

Abstract
Eukaryotic cells maintain an immense amount of genetic information by tightly wrapping their DNA around positively charged histones. While this strategy allows human cells to maintain more than 25,000 genes, histone binding can also block gene expression. Consequently, cells express histone acetyl transferases (HATs) to acetylate histone lysines and release DNA for transcription. Conversely, histone deacetylases (HDACs) are employed for restoring the positive charge on the histones, thereby silencing gene expression by increasing histone-DNA binding. It has previously been shown that histones bind and silence viral DNA, while hyperacetylation of histones via HDAC inhibition restores viral gene expression. In this study, we demonstrate that treatment with Entinostat, an HDAC inhibitor, enhances transgene (luciferase) expression by up to 25-fold in human prostate and murine bladder cancer cell lines when used with cationic polymers for plasmid DNA delivery. Entinostat treatment altered cell cycle progression, resulting in a significant increase in the fraction of cells present in the G0/G1 phase at low micromolar concentrations. While this moderate G0/G1 arrest disappeared at higher concentrations, a modest increase in the fraction of apoptotic cells and a decrease in cell proliferation were observed, consistent with the known anticancer effects of the drug. DNase accessibility studies revealed no significant change in plasmid transcriptional availability with Entinostat treatment. However, quantitative PCR studies indicated that Entinostat treatment, at the optimal dose for enhancing transgene expression, led to an increase in the amount of plasmid present in the nucleus in two cancer cell lines. Taken together, our results show that Entinostat enhances polymer-mediated transgene expression and can be useful in applications related to transient protein expression in mammalian cells.
Keywords: non-viral gene delivery, epigenetic silencing, transient protein expression, histones, acetylation
Introduction
If all of the DNAwithin a single human cell (~6 × 109 base pairs) was stretched out end to end, it would be nearly 2 m long (Annunziato, 2008). Since the diameter of most nuclei is only ~6 μm, this immense amount of DNA must be condensed by tightly wrapping it around histone proteins inside the nucleus. Humans have four core histones (H2A, H2B, H3, and H4) with positively charged tail domains that are rich in lysine and arginine residues that facilitate DNA binding. Histone octamers (H2A2H2B2H32H42) initiate DNA condensation by binding 147 base pairs of DNA to form nucleosomes. A special linker histone (H1) then connects the nucleosomes to produce chromatin fibers (30 nm) that are condensed even further into chromosomes (100–400 nm) during mitosis (Wolffe, 1999).
While DNA condensation allows cells to maintain vast amounts of genetic information, histone binding can also physically block gene expression. Cells address this problem by expressing a wide variety of enzymes that phosphorylate (Rossetto et al., 2012), methylate (Kouzarides, 2002), ubiquitinylate (Bonnet et al., 2012), or acetylate histones to regulate DNA binding and transcription (Bannister and Kouzarides, 2011). For example, histone acetyl transferases (HATs) use acetyl CoA to acetylate and neutralize the charge of ε-amino groups on lysine residues, thereby releasing DNA for transcription. In contrast, histone acetylation may be reversed by histone deacetylases (HDACs), which remove acetyl groups to restore the positive charge and DNA binding activity of histones (Xhemalce et al., 2011; Yang and Seto, 2007). HATs and HDACs are generally non-specific (Bannister and Kouzarides, 2011), but HDACs have been shown to form complexes with other proteins that target deacetylase activity to specific DNA sequences (Hayakawa and Nakayama, 2011; Nan et al., 1998). HDACs can also influence gene expression by deacetylating non-histone proteins like NF-κB and other transcription factors (Glozak et al., 2005; Hasselgren, 2007). Consequently, aberrations in HAT/HDAC activity have been implicated in several neurodegenerative diseases (Kumar and Rinwa, 2012) and cancer (Ropero and Esteller, 2007).
Histone acetylation can also regulate expression of foreign DNA. Bishop et al. showed that viral DNA is efficiently delivered to the nucleus, but it is then quickly bound and silenced by histones within the densely packed centromeric heterochromatin. However, the HDAC inhibitor trichostatin A (TSA) was able to restore viral gene expression (Bishop et al., 2006; Poleshko et al., 2008). HDAC inhibitors have also been shown to bring viruses out of latency (Archin et al., 2009; Danaher et al., 2005), while some viruses express proteins to specifically inhibit HDACs (Gu and Roizman, 2007; Tang and Maul, 2003). Aside from viral DNA, histones also bind bacterial plasmids with high affinity (Yaneva et al., 1995) and form nucleosomes in vitro (Nakagawa et al., 2001). Purified histone proteins or synthetic peptides with the cationic histone tail sequence have also been used for non-viral gene delivery (i.e., histonefection)(Kaouass et al., 2006; Reilly et al., 2012).
Since histones appear to significantly inhibit viral gene expression and bind plasmid DNA (pDNA) in vitro, we hypothesized that HDAC inhibition could potentially enhance polymer-mediated transgene delivery and/or expression. We have previously shown that inhibition of the cytoplasmic HDAC6 with tubacin increases polymer-mediated transgene expression by influencing intracellular plasmid trafficking on stabilized micro-tubules (Barua and Rege, 2010). In this study, we investigated the effects of Entinostat, a selective inhibitor of class 1 HDACs 1 and 3 (Hu et al., 2003). Previous studies with Entinostat have demonstrated that it effectively inhibits HDACs in vivo, resulting in hyperacetylation of histones (Camphausen et al., 2004) and expression of genes that were previously silenced (Duque-Afonso et al., 2011; Kasman et al., 2007, 2012). Our results show that Entinostat significantly enhances polymer-mediated transgene expression in both prostate and bladder cancer cells with moderate effects on cell viability. Therefore, Entinostat treatment may be an effective way to enhance transgene expression levels in transient systems.
Materials and Methods
Polymer Synthesis
The 1,4C-1,4Bis and PA8 polymers were synthesized using methods similar to our previously published protocols (Barua et al., 2009; Potta et al., 2014; Vu et al., 2012). Briefly, the epoxide groups of diglycidyl ether (DGE) monomers 1,4 cyclohexanedimethanol DGE or ethylene glycol DGE, respectively, were reacted with polyamine monomers 1,4 bis(3-aminopropyl) piperazine and paromomycin, respectively, resulting in the formation of cationic polymers with molecular weights (MWs) >5,000 g/mol. Branched polyethyleneimine (PEI, MW = 25,000 g/mol) was purchased from Sigma (St. Louis, MO), and fresh stocks (50 ng/μL in HEPES buffer, pH 7.4) were prepared before every experiment to obviate any effects due to storage. Structures of monomers for synthesis of in-house polymers as well as the polymer structure for 25 kDa branched polyethylenemine are shown in Supporting Information Figures S1–3.
