7.Adkinson NF, Middleton E. Middleton’s Allergy: Principles & Practice. Mosby; Philadelphia, PA: 2003. [Google Scholar]
- 8.Wehr C, Kivioja T, Schmitt C, Ferry B, Witte T, Eren E, Vlkova M, Hernandez M, Detkova D, Bos PR, Poerksen G, von Bernuth H, Baumann U, Goldacker S, Gutenberger S, Schlesier M, Bergeron-van der Cruyssen F, Le Garff M, Debre P, Jacobs R, Jones J, Bateman E, Litzman J, van Hagen PM, Plebani A, Schmidt RE, Thon V, Quinti I, Espanol T, Webster AD, Chapel H, Vihinen M, Oksenhendler E, Peter HH, Warnatz K. The EUROclass trial: defining subgroups in common variable immunodeficiency. Blood. 2008;111:77–85. doi: 10.1182/blood-2007-06-091744. [DOI] [PubMed] [Google Scholar]
- 9.Zhang L, Radigan L, Salzer U, Behrens TW, Grimbacher B, Diaz G, Bussel J, Cunningham-Rundles C. Transmembrane activator and calcium-modulating cyclophilin ligand interactor mutations in common variable immunodeficiency: clinical and immunologic outcomes in heterozygotes. J Allergy Clin Immunol. 2007;120:1178–1185. doi: 10.1016/j.jaci.2007.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seshasayee D, Valdez P, Yan M, Dixit VM, Tumas D, Grewal IS. Loss of TACI causes fatal lymphoproliferation and autoimmunity, establishing TACI as an inhibitory BLyS receptor. Immunity. 2003;18:279–288. doi: 10.1016/s1074-7613(03)00025-6. [DOI] [PubMed] [Google Scholar]
- 11.Knight AK, Radigan L, Marron T, Langs A, Zhang L, Cunningham-Rundles C. High serum levels of BAFF, APRIL, and TACI in common variable immunodeficiency. Clin Immunol. 2007;124:182–189. doi: 10.1016/j.clim.2007.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cunningham-Rundles C. Autoimmune manifestations in common variable immunodeficiency. J Clin Immunol. 2008 doi: 10.1007/s10875-008-9182-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Warnatz K, Denz A, Drager R, Braun M, Groth C, Wolff-Vorbeck G, Eibel H, Schlesier M, Peter HH. Severe deficiency of switched memory B cells (CD27(+)IgM(−)IgD(−)) in subgroups of patients with common variable immunodeficiency: a new approach to classify a heterogeneous disease. Blood. 2002;99:1544–1551. doi: 10.1182/blood.v99.5.1544. [DOI] [PubMed] [Google Scholar]
- 14.Groom JR, Fletcher CA, Walters SN, Grey ST, Watt SV, Sweet MJ, Smyth MJ, Mackay CR, Mackay F. BAFF and MyD88 signals promote a lupuslike disease independent of Tcells. J Exp Med. 2007;204:1959–1971. doi: 10.1084/jem.20062567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee JJ, Ozcan E, Rauter I, Geha RS. Transmembrane activator and calcium-modulator and cyclophilin ligand interactor mutations in common variable immunodeficiency. Curr Opin Allergy Clin Immunol. 2008;8:520–526. doi: 10.1097/ACI.0b013e3283141200. [DOI] [PubMed] [Google Scholar]
- 16.Sanchez-Ramon S, Radigan L, Yu JE, Bard S, Cunningham-Rundles C. Memory B cells in common variable immunodeficiency: clinical associations and sex differences. Clin Immunol. 2008;128:314–321. doi: 10.1016/j.clim.2008.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chapel H, Lucas M, Lee M, Bjorkander J, Webster D, Grimbacher B, Fieschi C, Thon V, Abedi MR, Hammarstrom L. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood. 2008;112:277–286. doi: 10.1182/blood-2007-11-124545. [DOI] [PubMed] [Google Scholar]
- 18.Giovannetti A, Pierdominici M, Aiuti F. T-cell homeostasis: the dark(ened) side of common variable immunodeficiency. Blood. 2008;112:446. doi: 10.1182/blood-2008-03-145045. [DOI] [PubMed] [Google Scholar]
- 19.Cunningham-Rundles C, Bodian C, Ochs HD, Martin S, Reiter-Wong M, Zhuo Z. Long-term low-dose IL-2 enhances immune function in common variable immunodeficiency. Clin Immunol. 2001;100:181–190. doi: 10.1006/clim.2001.5052. [DOI] [PubMed] [Google Scholar]
- 20.Fevang B, Yndestad A, Sandberg WJ, Holm AM, Muller F, Aukrust P, Froland SS. Low numbers of regulatory T cells in common variable immunodeficiency: association with chronic inflammation in vivo. Clin Exp Immunol. 2007;147:521–525. doi: 10.1111/j.1365-2249.2006.03314.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Verhasselt V, Milcent V, Cazareth J, Kanda A, Fleury S, Dombrowicz D, Glaichenhaus N, Julia V. Breast milk-mediated transfer of an antigen induces tolerance and protection from allergic asthma. Nat Med. 2008;14:170–175. doi: 10.1038/nm1718. [DOI] [PubMed] [Google Scholar]
