

Abstract
Post-transcriptional gene expression is governed by the interaction of mRNAs with vast families of RNA-binding proteins (RBPs) and noncoding (nc)RNAs. RBPs and ncRNAs jointly influence all aspects of post-transcriptional metabolism, including pre-mRNA splicing and maturation, mRNA transport, editing, stability, and translation. Given the impact of mRNA-interacting molecules on gene expression, there is great interest in identifying mRNA-binding factors comprehensively. Here, we provide a detailed protocol to tag mRNAs with MS2 hairpins and then affinity-purify trans-binding factors (RBPs, ncRNAs) associated with the MS2-tagged mRNA. This method, termed MS2-TRAP, permits the systematic characterization of ribonucleoprotein (RNP) complexes formed on a given mRNA of interest. We describe how to prepare the mRNA-MS2 expression vector, purify the MS2-tagged RNP complexes, and detect bound RNAs and RBPs, as well as variations of this methodology to address related questions of RNP biology.
Keywords: RBP, ribonucleoprotein complexes, lncRNA, RNA affinity pull down
1. Introduction
Post-transcriptional gene regulation is mainly controlled by two classes of factors associating with mRNAs, RNA-binding proteins (RBPs) and noncoding RNAs (ncRNAs) [1,2]. RBPs are involved in all steps of post-transcriptional gene regulation, including pre-mRNA splicing and maturation, mRNA export, degradation, editing, localization, storage and translation [3]. Among the various small ncRNAs (miRNAs, piRNAs, siRNAs, tiRNAs, snoRNAs, etc.), microRNAs have been studied most extensively and function by promoting mRNA degradation and/or translation repression [4,5]. Long (l)ncRNAs also control many post-transcriptional steps through their interaction with target mRNAs [6]. Together, these families of ncRNAs and RBPs regulate the post-transcriptional fate of the mRNA in a robust and dynamic manner, allowing gene expression patterns to respond adequately to developmental, metabolic, and environmental cues [2]. Accordingly, studying the composition of mRNA RNPs is critical for understanding the ultimate outcome of mRNAs.
Interactions between RBPs and RNAs have been studied for decades (reviewed comprehensively in [7] and chapters therein). The binding of a specific RBP to a specific RNA has been studied using classic in vitro methods like RNA electrophoretic mobility shift, biotin pulldown, and agarose gel retardation assays [7–9]. Systematic methods to detect binding of one RBP to collections of target mRNAs are more recent and include binding assays to measure native complexes (ribonucleoprotein immunoprecipitation or RIP) and crosslinked complexes (crosslinking immunoprecipitation or CLIP) [10]. The physical and functional interactions of specific mRNAs and microRNAs have also been studied using a variety of analytic methods. However, few high-throughput approaches are available to identify the collection of factors that interact with a given mRNA in the cell. One such approach, affinity pulldown using antisense oligonucleotides complementary to the mRNA. is feasible in cell-free systems, but is not a dependable method for targeting intracellular RNP complexes in vivo.
MS2 hairpin sequences have long been utilized for studying mRNAs in eukaryotic cells [11]. The high-affinity interaction of the MS2 RNA hairpin and its binding protein, the viral coat protein MS2-BP, has long been exploited for visualizing and purifying RNP complexes without introducing purified proteins or RNAs into cells. The existence of many chimeric variants of MS2-BP, bearing different tags for imaging and for biochemical detection gives this approach additional advantages. Here, we describe a detailed protocol for a methodology we have named MS2-TRAP (MS2-tagged RNA affinity purification) to purify RNP complexes that include RBPs and ncRNAs bound to an mRNA of interest. Detailed directions to design target plasmids expressing MS2-tagged RNA, step-by-step procedures for complex purification, detection of co-purified proteins and RNAs, troubleshooting suggestions, and additional notes are provided.