Transfections in the Presence of Entinostat
Entinostat was kindly provided by Syndax Pharmaceuticals of Waltham, MA through an agreement with the Cancer Therapeutics Evaluation Program (CTEP) at NIH. Stocks were prepared in DMSO at concentrations ranging from 60 μM–20 mM and frozen at −80°C until needed. Human prostate (PC3 and PC3-PSMA) and murine bladder (MB49) cancer cells were seeded onto 24-well plates at a density of 50,000 cells per well with 500 μL RPMI (PC3 and PC3-PSMA) or DMEM (MB49) containing 10% heat-inactivated fetal bovine serum (FBS), 100 units/mL penicillin, and 100 mg/mL streptomycin. The PC3-PSMA cell line, derived by transducing PC3 cells for stable expression of the Prostate Specific Membrane Antigen (PSMA) receptor, was a generous gift from Dr. Michel Sadelain (Memorial Sloan Kettering Cancer Center, New York, NY) (Gong et al., 1999). All cell lines were incubated overnight (~18–20 h) at 37°C, and the serum-containing media (SCM) was replaced with serum-free media (SFM) immediately prior to transfection (except in the case of transfections performed in the presence of SCM). Polyplexes were prepared by incubating cationic polymers (1,4C-1,4Bis, PEI, or PA8) with pGL3-Control (luciferase reporter gene, Promega, Madison, WI) or pEGFP-C1 (enhanced green fluorescent protein or EGFP reporter gene, Clontech, Mountain View, CA) plasmid DNA. The concentration of plasmid DNA was kept constant at 200 ng/well, while the polymer:pDNA mass ratio varied for each polymer (PEI = 1:1, 1,4C-1,4bis = 10:1, PA8 = 50:1), depending on their previously determined optimal concentrations (Potta et al., 2014). Polyplexes and different doses (0, 0.33, 1, 3.3, 10, 33, and 100 μM) of the HDAC inhibitor Entinostat were simultaneously added to the cells while a constant DMSO (for solubilizing the drug) concentration of 0.5% (v/v) was employed in all cases. Following 6 h of incubation at 37°C with the polyplex and drug, serum-free media was exchanged with serum-containing media containing the corresponding Entinostat concentrations. The cells were then incubated at 37°C for an additional 48 h to allow for transgene expression. Transfections with 0 μM Entinostat with or without 0.5% DMSO were also performed as controls, and 0.5% DMSO was found to not have any significant effects on transgene expression efficacy (data not shown).
Luminescence Assay and Fluorescence Microscopy
Luciferase expression was quantified using the Luciferase Assay Kit from Promega. At 48 h after transfection, cell culture media was removed from each well, and the cells were washed once with phosphate- buffered saline (PBS) before adding 150 μL of the cell culture lysis reagent (Promega) to each well. The wells were then incubated at 37°C for 20 min to ensure complete cell lysis. Cell lysates (15 μL) were then mixed with luciferin solution (30 μL) and luminescence (LUM) was immediately measured using a Synergy 2 plate reader (Biotek, Winooski, VT). Luminescence values were divided by cell viability to obtain LUV values to account for differences in cell viability. Relative LUV (RLUV) values were then obtained by dividing by the LUV of each sample by the LUV of the polyplex control, which only consisted of the corresponding polymer (PEI, 1,4C-1,4Bis, or PA8) and plasmid DNA (i.e., 0 mM Entinostat). Therefore, the RLUV values presented here account for changes in cell density (e.g., a condition with luminescence similar to the control but with 50% viability will be multiplied by a factor of two) and illustrate the degree of enhancement for each condition relative to the control. Following transfections with the pEGFP-C1 plasmid, cells were examined with a Zeiss fluorescence microscope to visualize EGFP expression. All images were acquired within areas of 90–100% confluence near the center of each well.
MTT Cell Viability Assay
Cell viability was quantified using 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), a yellow reagent which is converted to formazan (a purple dye) by living cells. This assay is a commonly used indicator of metabolic activity, which indirectly reports for cell viability. The MTT reagent was added to the cells (37°C for 2 h) and then a detergent from the kit (ATCC, Manassas, VA) was used to lyse the cells (additional 2 h). The formazan concentration was then quantified using by measuring the absorbance of the sample at 570 nm (A570), and cell viability was calculated by dividing the A570 value of each sample by the A570 value of the live cell control (no drug or polyplex added).
Cell Cycle Analysis
Cell cycle analysis was carried out by staining genomic DNA with propidium iodide (PI) with a few modifications of methods previously described in the literature (Krishan, 1975; Pozarowski and Darzynkiewicz, 2004). Briefly, PC3-PSMA human prostate cancer cells were seeded in 6-well plates at a density of 150,000 cells/well and cultured in the presence of 0.5% DMSO. The cells were treated with 0, 3.3, or 33 μM Entinostat for 48 h. Cells were harvested for flow cytometry analysis via trypsinization, rinsed once with PBS, and fixed with 70% EtOH. Cells were then permeabilized in a 0.001% Triton × solution, washed again in a PBS/FBS solution, and resuspended in a staining solution containing 5% FBS, 50 μg/mL PI, and 100 μg/mL RNase A for final flow cytometry analysis using an Attune® Acoustic Focusing Cytometer (Thermo Fisher Scientific, Waltham, MA). The distribution of cells in each phase of the cell cycle was then determined by measuring the intensity of PI fluorescence within each cell (<1 × fluorescence = apoptotic/Sub G, 1 × fluorescence = G0/G1, 1–2 × fluorescence = S, 2 × fluorescence = G2/M). Dead cells, as determined via a forward scatter versus side scatter plot, were gated out of the analysis. Thus, cells denoted as apoptotic were alive at the time of analysis. Fluorescence intensity was detected using the BL3 channel, through a 640 nm long-pass filter.
Quantification of DNase Accessibility and Plasmid DNA in Target Cell Nuclear Fraction
We investigated the presence and availability of plasmid DNA in the nuclear fraction of cells using methods described below.
Genomic DNA Extraction
Genomic DNA (gDNA) was extracted from PC3-PSMA cells to test primer specificity against pGL3 plasmid DNA, in the presence of target cell genomic DNA. Extraction was carried out using a phenol: chloroform:isoamyl alcohol (iaa) technique. Briefly, cells were trypsinized, washed and lysed. Cell content was then isolated via extraction with a 25:24:1 phenol-chloroform-iaa buffer (Sigma). A second extraction was carried out with chloroform, and trace amounts of chloroform were subsequently evaporated at 55°C. DNA was precipitated overnight using a sodium acetate/EtOH mixture, and resuspended in TE buffer (10 mM Tris-HCl pH 8.0, 1 mM EDTA).
All primers, designed against the promoter or luciferase (luc+) gene region of the pGL3 plasmid, were purchased from Integrated DNA Technologies (Coralville, IA). The sequences of the forward and reverse primers for the four regions on the plasmid, denoted 1–4, are as follows (5′–3′):
Forward 1—GGTACCGAGCTCTTACGCGTGC
Reverse 1—CGGGATGGGCGGAGTTAGGG
Forward 2—CAGAAGTAGTGAGGAGGCTTTTTTGGAC
Reverse 2—TATGTTTTTGGCGTCTTCCATGGTGGC
Forward 3—GCTTTTACAGATGCACATATCGAGGTGG
Reverse 3—GTATTCAGCCCATATCGTTTCATAGCTTC
Forward 4—CGAAATGTCCGTTCGGTTGGCAGAAG
Reverse 4—GCATAAAGAATTGAAGAGAGTTTTCACTGCATAC.
Region 1 mostly consists of the upstream portion of the SV40 promoter, region 2 bridges the promoter region as well as the upstream region of the luc+ gene, covering portions of both, and regions 3 and 4 are located entirely on the luc+ gene. Endpoint PCR was carried out against both the pure pGL3 plasmid template as well as against PC3-PSMA gDNA using all pGL3 primer pairs. Agarose gel electrophoresis analyses were carried out on PCR products to validate the efficacy and specificity of the pGL3 primers.