- 22.Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775–787. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 23.Torgerson TR, Ochs HD. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked: forkhead box protein 3 mutations and lack of regulatory T cells. J Allergy Clin Immunol. 2007;120:744–750. doi: 10.1016/j.jaci.2007.08.044. quiz. 751–2. [DOI] [PubMed] [Google Scholar]
- 24.Huter EN, Punkosdy GA, Glass DD, Cheng LI, Ward JM, Shevach EM. TGF-beta-induced Foxp3+ regulatory T cells rescue scurfy mice. Eur J Immunol. 2008;38:1814–1821. doi: 10.1002/eji.200838346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crispin JC, Martinez A, Alcocer-Varela J. Quantification of regulatory Tcells in patients with systemic lupus erythematosus. J Autoimmun. 2003;21:273–276. doi: 10.1016/s0896-8411(03)00121-5. [DOI] [PubMed] [Google Scholar]
- 26.Viglietta V, Baecher-Allan C, Weiner HL, Hafler DA. Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med. 2004;199:971–979. doi: 10.1084/jem.20031579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saruta M, Yu QT, Fleshner PR, Mantel PY, Schmidt-Weber CB, Banham AH, Papadakis KA. Characterization of FOXP3+CD4+ regulatory T cells in Crohn’s disease. Clin Immunol. 2007;125:281–290. doi: 10.1016/j.clim.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 28.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 29.Grossman WJ, Verbsky JW, Barchet W, Colonna M, Atkinson JP, Ley TJ. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity. 2004;21:589–601. doi: 10.1016/j.immuni.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 30.Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. 2008;8:523–532. doi: 10.1038/nri2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cerdan C, Devilard E, Xerri L, Olive D. The C-class chemokine lymphotactin costimulates the apoptosis of human CD4(+) T cells. Blood. 2001;97:2205–2212. doi: 10.1182/blood.v97.8.2205. [DOI] [PubMed] [Google Scholar]
- 32.Cohen AC, Nadeau KC, Tu W, Hwa V, Dionis K, Bezrodnik L, Teper A, Gaillard M, Heinrich J, Krensky AM, Rosenfeld RG, Lewis DB. Cutting edge: decreased accumulation and regulatory function of CD4+ CD25(high) T cells in human STAT5b deficiency. J Immunol. 2006;177:2770–2774. doi: 10.4049/jimmunol.177.5.2770. [DOI] [PubMed] [Google Scholar]
- 33.McHugh RS, Whitters MJ, Piccirillo CA, Young DA, Shevach EM, Collins M, Byrne MC. CD4(+)CD25(+) immunoregulatory Tcells: gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity. 2002;16:311–323. doi: 10.1016/s1074-7613(02)00280-7. [DOI] [PubMed] [Google Scholar]
- 34.Jago CB, Yates J, Camara NO, Lechler RI, Lombardi G. Differential expression of CTLA-4 among T cell subsets. Clin Exp Immunol. 2004;136:463–471. doi: 10.1111/j.1365-2249.2004.02478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, Nomura T, Sakaguchi S. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008;322:271–275. doi: 10.1126/science.1160062. [DOI] [PubMed] [Google Scholar]
- 36.Nguyen KD, Fohner A, Booker JD, Dong C, Krensky AM, Nadeau KC. XCL1 enhances regulatory activities of CD4+ CD25(high) CD127(low/-) T cells in human allergic asthma. J Immunol. 2008;181:5386–5395. doi: 10.4049/jimmunol.181.8.5386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ko J, Radigan L, Cunningham-Rundles C. Immune competence and switched memory B cells in common variable immunodeficiency. Clin Immunol. 2005;116:37–41. doi: 10.1016/j.clim.2005.03.019. [DOI] [PubMed] [Google Scholar]
- 38.Tsaknaridis L, Spencer L, Culbertson N, Hicks K, LaTocha D, Chou YK, Whitham RH, Bakke A, Jones RE, Offner H, Bourdette DN, Vandenbark AA. Functional assay for human CD4+CD25+ Treg cells reveals an age-dependent loss of suppressive activity. J Neurosci Res. 2003;74:296–308. doi: 10.1002/jnr.10766. [DOI] [PubMed] [Google Scholar]
- 39.Aspalter RM, Sewell WA, Dolman K, Farrant J, Webster AD. Deficiency in circulating natural killer (NK) cell subsets in common variable immunodeficiency and X-linked agammaglobulinaemia. Clin Exp Immunol. 2000;121:506–514. doi: 10.1046/j.1365-2249.2000.01317.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bayry J, Hermine O, Webster DA, Levy Y, Kaveri SV. Common variable immunodeficiency: the immune system in chaos. Trends Mol Med. 2005;11:370–376. doi: 10.1016/j.molmed.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 41.Brouet JC, Chedeville A, Fermand JP, Royer B. Study of the B cell memory compartment in common variable immunodeficiency. Eur J Immunol. 2000;30:2516–2520. doi: 10.1002/1521-4141(200009)30:9<2516::AID-IMMU2516>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 42.Goldacker S, Warnatz K. Tackling the heterogeneity of CVID. Curr Opin Allergy Clin Immunol. 2005;5:504–509. doi: 10.1097/01.all.0000191888.97397.b3. [DOI] [PubMed] [Google Scholar]