2. Materials
2.1. Cell culture and transfection
2.2. Immunoprecipitation
Glutathione agarose beads (GE Healthcare)
Protein-A or -G sepharose beads (GE Healthcare)
Anti-YFP antibody (Santa Cruz Biotechnology)
Anti-Flag antibody (M2, Sigma)
Normalized IgG (Santa Cruz Biotechnology)
Proteinase K (Roche)
DNase I (Ambion): 2 U/μL
Acid-phenol:chloroform, pH 4.5 (with IAA, 25:24:1) (Ambion)
GlycoBlue™ coprecipitant (15 mg/mL) (Ambion) ice-cold phosphate-buffered saline (PBS): 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 1.8 mM KH2PO4
NP-40 lysis buffer: 20 mM Tris–HCl at pH 7.5, 100 mM KCl, 5 mM MgCl2, 0.5% NP-40, 1X protease inhibitors (Roche), RiboLock RNase inhibitor (Fermentas, 40 U/mL), and 1 mM DTT
NT2 buffer: 50 mM Tris–HCl at pH 7.5, 150 mM NaCl, 1 mM MgCl2, and 0.05% NP-40
RIPA buffer: 10 mM Tris–HCl, pH 7.4, 150 mM NaCl, 1% (v/v) Nonidet P-40, 1 mM EDTA, 0.1% (w/v) SDS, and 1 mM DTT
4X SDS sample buffer: 40% glycerol, 240 mM Tris/HCl pH 6.8, 8% SDS, 0.04% bromophenol blue, and 5% β-mercaptoethanol
2.3. Reverse transcription and real-time quantitative (q)PCR
dNTP mix (10 mM)
Random hexamers (150 ng/μl)
Reverse transcriptase (Fermentas): Maxima Reverse Transcriptase (200 U/μL)
Gene-specific primer sets
KAPA SYBR® FAST qPCR Kits QuantiMir™ RT kit (System Biosciences)
3. Methods
3.1. Plasmid construction, cell culture, and transfection
Plasmid pMS2 has a pcDNA3 backbone containing 24 repeats of MS2 sequences between the EcoRI and EcoRV sites. Cloning sites at HindIII, KpnI, and EcoRI are available for attaching complementary DNA (cDNA, prepared from the mRNA of interest) upstream of the 24 MS2 repeats (Figure 1). Alternatively, MS2 can be transferred to the parental plasmid expressing the mRNA of interest by standard enzymatic digestion or PCR amplification (see Note 1).
Once the p(cDNA)-MS2 plasmid construct is ready, prepare a mammalian cell culture (e.g., human embryonic kidney 293 cells, HeLa cells or mouse embryonic fibroblasts) at 80% confluence in 100-mm culture plates in DMEM supplemented with 10% fetal bovine serum and appropriate antibiotics.
Transfect 3 μg of p(cDNA)-MS2 or pMS2 empty vector, together with 1 μg of pMS2-GST (alternatively pMS2-YFP or pcDNA3-FLAG-MS2) diluted in 200 μl of OPTI-MEM, mixed with 5 μl lipofectamine 2000 diluted with 200 μl of OPTI-MEM (see Note 2).
After incubation for 20 min at room temperature, rinse cells with phosphate-buffered saline (PBS) once, replace with OPTI-MEM, add the transfection mixture.
Incubate cells for an additional 5 hours at 37 °C in 5% CO2, rinse cells once with PBS, and replace DMEM supplemented with 10% FBS without antibiotics.
Forty-eight hours later, wash the cells with PBS once and lyse them in 500 μl NP-40 lysis buffer for 10 min on ice.
Scrape the cells, collect the lysate, and centrifuge them at 10,000 g for 30 min at 4 °C.
Transfer the supernatant to a new tube and use 2 mg of lysate in 1 mL for pulldown or immunoprecipitation.
Figure 1.