Nucleosomal DNA Purification
PC3-PSMA and PC3 cell nuclei were extracted using the EZ Nucleosomal DNA Prep Kit (Zymo) and purified nuclear DNA was treated with DNase following 48 h of transfection. Transfections were carried out with the pGL3 plasmid complexed with 1,4C-1,4Bis polymer, in the presence or absence of 33 μM Entinostat, as described above. Isolated nuclei were then either treated with Atlantis dsDNase (0.4 Units/100 μL, per kit instructions, resulting in cut DNA), or equivalent volume 1×PBS (resulting in uncut DNA). Nucleosomal DNA (cut and uncut) were then isolated using spin columns, supplied with the EZ Nucleosomal DNA Prep kit. The spin columns efficiently (70–90%) capture DNA 75 bp to 10 kb, which is a relevant size range for the pGL3 plasmid.
qPCR Experiments and Analysis
Quantitative PCR (qPCR) experiments were carried out on DNA isolated from the nuclear fraction of PC3-PSMA or PC3 cells, using the primers described above. Detection of fluorescence accumulated by amplified, double stranded DNA was carried out using SYBR® Green Master Mix (Life Technologies). PCR reactions (15 mL volumes) were prepared in triplicate for each independent experiment as follows: 2 μL template DNA (1/75 total cellular nucleosomal DNA extraction, or roughly 3.8 × 104 nuclei for vehicle-control treated PC3-PSMA cells and 1.0 × 104 nuclei for Entinostat-treated PC3-PSMA cells; PC3 cell counts were not quantified), 7.5 μL 2× SYBR Green Master Mix, 1.5 μL forward primer (0.57 pmol), 1.5 μL reverse primer (0.57 pmol), and 2.5 μL ddH2O. Reactions were carried out in opaque white 96-well plates (Roche 04 729 692 001), and thermal cycling was conducted in a Light Cycler® 480 PCR instrument (Roche). A pre-incubation step was carried out for 5 min. at 95°C. Next, 45 amplification cycles were run: denaturation at 95°C for 10 s, annealing at 58°C for 10 s, extension at 72°C for 10 s. Fluorescence was recorded after each extension step. Melting analysis (95°C for 60 s, 40°C for 120 s, 95°C at 0.19°C (sec-1) with fluorescence readings at 3 (sec−1)) of reactions with and without DNA templates confirmed that 95–100% of the fluorescence signal was associated with PCR amplicons rather than primer dimers. The Roche Light Cycler® 480 software was used for subsequent calculations of DNase accessibility and relative plasmid content in the nuclear fraction.
Cp values (maximum y-value of the second derivative of fluorescence = y/cycle number = x) for cells treated with Entinostat (E) or 0.2% DMSO (D) were used in DNase accessibility calculations. Calculations were performed separately for every PCR-amplified region (Fig. 6). Cp (crossing point) values indicate the PCR cycle number at which fluorescence due to amplification exceeds background fluorescence; thus, a lower Cp value indicates a greater amount of target DNA template since fewer numbers of cycles are required to produce a detectable fluorescence signal. The effect of DNase treatment on the purified nuclear DNA was quantified for cases with vehicle control (DMSO) and Entinostat treatment, simply as the difference in Cp for DNA treated with DNase (C = cut) and without (UC = uncut):
where each delta value represents the difference in intact DNA before and after DNase treatment. A larger delta value indicates greater sensitivity to DNase, and thus indicates greater accessibility. All delta values were normalized using the average DMSO control value to test the hypothesis that relative to the DMSO control, Entinostat treatment increases DNA accessibility:
where ΔCP,D_avg is an average of ΔCP,D across all replicates. DNase accessibility can be expressed as a fold-difference in the template DNA that remains after cutting by DNase for all samples, relative to the DMSO control.
Figure 6.

DNase accessibility data normalized to DMSO treatment (n = 3) in PC3-PSMA cells. The y-axis indicates fractional DNase accessibility relative to DMSO-treated cells. Data shown indicate mean values ± one standard deviation. For all four regions, as indicated by primer pairs 1–4 in the promoter/gene map, the difference in DNase accessibility between DNA harvested from DMSO and Entinostat (33 μM) treated cells was not found to be statistically significant (P < 0.05 threshold, Student’s t-test).
“Normalized DNase accessibility, DMSO” has an average value close to 1, since it is normalized by its own average value (Fig. 6). “Normalized DNase accessibility, Entinostat” values greater than 1 support the hypothesis that an increase in DNase accessibility is associated with Entinostat treatment, while other values reject the hypothesis (Fig. 6).
Next, we used Cp values from the uncut DNA samples to compare the amount of plasmid DNA in the nuclear fraction of Entinostat-treated versus untreated (DMSO) cells. All values were normalized using the average Cp value for the DMSO control sample
with fold-increase of plasmid in the nuclear fraction calculated as:
“Normalized plasmid in nuclear fraction, Entinostat” values greater than 1 support the hypothesis that greater plasmid uptake is associated with Entinostat treatment, while other values reject the hypothesis (Fig. 7).
Figure 7.

Normalized plasmid content in the nuclear fraction, indicating the relative amount of exogenously delivered plasmid DNA present in the nucleus following Entinostat treatment relative to vehicle control (DMSO) treatment (n = 3). Data are reported for (A) PC3-PSMA (33 μM Entinostat) and (B) PC3 cells (10 μM Entinostat), and are represented as mean ± one standard deviation. Asterisks (*) denote P < 0.05 comparing pGL3 levels in nuclear fraction in Entinostat-treated cells relative to DMSO treated cells (Student’s t-test). The base pairs are labeled such that position 1 is the first base pair amplified by our primers. The numbering system was defined for convenience, and is not related to that provided by the vendor (Promega).
Results
Enhancement of Luciferase Transgene Expression by Entinostat
The effects of Entinostat on plasmid transgene expression were investigated in three different cell lines: PC3 and PC3-PSMA human prostate cancer cells and MB49 murine bladder cancer cells. Enhancement of luciferase expression (relative luminescence, RLUV) by Entinostat is shown on the left-hand side of Figure 1 and numerically in Table I. Note: In terms of conventional RLU/mg units, all polyplex controls were approximately similar in MB49 cells (1–5 × 106 RLU/mg), while PEI and PA8 polyplexes consistently resulted in values that were 5–10 fold higher (1–10 × 106 RLU/mg) than 1,4C-1,4Bis polyplexes (0.2–1 × 106 RLU/mg) in both PC3 and PC3-PSMA cells under these conditions.
Figure 1.

Relative luciferase expression (left column) and cell viability (right column) in PC3 human prostate cancer (top row), PC3-PSMA human prostate cancer (middle row), and MB49 murine bladder cancer (bottom row) cells. Asterisks (*) denote statistically significant (P < 0.05) enhancement of luciferase expression or reduction of viability relative to the corresponding polyplex controls. Data shown indicate mean values ± one standard deviation. Note: luminescence and viability were not measured at 100 μM Entinostat in MB49 with PA8 polymer.
Table I.
Fold-enhancement of luciferase transgene expression by Entinostat with various polymers and cell lines.
| Cell lines | |||
|---|---|---|---|
|
|
|||
| Polymers (P:D) | PC3 | PC3-PSMA | MB49 |
| 1,4C-1,4Bis (10:1) |
11.2±7.6 (33 μM) | 11.6±1.9 (33 μM) | 8.0±3.0 (3.3 μM) |
| PEI (1:1) | 24.8±4.8 (33 μM) | 21.3±3.3 (33 μM) | 6.8±2.5 (3.3 μM) |
| PA8 (50:1) | 7.6±3.0 (10 μM) | 17.8±6.0 (10 μM) | 5.9±0.6 (3.3 μM) |
In each cell, the top value is the mean degree of enhancement±standard deviation, while the value listed in brackets below indicates the corresponding optimum concentration of Entinostat.
While Entinostat consistently enhanced luciferase expression for all conditions tested, some significant differences were observed between polymer delivery vehicles (PEI, 1,4C-1,4Bis, and PA8) and cell lines. For example, the highest enhancement of luciferase expression by Entinostat was observed with PEI in PC3 cells (24.8 ± 4.8 fold enhancement). In the same cell line (PC3), however, enhancement was 2–3 times lower with 1,4C-1,4Bis (11.2 ± 7.6 fold) and PA8 (7.6 ± 3.0 fold) polymers. Similar results were observed in the PC3-PSMA cell line, with a twofold enhancement in luciferase expression observed in case of PEI (21.3 ± 3.3 fold) compared to the 1,4C-1,4Bis polymer (11.6 ± 1.9 fold). However, enhancement of PA8 polyplex transfection by Entinostat was much higher in PC3-PSMA cells (17.8 ± 6.0 fold) than in PC3 cells (7.6 ± 3.0 fold). In contrast, maximum enhancement in transgene expression by Entinostat was reduced in MB49 cells to approximately similar levels (5.9–8.0 fold) for all polymers.