- 43.Moratto D, Gulino AV, Fontana S, Mori L, Pirovano S, Soresina A, Meini A, Imberti L, Notarangelo LD, Plebani A, Badolato R. Combined decrease of defined B and T cell subsets in a group of common variable immunodeficiency patients. Clin Immunol. 2006;121:203–214. doi: 10.1016/j.clim.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 44.Taubenheim N, von Hornung M, Durandy A, Warnatz K, Corcoran L, Peter HH, Eibel H. Defined blocks in terminal plasma cell differentiation of common variable immunodeficiency patients. J Immunol. 2005;175:5498–5503. doi: 10.4049/jimmunol.175.8.5498. [DOI] [PubMed] [Google Scholar]
- 45.Eibl MM, Wolf HM. Common variable immunodeficiency: clinical aspects and recent progress in identifying the immunological defect(s) Folia Microbiol (Praha) 1995;40:360–366. doi: 10.1007/BF02814744. [DOI] [PubMed] [Google Scholar]
- 46.Punnonen J, Kainulainen L, Ruuskanen O, Nikoskelainen J, Arvilommi H. IL-4 synergizes with IL-10 and anti-CD40 MoAbs to induce B-cell differentiation in patients with common variable immunodeficiency. Scand J Immunol. 1997;45:203–212. doi: 10.1046/j.1365-3083.1997.d01-381.x. [DOI] [PubMed] [Google Scholar]
- 47.Reinherz EL, Geha R, Wohl ME, Morimoto C, Rosen FS, Schlossman SF. Immunodeficiency associated with loss of T4+ inducer T-cell function. N Engl J Med. 1981;304:811–816. doi: 10.1056/NEJM198104023041403. [DOI] [PubMed] [Google Scholar]
- 48.Boncristiano M, Majolini MB, D’Elios MM, Pacini S, Valensin S, Ulivieri C, Amedei A, Falini B, Del Prete G, Telford JL, Baldari CT. Defective recruitment and activation of ZAP-70 in common variable immunodeficiency patients with T cell defects. Eur J Immunol. 2000;30:2632–2638. doi: 10.1002/1521-4141(200009)30:9<2632::AID-IMMU2632>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 49.Majolini MB, D’Elios MM, Boncristiano M, Galieni P, Del Prete G, Telford JL, Baldari CT. Uncoupling of T-cell antigen receptor and downstream protein tyrosine kinases in common variable immunodeficiency. Clin Immunol Immunopathol. 1997;84:98–102. doi: 10.1006/clin.1997.4372. [DOI] [PubMed] [Google Scholar]
- 50.Farrington M, Grosmaire LS, Nonoyama S, Fischer SH, Hollenbaugh D, Ledbetter JA, Noelle RJ, Aruffo A, Ochs HD. CD40 ligand expression is defective in a subset of patients with common variable immunodeficiency. Proc Natl Acad Sci U S A. 1994;91:1099–1103. doi: 10.1073/pnas.91.3.1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.North ME, Webster AD, Farrant J. Primary defect in CD8+ lymphocytes in the antibody deficiency disease (common variable immunodeficiency): abnormalities in intracellular production of interferon-gamma (IFN-gamma) in CD28+ (‘cytotoxic’) and CD28−(‘suppressor’) CD8+ subsets. Clin Exp Immunol. 1998;111:70–75. doi: 10.1046/j.1365-2249.1998.00479.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tangye SG, Liu YJ, Aversa G, Phillips JH, de Vries JE. Identification of functional human splenic memory B cells by expression of CD148 and CD27. J Exp Med. 1998;188:1691–1703. doi: 10.1084/jem.188.9.1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Haas J, Fritzsching B, Trubswetter P, Korporal M, Milkova L, Fritz B, Vobis D, Krammer PH, Suri-Payer E, Wildemann B. Prevalence of newly generated naive regulatory Tcells (Treg) is critical for Treg suppressive function and determines Treg dysfunction in multiple sclerosis. J Immunol. 2007;179:1322–1330. doi: 10.4049/jimmunol.179.2.1322. [DOI] [PubMed] [Google Scholar]
- 54.Warnatz K, Bossaller L, Salzer U, Skrabl-Baumgartner A, Schwinger W, van der Burg M, van Dongen JJ, Orlowska-Volk M, Knoth R, Durandy A, Draeger R, Schlesier M, Peter HH, Grimbacher B. Human ICOS deficiency abrogates the germinal center reaction and provides a monogenic model for common variable immunodeficiency. Blood. 2006;107:3045–3052. doi: 10.1182/blood-2005-07-2955. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.