

Abstract
Mammalian cells express a wide range of transcripts, some protein-coding RNAs (mRNA) and many noncoding (nc) RNAs. Long (l)ncRNAs can modulate protein expression patterns by regulating gene transcription, pre-mRNA splicing, mRNA export, mRNA degradation, protein translation, and protein ubiquitination. Given the growing recognition that lncRNAs have a robust impact upon gene expression, there is rising interest in elucidating the levels and regulation of lncRNAs. A number of high-throughput methods have been developed recently to map the interaction of lncRNAs and RNA-binding proteins (RBPs). However, few of these approaches are suitable for mapping and quantifying RBP-lncRNA interactions. Here, we describe the recently developed method CLIP-qPCR (crosslinking and immunoprecipitation followed by reverse transcription and quantitative PCR) for mapping and quantifying RBP-lncRNA interactions.
Keywords: CLIP, lncRNA, RBP, ribonucleoprotein complexes, qPCR
1 Introduction
Gene expression programs are influenced transcriptionally via processes such as chromatin remodeling and transcription factor mobilization, and posttranscriptionally through processes like pre-mRNA splicing, 5′ capping, 3′ polyadenylation, editing, mRNA export, localization, and translation. These processes are governed by DNA-binding proteins, RNA-binding proteins, and noncoding RNAs [1–4]. In recent years, lncRNAs have emerged as major regulators of transcription, mRNA fate, and protein stability [4–6]. Although they can perform these gene regulatory functions directly via sequence complementarity with target DNA and RNA sequences, most lncRNA functions identified to-date involve their interaction with partner DNA-binding proteins and RNA-binding proteins (RBPs) [4, 6].
RBP-RNAs complexes have been studied using a variety of methods [7]. Interactions of specific RBPs and specific RNAs have been examined using traditional procedures such as the analysis of ribonucleoprotein immunoprecipitation (RIP) complexes, biotinylated RNA pulldown complexes, and RNA electrophoretic mobility shifts assays [8]. Global analyses of mRNAs interacting with RBP were developed using RIP followed by cDNA microarray analysis [9] or by RIP followed by RNA sequencing (RNA-seq [10]). Subsequently, RBP crosslinking using UVC light (254 nm) and immunoprecipitation (CLIP) was developed, initially combined with Sanger Sequencing method [11] and later with high-throughput sequencing (RNA-seq) of the CLIP cDNA library [12, 13]. Further advances were achieved by photoactivatable ribonucleoside-enhanced (PAR)-CLIP analysis, using nucleotide analogs such as 4-thiouridine (4-SU) or 6-thioguanosine (6-SG) following exposure to UVA light (365nm) [14]. Alternative CLIP methods are also known as individual nucleotide resolution CLIP [15] using reverse transcriptase stall and crosslinking, ligation and sequencing of hybrids (CLASH), which monitors inter-RNA interactions within tripartite complexes [16].
Despite such enormous advances in RBP-RNA detection, high-throughput analyses require the generation of a small RNA library, and thus a significant amount of effort, cost, time, and optimization. In addition, a substantial amount of bioinformatic analysis is required to map the sequence reads and identify the binding sites. Therefore, we have developed a protocol that combines PAR-CLIP with qPCR analysis to map RBP binding sites at 100-nt intervals within an lncRNA of interest. This method is based on the novel introduction of partial RNase digestion and the detection of serial, progressive scanning of the target RNA by reverse transcription and qPCR.
2 Materials
2.1 Mammalian cell culture and RNA labeling
DMEM, 10% FBS, 2 mM L-Glutamine, 100 U/ml Penicillin/streptomycin
1 M 4-thiouridine (4-SU) in DMSO
2.2 UVA crosslinking and cell harvesting
UV cross linker
Stratalinker 1800, 365 nm bulb
Ice-cold phosphate-buffered saline (PBS)
2.3 RNase digestion and immunoprecipitation
-
1
RNase T1
-
2
Protein A or G Sepharose beads
-
3
Antibodies to detect an endogenous protein of interest
-
5
Normalized IgG
-
4
Proteinase K
-
5
DNase I
-
6
Acidic phenol
-
7
Glycoblue
-
8
NP-40 lysis buffer: 20 mM Tris–HCl at pH 7.5, 100 mM KCl, 5 mM MgCl2, and 0.5% NP-40
-
9
NT2 buffer: 50 mM Tris–HCl at pH 7.5, 150 mM NaCl, 1 mM MgCl2 and 0.05% NP-40
2.4 Reverse transcription and qPCR
dNTP mix (10 mM)
Random hexamer (150 ng/μl)
Reverse transcriptase
Gene-specific primer sets
SYBR mix
3 Methods
3.1 Mammalian cell culture and RNA labeling
Expand Human Embryonic Kidney (HEK) or Human cervical cancer (HeLa) cells in 150-mm culture plates, cultured in DMEM supplemented with 10% (v/v) fetal bovine serum and antibiotics. Grow cells up to 80% confluence.
Sixteen hours before UVA exposure, add 4SU (1 M stock solution in DMSO) to a final concentration of 100 μM in culture medium. Alternatively, 100 μM 6SG can be used, but the crosslinking efficiency is somewhat lower.
3.2 UVA crosslinking and harvesting cells
Aspirate the culture medium, wash cells once with 15 ml ice-cold PBS and aspirate PBS completely.
Set up a tray containing ice and place the plate on the ice.
Uncover the plate and irradiate it with 150 mJ/cm2 of UVA (365 nm) in Stratalinker or similar device.