It is also interesting to note that the optimum concentration of Entinostat for transgene expression was dependent on both the polymer and the cell line used. In the prostate cancer cell lines, Entinostat demonstrated the highest enhancement for PEI and 1,4C-1,4Bis at a concentration of 33 μM, while the highest average enhancement of PA8 polyplexes was observed with only 10 μM Entinostat. The optimum concentration of Entinostat was even lower in MB49 cells, but similar for all polymers tested (3.3 μM). Despite the minor differences in activity, these results still clearly demonstrate that Entinostat is able to enhance luciferase expression in different cell types and when different polymers are used for plasmid DNA delivery.
Effect of Entinostat on Cell Viability
The right-hand side column of Figure 1 shows the effects of Entinostat on cell viability relative to live cell controls, which were not treated with any polyplexes or drug. As expected, all polyplex controls (0 μM Entinostat) decreased viability by about 10–20% (with the exception of PEI in MB49, which had almost no effect on cell viability under the conditions employed). Significant differences in Entinostat-induced reduction in viability were observed between cell lines. PC3 cells only showed slight decreases in cell viability, except at the highest concentrations of Entinostat (10–100 μM) with certain polymers (PA8). In contrast, almost all concentrations of Entinostat significantly reduced PC3-PSMA cell viability. In MB49 cells, Entinostat reduced cell viabilities to below 50% at concentrations above 10 μM. In addition, higher concentrations of Entinostat (33–100 μM) caused significant MB49 cell detachment, and even induced changes in PC3-PSMA cell morphology (cells appeared thinner and stretched out). However, the type of polymer did not appear to have any effect on Entinostat toxicity, indicating that synergistic toxicity between the drug and polymers used in this study is unlikely.
Enhancement of Polymer-Mediated EGFP Expression by Entinostat
Figure 2 clearly shows enhancement of EGFP expression at 3.3–100 μM Entinostat in both PC3 and PC3-PSMA cells. In the PEI polyplex control images, only a few PC3 and PC3-PSMA cells are fluorescent; some dimly fluorescent cells are also present, but are difficult to visualize. However, a drastic increase in the number of GFP+ cells is observed at Entinostat concentrations above 1.0–3.3 μM. It is also interesting to note that transgene expression enhancement by Entinostat can be observed after only 24 h, with similar effects at 48 h. These results indicate that Entinostat can be used to enhance expression of different plasmids and transgenes in cultured cells.
Figure 2.

Enhancement of EGFP expression by Entinostat in PC3 and PC3-PSMA human prostate cancer cells at 24 and 48 h post-transfection with PEI (polymer:plasmid DNA mass ratio of 1:1). These representative images are consistent with n = 3 independent experiments.
Effects of Entinostat on Polymer-Mediated Transgene Expression in Serum-Containing Media
A comparison of the effects of Entinostat on luciferase expression in both serum-containing media (SCM) and serum-free media (SFM) for PC3-PSMA cells is shown in Figure 3. The baseline expression of luciferase with the PEI polyplex control decreased approximately threefold in SCM relative to SFM, but statistically significant enhancement was still observed at all concentrations of Entinostat tested (0.33–33 μM). The highest enhancement by Entinostat in SCM (17-fold) was not significantly lower than the 23-fold enhancement observed at the same Entinostat concentration in SFM. However, the toxicity of the PEI polyplex was significantly lower in SCM relative to SFM. For example, the PC3-PSMA cells were virtually unaffected by PEI polyplexes in SCM (100% viability), while the viability of cells exposed to PEI polyplexes in SFM decreased to 88%. Likewise, cell viabilities in SCM were approximately 12% higher than in SFM, but still significantly lower than control viabilities for all concentrations of Entinostat.
Figure 3.

Effect of Entinostat on luciferase expression (left) and cell viability (right) in PC3-PSMA cells, following transfections with PEI in serum (10% FBS)-containing media, that is, SCM or serum free media, that is, SFM (n = 3). Data shown indicate mean values + one standard deviation. Asterisks (*) denote significant differences relative to the polyplex control (0 μM Entinostat).
Enhancement of Transgene Expression Relative to Change in Global Protein Expression
Due to the effects of Entinostat on chromosomal structure, it is possible that Entinostat treatment increases global protein production. This necessitates an investigation into how increases in transgene expression might vary corresponding to any changes in global protein expression levels. Interestingly, the total protein content in PC3-PSMA cells transfected with 1,4C-1,4Bis:pGL3 polyplexes did not change significantly with optimal Entinostat treatment (33 μM) compared to DMSO (vehicle)-treated cells (i.e., absence of Entinostat). However, it is important to note that with fewer cells present following 33 μM Entinostat treatment, the total protein content per cell escalated as shown in Figure 4. This could be due to increased acetylation of chromatin or possibly reduced cell growth rate and a concomitant alteration in cell metabolism. However, the fold-increase in protein content per cell (approximately 2.8-fold) with 33 μM Entinostat treatment did not nearly account for the fold-enhancement in transgene expression observed per cell with inhibitor treatment (approximately 28-fold; cell counts, RLU values, and protein content in Supporting Information Table I) (Fig. 4). This result indicates that mechanisms in addition to changes in total protein content are likely responsible for enhancement in transgene expression with 33 μM Entinostat treatment.
Figure 4.
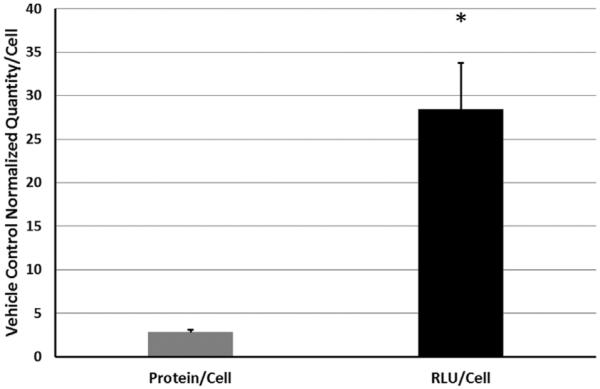
Effect of Entinostat on global protein expression (as measured via BCA assay) and transgene (luciferase) expression in PC3-PSMA cells transfected with 10:1 (w/w) 1,4C-1,4Bis pGL3 polyplexes in serum free-media, that is, SFM (n = 3). The protein/cell and RLU/cell reported for Entinostat in this graph are normalized with those observed for the DMSO (i.e., vehicle control in absence of Entinostat). Data shown indicate mean values ± one standard deviation. Asterisks (*) denote significant increase in RLU/cell relative to protein/cell with 33 mM Entinostat treatment.
Cell Cycle Analysis
To determine the nature of the effects of Entinostat on cell viability and transgene expression, we examined the effects of Entinostat on cell cycle progression in PC3-PSMA cells (Fig. 5 and Table II). Cells in the G0 and G1 (i.e., resting) phases of the cell cycle have two sets of chromosomes, and have a fluorescence intensity of 2 × 106 following staining with the DNA stain propidium iodide. Since cells in the G2 and M phases have four sets of chromosomes, they bind twice as much PI stain and exhibit fluorescence intensities of 4 × 106. Apoptotic cells with fragmented nuclei are seen in the sub Go/G1 population with a fluorescent intensity less than 2 × 106.
Figure 5.

Cell cycle analysis of PC3-PSMA cells treated with 0.5% DMSO, 3.3 μM Entinostat, and 33 μM Entinostat (n = 3); results from one representative experiment are shown. The y-axis indicates the number of cells with the specific fluorescence intensity shown on the x-axis.
Table II.