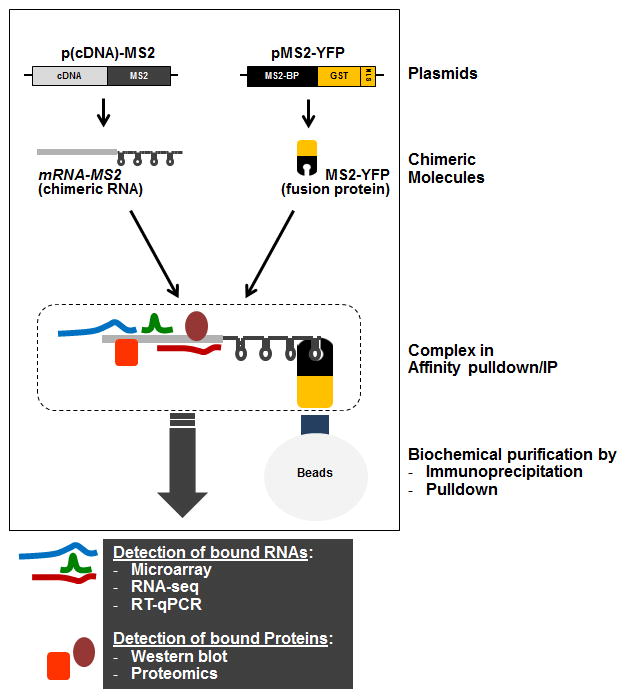
Schematic representation of the MS2-TRAP methodology, including the plasmids required (‘Plasmids’), the fusion RNA and protein expressed from the plasmids (‘Chimeric Molecules’) and the resulting RNP complex (‘Complex in Affinity pulldown/IP’). After pulldown or immunoprecipitation using the appropriate conjugated beads, RNAs and proteins bound to the chimeric transcript can be identified using are methodologies listed (gray box).
3.2. Complex purification and RNA isolation
Prepare the beads (glutathione agarose, Protein-A or -G sepharose) by washing them with ice-cold PBS three times and resuspending them with an equal volume of PBS. The 40-μl slurry of glutathione beads can be used directly for pulldown analysis.
For coating anti-Flag or anti-YFP antibody, incubate 40 μl of the slurry with 3 μg of normalized IgG or anti-Flag (or anti-YFP) antibody for 3 hours at 4 °C in NT2 buffer, centrifuge the beads at 2,000 × g for 2 min at 4 °C, and wash five times using NT2 buffer.
Add 1 mL of cell lysate to the beads (GSH agarose or antibody-coated sepharose) and incubate for 2 hours at 4 °C (see Note 3).
Centrifuge samples at 2,000 × g for 2 min at 4 °C, and wash three times with NP-40 lysis buffer.
Add 20 units of RNase-free DNase I (Ambion) in 100 μl NP-40 lysis buffer and incubate them for 15 min at 37 °C.
Add 700 μl NT2 buffer and centrifuge at 2,000 × g for 2 min at 4 °C.
For protein analysis, go to section 3.4 (protein detection).
For RNA analysis, add 0.1% SDS/0.5 mg/ml Proteinase K and incubate for 20 min at 55 °C.
Centrifuge at 10,000 × g for 10 min at 4 °C and collect the supernatant.
Add 500 μl RNase-free water/500 μl of acidic phenol and vortex them for 3 min.
Centrifuge at 10,000 × g for 30 min at 4 °C and transfer 400 μl of the supernatant.
Add 800 μl of 100% ethanol/40 μl of 3 M sodium acetate/2 μl of glycoblue and incubate at −80 °C for 1 hour or overnight.
Centrifuge at 10,000 × g at 4 °C for 30 min, wash with 400 μl of 70% ethanol, and centrifuge at 10,000 × g, for 15 min 4 °C.
Dry the pellets and resuspend in 12 μl of RNase-free water.
3.3. RNA detection (mRNAs, lncRNAs, and miRNAs)
For detecting mRNA or lncRNA in the pulldown/IP sample, add 1 μl dNTP mix (10 mM) and 1 μl of random hexamers (150 ng/μl) with 12 μl RNAs.
Incubate at 65°C for 5 min and 4°C for 5 min.
Add 1 μl reverse transcriptase (200 U/μl), 1 μl RNase Inhibitor (40 U/μl), and 4 μl 5× reverse transcription reaction buffer.