Fractions of PC3-PSMA cells in each phase of the cell cycle following treatment with Entinostat.
| Sample | Sub G0 (%) | G0/G1 (%) | S (%) | G2/M (%) |
|---|---|---|---|---|
| DMSO Control | 0.7±0.4 | 61.9±7.8 | 9.9±2.7 | 27.6±7.3 |
| 3.3 μM Entinostat | 1.3±0.9 | 86.3±2.3* | 2.2±0.8* | 10.2±2.7* |
| 33 μM Entinostat | 6.2±1.3* | 69.7±10.5 | 3.7±1.2* | 20.5±9.2 |
P < 0.05, for each phase of the cell cycle for Entinostat treatments relative to DMSO control (Two-tailed Student’s T-test).
A majority of the control cells (treated with only 0.5% DMSO) were found to be in the G0/G1 phase of the cell cycle (62%), while 28% of the cells were in the G2/M phase and 10% were in the S phase. Treatment with 3.3 μM Entinostat resulted in significant accumulation (86%) of PC3-PSMA cells in the G0/G1 phase, with a concomitant decline in both S and G2/M cells. Treatment with 33 μM Entinostat (the optimal tested dose for transgene expression in PC3-PSMA cells) showed no significant G0/G1 arrest, although a modest (6.2%) increase in the apoptotic cell population was observed. These results have some similarity to a previous study in which 1 μM Entinostat resulted in G0/G1 arrest of U397 human leukemic monocyte lymphoma cells, while a higher dose (5 μM) failed to induce G0/G1 arrest, but did lead to a significant increase in apoptotic cell population (Rosato et al., 2003). However, these results differ from a study in closely related PC-3 cells, in which, G2/M arrest was reported by Entinostat (Khandelwal et al., 2008). Our results indicate that the observed increase in transgene expression observed with 33 μM Entinostat treatment is unlikely to be explained by cell cycle effects alone, but the G0/G1 arrest observed at 3.3 μM and the modest increase in apoptotic cells at 33 μM correlate with the observed decreases in cell viability at these concentrations.
DNase Accessibility With Entinostat Treatment
Positively charged deacetylated histones are tightly wrapped with negatively charged DNA, while acetylation of histones removes the positive charge, disrupts DNA-histone binding, and facilitates increased DNA availability for transcription. We hypothesized that treatment with Entinostat would, in part, increase transgene expression by reducing the binding of DNA to histones, thereby increasing the availability for transcription. One way to indirectly measure the availability of a region of DNA is by treatment with DNase, which cuts “unprotected” or transcriptionally “accessible” DNA. This includes portions of the plasmid that do not interact with histones in the nucleus. Thus, determination of amount/extent of “cut” portions, for example, using qPCR, is an indication of how accessible the plasmid is for transcription inside host cell nuclei. This approach has been previously applied to elucidate the availability of promoter and other regions of chromosomal DNA (Anthony et al., 2014; Lu et al., 2012; Shu et al., 2013). In our case, we applied this same principle, but to exogenously delivered plasmid DNA, as opposed to host cell chromosomal DNA assayed in these previous investigations. As stated previously, evidence suggests that plasmids do form nucleosomes extracellularly, which motivated the use of this strategy for elucidation of DNase accessibility in vitro.
We carried out qPCR analyses on purified PC3-PSMA nuclear DNA fractions for which, we used primer pairs to amplify different regions around the luc+ gene on the pGL3 plasmid; control experiments with nuclei from untransfected cells were carried out in order to verify that the PCR signal was exclusively from pGL3 plasmid DNA. The regions amplified using qPCR include: a region on the luc+ gene promoter (primer pair 1), a region bridging the promoter and luc+ gene (primer pair 2), and regions on the luc+ gene (primer pairs 3 and 4) (Fig. 6). Normalized DNase accessibility for each of the four explored regions in the promoter and gene area are shown in Figure 6. The results are similar for each of the four regions assayed in that no statistically significant change in DNase accessibility was observed due to Entinostat treatment.
It is possible that plasmid DNA forms nucleosomes inside cells, and that Entinostat does not strongly influence the transcriptional availability in the luc+ promoter/gene region. It is also possible that nucleosomes are not formed in cells, and the increase in transgene expression observed with Entinostat treatment is due to changes in chromosomal availability (discussed below). Additionally, there may be regions, upstream of the promoter that were not included in the qPCR analyses, but may play a role in transcription factor binding.
Evaluation of Plasmid DNA Content in the Nucleus
In addition to analyzing the effect of Entinostat on DNase accessibility, we quantified the relative fold change in plasmid content in the nuclear fraction of transfected PC3-PSMA and PC3 cells following treatment with 33 μM and 10 μwith the latter including a portion of theM Entinostat, respectively (Fig. 7). In PC3-PSMA cells (Fig. 7A), normalized plasmid in nuclear fraction values of 2.8 ± 0.6 and 2.5 ± 0.4 were observed with primer pair 1 and primer pair 2 for the plasmid in the nuclear fraction; both primer pairs amplify regions containing part of the SV40 promoter, with the latter including a portion of the luc+ gene. qPCR using primer pair 3 for amplification indicated the normalized plasmid levels to be 3.0 ± 0.7, while primer pair 4 yielded a value of 3.9 ± 2.0 compared to the control; both of these primer pairs amplify portions of the luc+ gene (Fig. 7). In addition to PC3-PSMA cells, these experiments were carried out in PC3 cells, in which a modest increase in pGL3 plasmid was detected in the nuclear fraction (Fig. 7B). As shown through PCR amplification of several portions close to/of the pGL3 SV40-luc+ region, the presence of the exogenous plasmid DNA in the nucleus is significantly enhanced by Entinostat treatment in two different cell lines. These results indicate that the modest increase in plasmid present in cell nuclei (~3.1-fold in PC3-PSMA cells and ~1.7 in PC3 cells, from the average value for all primer pairs) likely contributes to the enhancement in transgene expression observed with Entinostat (Fig. 1).
Discussion
The results shown in Figures 1, 2, and 3 demonstrate that Entinostat significantly enhances transgene (luciferase and EGFP) expression by up to 25-fold in human prostate and murine bladder cancer cells at an optimum concentration range of 3.3–33 μM, even in the presence of serum. Statistically significant enhancement can be seen at concentrations as low as 1 μM of the drug in some cases (Fig. 1). Since the luciferase and EGFP plasmids used in this study rely on different promoters (SV40 and CMV, respectively), these results suggest that Entinostat may be able to enhance a variety of different therapeutic genes and promoters. These results agree with previous findings that the pan-HDAC (HDAC 1, 3, 4, 6, and 10) inhibitor Trichostatin A significantly enhances expression from a variety of viral promoters, including the SV40 and CMV promoters used in this study (Barua and Rege, 2010; Hayashi et al., 2011). The highest average luciferase expression values were obtained when Entinostat was used with PEI, but the other polymers tested (1,4C-1,4Bis and PA8) also showed significant degrees of enhancement. Therefore, Entinostat-mediated enhancement is likely a general phenomenon, and may also enhance the efficacy of other gene delivery vehicles. Indeed, it has been previously shown that Entinostat also enhances adenovirus-mediated transgene expression (Kasman et al., 2007).
The most significant differences in Entinostat enhancement were observed between the different cell lines. Instead of the >20-fold enhancement observed with the prostate cancer cell lines at 33 μM Entinostat, the drug was only able to enhance luciferase expression by approximately seven-fold in MB49 cells at a much lower optimum concentration of 3.3 μM. This reduction in enhancement may be related to the sharp decrease in MB49 cell viability at Entinostat concentrations above 3.3 μM. This decrease in enhancement may also reflect differences between the interactions of human and murine HDACs with Entinostat, although additional cell lines of both species would need to be included in this study to verify this hypothesis. Regardless of the nature of these differences, it is still clear that Entinostat significantly enhanced transgene expression in each of the cell lines tested in a dose range from 3.3 μM to 33 μM. It is interesting to note that the optimum concentration of Entinostat observed in our experiments (3.3–33 μM) is much higher than the previously published submicromolar (<1 μM) IC50 values for the anti-proliferative effects of Entinostat (Rosato et al., 2003). However, these concentrations are consistent with other studies showing enhancement of viral gene therapy at 3.3–10 μM (Kasman et al., 2007, 2012). This optimum concentration range corresponds well with inhibition of HDAC 3 (IC50 = 8 μM) but is much higher than that required for inhibiting HDAC 1 (IC50 = 0.3 μM) (Hu et al., 2003). Thus, it appears that inhibition of HDAC3 may be at least partly responsible for the observed enhancement of transgene expression, although more studies would be required to determine this conclusively.
Previous studies have shown that HDAC inhibitors modulate cell cycle progression, inducing cell cycle arrest in some cases (Rosato et al., 2003; Sambucetti et al., 1999; Sikandar et al., 2010). This activity may explain the reduction in PC3-PSMA and MB49 cell proliferation/viability shown in Figure 1. Indeed, our analysis shows that Entinostat does modulate cell cycle progression, increasing the fraction of cells present in the G0/G1 phase from 61.9% for the control (0 μM Entinostat) to 86.3% for cells treated with 3.3 mM Entinostat. This G0/G1 arrest may be responsible for enhanced transgene expression, since G0/G1 arrest has been previously shown to increase overall protein production (Sunley and Butler, 2010). However, since G0/G1 arrest was not observed at the optimum concentration of 33 μM, other mechanisms may also cause this enhancement, especially at higher concentrations of the drug.
While a positive correlation between DNase accessibility (sometimes used interchangeably with chromatin openness) and gene expression may be anticipated, there is recent evidence that indicates a more complex relationship between these factors. For example, high rates of DNase I hypersensitivity were detected on cis-elements associated with low-expression genes in HeLa cells, suggesting DNase accessibility alone does not indicate high expression. Counter-intuitively, a positive correlation between silenced genes and chromatin relaxation was observed (Wang et al., 2012).
While our data do not indicate that Entinostat treatment increases DNase accessibility to the promoter or gene regions assayed on the pGL3 plasmid, they indicate that higher amounts of the pGL3 plasmid are present in the nuclear fraction of Entinostat-treated PC3 and PC3-PSMA cells when compared to those treated with DMSO. It is possible that Entinostat treatment disrupts nucleosomal structure at a position that we have not tested on the plasmid. Additionally, remodeling of the chromatin in the nucleus of the target cells could be (and likely is) induced. This, in turn, might influence the expression of endogenous genes that increase nuclear plasmid concentration, and thus increase transgene expression. Specifically, Entinostat inhibits class 1 HDACs, thereby causing an increase in histone hyperacetylation and a decrease in histone-DNA binding (Camphausen et al., 2004). Since HDACs are known to bind several transcription factors that influence the expression of other genes, it is also possible that Entinostat could indirectly influence several other pathways that are involved in transgene delivery and/or expression (Glozak et al., 2005; Hasselgren, 2007). HDAC inhibition could also directly influence gene delivery and expression in a variety of unforeseen ways, since over 1700 proteins are known to have one or more acetylation sites (Choudhary et al., 2009). Entinostat only affects a fraction of this “acetylome” since it is known to inhibit the nuclear HDACs 1 and 3 (Hu et al., 2003), but HDAC 1 inhibition has been shown to enhance viral gene delivery by enhancing endosomal maturation/escape and trafficking to the nucleus (Yamauchi et al., 2011). Nuclear isolation and qPCR experiments showed an increase in exogenous plasmid content in the nuclei following Entinostat treatment in both PC3-PSMA and PC3 cells. We have previously shown significant differences in trafficking of exogenous nanoscale cargo (plasmid DNA and fluorescent quantum dots) in these two cell lines. Quantum dots or polyplexes localize in a perinuclear recycling compartment (PNRC) in PC3-PSMA cells, these nanoparticles demonstrate a punctate distribution throughout the cytoplasm in PC3 cells (Barua and Rege, 2009, 2010). The likelihood that Entinostat treatment affects endosomal maturation and subsequent cargo trafficking may be responsible for the observed differences in plasmid levels nuclei of these two cell lines. Entinostat is known to inhibit cytokine signaling and phosphorylation of the Signal Transducer and Activator of Transcription 3 (STAT-3) (Liu et al., 2013), a pathway that is known to bind and silence viral and bacterial DNA sequences (Harms and Splitter, 1995). Further studies (e.g., ChIP assays) are required to determine if histone hyperacetylation via HDAC inhibition is responsible for the observed enhancement of transgene expression following Entinostat treatment.
Conclusions
Our results demonstrate that Entinostat enhances polymer-mediated transgene expression in different cancer cell lines, and that the drug can be used with different polymeric delivery vehicles. The increased presence of the pGL3 plasmid in the nucleus is a likely contributor to the observed increase in transgene expression upon treatment with the HDAC inhibitor. However, the mechanisms responsible for the increased plasmid DNA content in the nucleus are not entirely clear at this point. Analysis of known epigenetic regulators mutated in cancers reveals that acetylation and methylation are the two epigenetic pathways most commonly affected (Dawson and Kouzarides, 2012). Although no individual HDACs have been identified to be commonly mutated in cancer to date (Dawson and Kouzarides, 2012), HDACs 1-3 are overexpressed in many cancer types (Marks et al., 2001). Entinostat has demonstrated anticancer activity when used in combination with another epigenetic inhibitor in a lung adenocarcinoma orthotopic rat model (Belinsky et al., 2011), and this same strategy has shown efficacy in a clinical trial involving patients with refractory advanced non-small cell lung cancer (Juergens et al., 2011). Additionally, Entinostat has been shown to block cell cycle and induce apoptosis of bladder cancer cells in vitro (Qu et al., 2010), as well as prostate cancer cells in vivo (Perry et al., 2010). Entinostat can therefore likely be used as both an anti-cancer drug and as an enhancer of gene therapies in cancer diseases.
In addition to cancer gene therapy, Entinostat may have utility in enhancing transient therapeutic recombinant protein production in mammalian bioprocesses. We have shown that inhibition of HDACs with Entinostat enhances protein expression per cell in PC3-PSMA prostate cancer cells. The use of Entinostat may be applicable to other mammalian systems commonly used in biological protein production (e.g., Chinese hamster ovary cells), in which it is desirable to control cell proliferation and maximize the protein production per cell (Sunley and Butler, 2010). Interestingly, a common method for carrying this out is by inducing G1 cell cycle arrest (for instance using sodium butyrate), which is possible with low micromolar dosage of Entinostat.
The Entinostat used in this study was generously provided by Syndax Pharmaceuticals, Inc., through the Cancer Therapeutics Evaluation Program (CTEP) and the National Cancer Institute, NIH. The authors thank Professor Christina Voelkel–Johnson at the Medical University of South Carolina, Charleston, SC for several helpful discussions.
Supplementary Material
Acknowledgments
Contract grant sponsor: National Institute of General Medical Sciences
Contract grant sponsor: NIH
Contract grant number: 1R01GM093229-01A1
Contract grant sponsor: Synthetic Biology Engineering Research Center (Synberc)
Contract grant sponsor: NSF
Contract grant number: EEC 0540879
Contract grant sponsor: Women and Philanthropy, ASU Foundation
Abbreviations
- DMSO
dimethyl sulfoxide
- EGFP
enhanced green fluorescent protein
- HAT
histone acetyl transferase
- HDAC
histone deacetylase
- MTT
3 4,5-dimethylthiazol-2-yl-2,5-diphenyltetrazolium bromide
- PEI
polyethyleneimine
- SCM
serum containing media
- SFM
serum free media
Footnotes
Disclosure statement: Dr. Rege was an invited speaker at PepTalk: The Protein Science Week in 2015. All expenses for Dr. Rege were borne by Cambridge Healthtech Institute.