Incubate reactions at 25°C for 10 min, at 50°C for 30 min, and at 85°C for 5 min in thermo cycler.
Take 2.5 μl of cDNAs and add 2.5 μl forward and reverse gene specific primers (2.5–10 μM) and 5 μl of SYBR green master mix.
Once PCR is performed, calculate Ct values of each sample. For GSH pull down, Ct values from pMS2 empty vector sample can be utilized for normalization. For immunoprecipitation, normalization can be performed with Ct values from pMS2 empty vector sample or from p(cDNA)-MS2 sample with IgG immunoprecipitation. Normalization of non-specific binding can be performed using Ct values of mRNAs encoding housekeeping proteins such as GAPDH, ACTB, UBC or SDHA.
For detecting miRNAs, take 5 μl of RNAs from precipitation and add 2 μl of 5× Poly(A) buffer, 1 μl of 25 mM MnCl2, 1.5 μl of 5 mM ATP and 0.5 μl Poly(A) polymerase included in the QuantiMir kit.
Incubate reaction for 30 min at 37 °C and add 0.5 μl of Oligo(dT) adaptor (QuantiMir kit).
Incubate reaction for 5 min at 60 °C and cool it down at room temperature for 2 min.
Add 4 μl of 5× RT buffer, 2 μl of dNTP mix, 1.5 μl of 0.1 M DTT, 1.5 μl of RNase-free water, and 1 μl of reverse transcriptase (QuantiMir kit).
Incubate reactions for 60 min at 42 °C and heat for 10 min at 95 °C.
Mix 2.5 μl of cDNAs with 2.5 μl of miRNA-specific primer (2.5 μM), universal primer (10 μM from QuantiMir Kit), and SYBR green master mix.
Perform PCR and calculate Ct values of each sample for normalization as described above for mRNA and lncRNA (see Note 4). Normalization of non-specific binding can be performed using Ct values of housekeeping small RNAs such as U6.
3.4. Protein detection
Forty-eight hours after transfection, whole-cell lysates can be prepared using RIPA buffer (500 μl) by incubating cells for 10 min on ice, scraping the cells, collecting the lysate, centrifuging it at 10,000 × g for 30 min at 4 °C, and transferring the supernatant to a new tube.
After step 3.2.7, the proteins present in the mRNA pulldown can be mixed with SDS sample buffer for further protein analysis.
After electrophoresis through SDS-containing polyacrylamide gels (SDS–PAGE), transfer samples onto nitrocellulose membranes (Invitrogen iBlot Stack) and probe membranes with primary antibodies of interest.
Use HRP-conjugated secondary antibodies to probe primary antibodies and detect the signals using chemiluminescence (Pierce) (see Note 5).
Acknowledgments
JHY and MG were supported by the National Institute on Aging Intramural Research Program, National Institutes of Health.
Footnotes
If the chimeric mRNA-MS2 transcript is not enriched relative to MS2 transcript alone, consider transfecting larger molar ratios of plasmid expressing the chimeric mRNA-MS2 than the detection plasmid (pMS2-GST, pMS2-YFP); for example 5:1 or 10:1. With this modification, non-specific binding of abundant cellular RNAs can be minimized.
If target mRNAs, lncRNAs, or miRNAs are not enriched in the pulldown or immunoprecipitation materials, reduce the time allowed for coating with antibody or for binding with beads. The amount of beads, antibody, and lysates can be reduced too.
If the construction of pMS2-mRNA plasmid is difficult, MS2 can be inserted before the mRNA. Challenges may arise from limited availability of restriction sites in the pMS2 plasmid and/or parental plasmid. Despite successful construction of the plasmid to express MS2-tagged mRNA, the chimeric transcript may not behave like the endogenous mRNA because of the 24 MS2 repeats at the 5’end. In this case, additional methods to pull down the fusion RNA can be considered by using biotinylated antisense DNA or RNA oligomers.
If plasmid DNA contamination is suspected during RNA precipitation, RT-minus PCR amplification can be performed. In order to remove residual plasmid, it may be necessary to increase the amount of DNase and/or lengthen the incubation time.
If the goal is to identify trans-factors interacting with a lncRNA (which also interact extensively with RBPs and ncRNAs [14–16]) or with a partial RNA fragment, the same general procedure can be adopted by tagging the lncRNA or RNA fragment with MS2. Additional RNA pulldown or IP can be performed as explained above.
References
- 1.Morris AR, Mukherjee N, Keene JD. Systematic analysis of posttranscriptional gene expression. Wiley Interdiscip Rev Syst Biol Med. 2010;2:162–180. doi: 10.1002/wsbm.54. [DOI] [PubMed] [Google Scholar]
- 2.Moore MJ. From birth to death: the complex lives of eukaryotic mRNAs. Science. 2005;309:1514–1518. doi: 10.1126/science.1111443. [DOI] [PubMed] [Google Scholar]
- 3.Glisovic T, Bachorik JL, Yong J, Dreyfuss G. RNA-binding proteins and post-transcriptional gene regulation. FEBS Lett. 2008;582:1977–1986. doi: 10.1016/j.febslet.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fabian MR, Sonenberg N, Filipowicz W. Regulation of mRNA translation and stability by microRNAs. Annu Rev Biochem. 2010;79:351–379. doi: 10.1146/annurev-biochem-060308-103103. [DOI] [PubMed] [Google Scholar]
- 6.Yoon JH, Abdelmohsen K, Gorospe M. Posttranscriptional gene regulation by long noncoding RNA. J Mol Biol. 2013;425:3723–3730. doi: 10.1016/j.jmb.2012.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin RJ, editor. Methods Mol Biol. Vol. 488. 2008. RNA-protein interaction protocols; pp. v–vii. [DOI] [PubMed] [Google Scholar]
- 8.Ryder SP, Recht MI, Williamson JR. Quantitative analysis of protein-RNA interactions by gel mobility shift. Methods Mol Biol. 2008;488:99–115. doi: 10.1007/978-1-60327-475-3_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abdelmohsen K, Tominaga K, Lee EK, Srikantan S, Kang MJ, Kim MM, Selimyan R, Martindale JL, Yang X, Carrier F, Zhan M, Becker KG, Gorospe M. Enhanced translation by Nucleolin via G-rich elements in coding and non-coding regions of target mRNAs. Nucleic Acids Res. 2011;39:8513–8530. doi: 10.1093/nar/gkr488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.König J, Zarnack K, Luscombe NM, Ule J. Protein-RNA interactions: new genomic technologies and perspectives. Nat Rev Genet. 2012;13:77–83. doi: 10.1038/nrg3141. [DOI] [PubMed] [Google Scholar]
- 11.Bertrand E, et al. Localization of ASH1 mRNA particles in living yeast. Mol Cell. 1998;2:437–445. doi: 10.1016/s1097-2765(00)80143-4. [DOI] [PubMed] [Google Scholar]
- 12.Dutko JA, et al. Inhibition of a yeast LTR retrotransposon by human APOBEC3 cytidine deaminases. Curr Biol. 2005;15:661–666. doi: 10.1016/j.cub.2005.02.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gong C, Maquat LE. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3′ UTRs via Alu elements. Nature. 2011;470:284–288. doi: 10.1038/nature09701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chu C, Qu K, Zhong FL, Artandi SE, Chang HY. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol Cell. 2011;44:667–678. doi: 10.1016/j.molcel.2011.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Quinn JJ, et al. Revealing long noncoding RNA architecture and functions using domain-specific chromatin isolation by RNA purification. Nat Biotechnol. 2014;32:933–940. doi: 10.1038/nbt.2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Engreitz JM, et al. RNA-RNA interactions enable specific targeting of noncoding RNAs to nascent Pre-mRNAs and chromatin sites. Cell. 2014;159:188–199. doi: 10.1016/j.cell.2014.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]