Supporting Information
Additional supporting information may be found in the online version of this article at the publisher’s web-site.
References
- Annunziato A. DNA packaging: Nucleosomes and chromatin. Nat Educ. 2008;1(1):26. [Google Scholar]
- Anthony K, More A, Zhang X. Activation of silenced cytokine gene promoters by the synergistic effect of TBP-TALE and VP64-TALE activators. PLoS ONE. 2014;9(4):e95790. doi: 10.1371/journal.pone.0095790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archin N, Espeseth A, Parker D, Cheema M, Hazuda D, Margolis D. Expression of latent hiv induced by the potent hdac inhibitor suberoylanilide hydroxamic acid. AIDS Res Hum Retroviruses. 2009;25(2):207–212. doi: 10.1089/aid.2008.0191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21(3):381–395. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barua S, Joshi A, Banerjee A, Matthews D, Sharfstein ST, Cramer SM, Kane RS, Rege K. Parallel synthesis and screening of polymers for nonviral gene delivery. Mol Pharm. 2009;6(1):86–97. doi: 10.1021/mp800151j. [DOI] [PubMed] [Google Scholar]
- Barua S, Rege K. Cancer-cell-phenotype-dependent differential intracellular trafficking of unconjugated quantum dots. Small. 2009;5(3):370–376. doi: 10.1002/smll.200800972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barua S, Rege K. The influence of mediators of intracellular trafficking on transgene expression efficacy of polymer-plasmid DNA complexes. Biomaterials. 2010;31(22):5894–5902. doi: 10.1016/j.biomaterials.2010.04.007. [DOI] [PubMed] [Google Scholar]
- Belinsky SA, Grimes MJ, Picchi MA, Mitchell HD, Stidley CA, Tesfaigzi Y, Channell MM, Liu Y, Casero RA, Baylin SB, et al. Combination therapy with vidaza and entinostat suppresses tumor growth and reprograms the epigenome in an orthotopic lung cancer model. Cancer Res. 2011;71(2):454–462. doi: 10.1158/0008-5472.CAN-10-3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop C, Ramalho M, Nadkarni N, Kong W, Higgins C, Krauzewicz N. Role for centromeric heterochrornatin and PML nuclear bodies in the cellular response to foreign DNA. Mol Cell Biol. 2006;26(7):2583–2594. doi: 10.1128/MCB.26.7.2583-2594.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet J, Devys D, Tora L. Histone H2B ubiquitination: Signaling not scrapping. Drug Discov Today Technol. 2012;12:e19–e27. doi: 10.1016/j.ddtec.2012.09.002. [DOI] [PubMed] [Google Scholar]
- Camphausen K, Scott T, Sproull M, Tofilon P. Enhancement of xenograft tumor radiosensitivity by the histone deacetylase inhibitor MS-275 and correlation with histone hyperacetylation. Clin Cancer Res. 2004;10(18):6066–6071. doi: 10.1158/1078-0432.CCR-04-0537. [DOI] [PubMed] [Google Scholar]
- Choudhary C, Kumar C, Gnad F, Nielsen M, Rehman M, Walther T, Olsen J, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325(5942):834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- Danaher R, Jacob R, Steiner M, Allen W, Hill J, Miller C. Histone deacetylase inhibitors induce reactivation of herpes simplex virus type 1 in a latency-associated transcript-independent manner in neuronal cells. J Neurovirol. 2005;11(3):306–317. doi: 10.1080/13550280590952817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson MA, Kouzarides T. Cancer epigenetics: From mechanism to therapy. Cell. 2012;150(1):12–27. doi: 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]
- Duque-Afonso J, Yalcin A, Berg T, Abdelkarim M, Heidenreich O, Lubbert M. The HDAC class I-specific inhibitor entinostat (MS-275) effectively relieves epigenetic silencing of the LAT2 gene mediated by AML1/ETO. Oncogene. 2011;30(27):3062–3072. doi: 10.1038/onc.2011.32. [DOI] [PubMed] [Google Scholar]
- Glozak M, Sengupta N, Zhang X, Seto E. Acetylation and deacetylation of non-histone proteins. Gene. 2005;363:15–23. doi: 10.1016/j.gene.2005.09.010. [DOI] [PubMed] [Google Scholar]
- Gong MC, Latouche JB, Krause A, Heston WD, Bander NH, Sadelain M. Cancer patient T cells genetically targeted to prostate-specific membrane antigen specifically lyse prostate cancer cells and release cytokines in response to prostate-specific membrane antigen. Neoplasia. 1999;1(2):123–127. doi: 10.1038/sj.neo.7900018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu H, Roizman B. Herpes simplex virus-infected cell protein 0 blocks the silencing of viral DNA by dissociating histone deacetylases from the CoREST-REST complex. Proc Natl Acad Sci USA. 2007;104(43):17134–17139. doi: 10.1073/pnas.0707266104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms JS, Splitter GA. Interferon-gamma inhibits transgene expression driven by SV40 or CMV promoters but augments expression driven by the mammalian MHC I promoter. Hum Gene Ther. 1995;6(10):1291–1297. doi: 10.1089/hum.1995.6.10-1291. [DOI] [PubMed] [Google Scholar]
- Hasselgren P. Ubiquitination, phosphorylation, and acetylation—Triple threat in muscle wasting. J Cell Physiol. 2007;213(3):679–689. doi: 10.1002/jcp.21190. [DOI] [PubMed] [Google Scholar]
- Hayakawa T, Nakayama J. Physiological roles of class I HDAC complex and histone demethylase. J Biomed Biotechnol. 2011;2011:129383. doi: 10.1155/2011/129383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi H, Ma Y, Kohno T, Igarashi M, Yasui K, Chua KJ, Kubo Y, Ishibashi M, Urae R, Irie S, Matsuyama T. Yongping Y, editor. Effective Transgene Constructs to Enhance Gene Therapy with Trichostatin A. Targets in gene therapy. 2011 ISBN: 978-953-307-540-2, InTech, DOI: 10.5772/18523. Available from: http://www.intechopen.com/books/targets-in-gene-therapy/effective-transgene-constructs-to-enhance-gene-therapy-with-trichostatin-a.
- Hu E, Dul E, Sung C, Chen Z, Kirkpatrick R, Zhang G, Johanson K, Liu R, Lago A, Hofmann G, et al. Identification of novel isoform-selective inhibitors within class I histone deacetylases. J Pharmacol Exp Ther. 2003;307(2):720–728. doi: 10.1124/jpet.103.055541. [DOI] [PubMed] [Google Scholar]
- Juergens RA, Wrangle J, Vendetti FP, Murphy SC, Zhao M, Coleman B, Sebree R, Rodgers K, Hooker CM, Franco N, et al. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer Discov. 2011;1(7):598–607. doi: 10.1158/2159-8290.CD-11-0214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaouass M, Beaulieu R, Balicki D. Histonefection: Novel and potent non-viral gene delivery. J Control Release. 2006;113(3):245–254. doi: 10.1016/j.jconrel.2006.04.013. [DOI] [PubMed] [Google Scholar]
- Kasman L, Lu P, Voelkel-Johnson C. The histone deacetylase inhibitors depsipeptide and MS-275, enhance TRAIL gene therapy of LNCaP prostate cancer cells without adverse effects in normal prostate epithelial cells. Cancer Gene Ther. 2007;14(3):327–334. doi: 10.1038/sj.cgt.7701017. [DOI] [PubMed] [Google Scholar]
- Kasman L, Onicescu G, Voelkel-Johnson C. Histone deacetylase inhibitors restore cell surface expression of the coxsackie adenovirus receptor and enhance CMV promoter activity in castration-resistant prostate cancer cells. Prostate Cancer. 2012;2012:137163. doi: 10.1155/2012/137163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandelwal A, Gediya L, Njar V. MS-275 synergistically enhances the growth inhibitory effects of RAMBA VN/66–1 in hormone-insensitive PC-3 prostate cancer cells and tumours. Br J Cancer. 2008;98(7):1234–1243. doi: 10.1038/sj.bjc.6604295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. Histone methylation in transcriptional control. Curr Opin Genet Dev. 2002;12(2):198–209. doi: 10.1016/s0959-437x(02)00287-3. [DOI] [PubMed] [Google Scholar]
- Krishan A. Rapid flow cytofluorometric analysis of mammalian cell cycle by propidium iodide staining. J Cell Biol. 1975;66(1):188–193. doi: 10.1083/jcb.66.1.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Rinwa P. Histone deacetylase (HDAC) inhibitors as potential drugs for neurodegernerative disorders: An update. World J Pharm Pharm Sci. 2012;1:486–498. [Google Scholar]
- Liu N, He S, Ma L, Ponnusamy M, Tang J, Tolbert E, Bayliss G, Zhao T, Yan H, Zhuang S. Blocking the class I histone deacetylase ameliorates renal fibrosis and inhibits renal fibroblast activation via modulating TGF-beta and EGFR signaling. PLoS ONE. 2013;8(1) doi: 10.1371/journal.pone.0054001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu ZM, Zhou J, Wang X, Guan Z, Bai H, Liu ZJ, Su N, Pan K, Ji J, Deng D. Nucleosomes correlate with in vivo progression pattern of de novo methylation of p16 CpG islands in human gastric carcinogenesis. PLoS ONE. 2012;7(4):e35928. doi: 10.1371/journal.pone.0035928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks P, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK. Histone deacetylases and cancer: Causes and therapies. Nat Rev Cancer. 2001;1(3):194–202. doi: 10.1038/35106079. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Bulger M, Muramatsu M, Ito T. Multistep chromatin assembly on supercoiled plasmid DNA by nucleosome assembly protein-1 and ATP-utilizing chromatin assembly and remodeling factor. J Biol Chem. 2001;276(29):27384–27391. doi: 10.1074/jbc.M101331200. [DOI] [PubMed] [Google Scholar]
- Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393(6683):386–389. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- Perry AS, Watson RW, Lawler M, Hollywood D. The epigenome as a therapeutic target in prostate cancer. Nat Rev Urol. 2010;7(12):668–680. doi: 10.1038/nrurol.2010.185. [DOI] [PubMed] [Google Scholar]
- Poleshko A, Palagin I, Zhang R, Boimel P, Castagna C, Adams P, Skalka A, Katz R. Identification of cellular proteins that maintain retroviral epigenetic silencing: Evidence for an antiviral response. J Virol. 2008;82(5):2313–2323. doi: 10.1128/JVI.01882-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potta T, Zhen Z, Grandhi TS, Christensen MD, Ramos J, Breneman CM, Rege K. Discovery of antibiotics-derived polymers for gene delivery using combinatorial synthesis and cheminformatics modeling. Biomaterials. 2014;35(6):1977–1988. doi: 10.1016/j.biomaterials.2013.10.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozarowski P, Darzynkiewicz Z. Analysis of cell cycle by flow cytometry. Methods Mol Biol. 2004;281:301–311. doi: 10.1385/1-59259-811-0:301. [DOI] [PubMed] [Google Scholar]
- Qu W, Kang YD, Zhou MS, Fu LL, Hua ZH, Wang LM. Experimental study on inhibitory effects of histone deacetylase inhibitor MS-275 and TSA on bladder cancer cells. Urol Oncol. 2010;28(6):648–654. doi: 10.1016/j.urolonc.2008.11.018. [DOI] [PubMed] [Google Scholar]
- Reilly M, Larsen J, Sullivan M. Histone H3 tail peptides and poly(ethylenimine) have synergistic effects for gene delivery. Mol Pharm. 2012;9(5):1031–1040. doi: 10.1021/mp200372s. [DOI] [PubMed] [Google Scholar]
- Ropero S, Esteller M. The role of histone deacetylases (HDACs) in human cancer. Mol Oncol. 2007;1(1):19–25. doi: 10.1016/j.molonc.2007.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosato RR, Almenara JA, Grant S. The histone deacetylase inhibitor MS-275 promotes differentiation or apoptosis in human leukemia cells through a process regulated by generation of reactive oxygen species and induction of p21CIP1/WAF1 1. Cancer Res. 2003;63(13):3637–3645. [PubMed] [Google Scholar]
- Rossetto D, Avvakumov N, Côté J. Histone phosphorylation: A chromatin modification involved in diverse nuclear events. Epigenetics. 2012;7(10):1098–1108. doi: 10.4161/epi.21975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambucetti LC, Fischer DD, Zabludoff S, Kwon PO, Chamberlin H, Trogani N, Xu H, Cohen D. Histone deacetylase inhibition selectively alters the activity and expression of cell cycle proteins leading to specific chromatin acetylation and antiproliferative effects. J Biol Chem. 1999;274(49):34940–34947. doi: 10.1074/jbc.274.49.34940. [DOI] [PubMed] [Google Scholar]
- Shu H, Gruissem W, Hennig L. Measuring Arabidopsis chromatin accessibility using DNase I-polymerase chain reaction and DNase I-chip assays. Plant Physiol. 2013;162(4):1794–1801. doi: 10.1104/pp.113.220400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikandar S, Dizon D, Shen X, Li Z, Besterman J, Lipkin S. The Class I Hdac inhibitor Mgcd0103 induces cell cycle arrest and apoptosis in colon cancer initiating cells by upregulating dickkopf-1 and non-canonical Wnt signaling. Oncotarget. 2010;1(7):596–605. doi: 10.18632/oncotarget.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunley K, Butler M. Strategies for the enhancement of recombinant protein production from mammalian cells by growth arrest. Biotechnol Adv. 2010;28(3):385–394. doi: 10.1016/j.biotechadv.2010.02.003. [DOI] [PubMed] [Google Scholar]
- Tang Q, Maul G. Mouse cytomegalovirus immediate-early protein 1 binds with host cell repressors to relieve suppressive effects on viral transcription and replication during lytic infection. J Virol. 2003;77(2):1357–1367. doi: 10.1128/JVI.77.2.1357-1367.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu L, Ramos J, Potta T, Rege K. Generation of a Focused Poly(amino ether) Library: Polymer-mediated Transgene Delivery and Gold-Nanorod based Theranostic Systems. Theranostics. 2012;2(12):1160–1173. doi: 10.7150/thno.4492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YM, Zhou P, Wang LY, Li ZH, Zhang YN, Zhang YX. Correlation between DNase I hypersensitive site distribution and gene expression in HeLa S3 cells. PLoS ONE. 2012;7(8):e42414. doi: 10.1371/journal.pone.0042414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolffe A. Chromatin: Structure and function. 3rd Academic Press; San Diego: 1999. [Google Scholar]
- Xhemalce B, Dawson M, Bannister A. Histone modifications. In: Meyers RA, editor. Encyclopedia of molecular cell biology and molecular medicine. Wiley-VCH Verlag GmbH & Co. KGaA; Weinheim, Germany: 2011. [Google Scholar]
- Yamauchi Y, Boukari H, Banerjee I, Sbalzarini I, Horvath P, Helenius A. Histone deacetylase 8 is required for centrosome cohesion and influenza A virus entry. PLoS Pathog. 2011;7(10) doi: 10.1371/journal.ppat.1002316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaneva J, Schroth GP, van Holde KE, Zlatanova J. High-affinity binding sites for histone H1 in plasmid DNA. Proc Natl Acad Sci USA. 1995;92(15):7060–7064. doi: 10.1073/pnas.92.15.7060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XJ, Seto E. HATs and HDACs: From structure, function, and regulation to novel strategies for therapy and prevention. Oncogene. 2007;26(37):5310–5318. doi: 10.1038/sj.onc.1210599. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.