Scrape cells in 5 mL PBS, transfer to 50 mL conical tube, centrifuge at 2,000 × g at 4 °C for 5 min, and aspirate the supernatant.
Cell pellets can be used immediately for lysis or stored at −80 °C for later use.
3.3 RNase digestion and immunoprecipitation
Resuspend cell pellets by adding 3 volumes of NP-40 lysis buffersupplemented with protease inhibitors, and 1 mM DTT.
Incubate on ice for 10 min and centrifuge at 10,000 × g for 15 min at 4 °C.
Collect supernatants and add RNase T1 to 1 U/μl final concentration, incubate at 22 °C for 2, 4, 6, 8, 10, and 15 min.
Spare 100 μl lysate, add 400 μl water and 500 μl acidic phenol, vortex for 1 min, and centrifuge at 10,000 × g for 20 min at 4 °C.
Collect 400 μl of supernatant, add 800 μl 100% ethanol, 40 μl 3 M Sodium acetate, and 1 μl glycoblue. Incubate it at −80 °C for 1 h (or overnight).
Centrifuge at 10,000 × g for 20 min at 4 °C, aspirate the supernatant, and add 500 μl of 70% ethanol.
Centrifuge at 10,000 × g for 10 min at 4 °C, aspirate the supernatant, dry pellet at room temperature, and dissolve the pellet with RNase-free water.
Run the RNA samples in 1.5% formaldehyde agarose gel to verify that RNAs are digested in 100- to 300-nt range.
Select the samples having RNAs partially digested in the 100- to 300-nt range.
To prepare sepharose beads, wash beads with ice-cold PBS three times and resuspend them with equal volume of ice-cold PBS to create a 50% slurry.
Incubate 40 μl of the beads slurry with 10 μg of normalized IgG or antibody of interest for 2 h at 4 °C in NT2 buffer.
Centrifuge the beads at 2,000 × g for 1 min at 4 °C, wash three times with NT2 buffer.
Add 1 mL of cell lysates to the sepharose beads coated with antibody and incubate them for 3 h at 4 °C.
After centrifugation at 2,000 × g for 1 min at 4 °C, wash beads three times with NP-40 lysis buffer.
Incubate the pellets with 20 units of RNase-free DNase I in 100 μl NP-40 lysis buffer for 15 min at 37 °C.
Add 700 μl of NP-40 lysis buffer and centrifuge at 2,000 × g for 1 min at 4 °C.
Incubate the pellets with 0.1% SDS and 0.5 mg/ml Proteinase K for 15 min at 55 °C.
Collect the supernatant after centrifugation at 10,000 × g at 4 °C for 5 min.
Add 500 μl of RNase-free water and 500 μl of acidic phenol, then vortex for 5 min.
Centrifuge at 10,000 × g, 4 °C for 20 min, collect 400 μl supernatant, add 800 μl 100% ethanol, 40 μl 3 M sodium acetate, and 1 μl glycoblue, and incubate it at −80 °C for 1 h (or overnight).
Centrifuge at 10,000 × g for 20 min at 4 °C, remove the supernatant, and add 500 μl of 70% ethanol.
Centrifuge at 10,000 × g for 10 min at 4 °C, remove the supernatant, dry pellet at room temperature, and dissolve the pellet with 12 μl RNase-free water.
3.4 Reverse transcription and qPCR
Mix 1 μl dNTP mix (10 mM) and 1μl random hexamer (150 ng/μl) with12 μl purified RNAs.
Incubate them at 65°C for 5 min and 4°C for 5 min using a thermo cycler.
Add 1 μl Reverse transcriptase (200 U/μl), 1 μl RNase Inhibitor (40 U/μl), and 4 μl 5× reaction buffer.
Incubate samples at 25°C for 10 min, at 50°C for 30 min, and at 85°C for 5 min using a thermo cycler.
Mix 2.5 μl of cDNAs, 2.5 μl forward and reverse gene-specific primers (2.5–10 μM) designed to amplify PCR products in 200 nt intervals, and 5 μl of SYBR green master mix.
After completion of qPCR, calculate Ct values of IgG IP and specific antibody IP normalized with Ct values of mRNAs encoding housekeeping proteins like GAPDH, ACTB, UBC, SDHA, etc.
4 Notes
If a method without RNA labeling is preferred, UVC at 254 nm can be utilized instead of UVA at 365 nm and preincubation with 4-SU or 4-SG can be omitted.
If non-canonical RBPs are of interest, RBP and RNA crosslinking may not be successful upon UV exposure. In this case, formaldehyde crosslinking can be utilized instead.
It is critical to titrate the amount of RNase T1 (or other RNase) and the time of incubation. Optimization of these parameters in order to obtain 100–300-nt RNA fragments (mainly from 18S and 28S ribosomal RNAs) is a key in step in CLIP-qPCR analysis. For highly abundant RNAs (e.g., MALAT1 and NEAT1), higher amount of RNase and longer incubation can be utilized.
For primer design, divide the RNA of interest in 200-nt overlapping intervals (e.g., spanning positions 1–200, 101–300, 201–400, 301–500, etc). This way, each gene-specific primer will cover all of the full length transcripts after qPCR. If the full-length target RNA is not in a public database, primer extension or rapid amplification of cDNA ends (RACE) can be done first.
Depending on the RBP and RNAs localized in specific cellular compartments, NP-40 lysis buffer can be modified by increasing the concentration of NP-40 (e.g., from 0.5% to 2%) or by adding more stringent detergents such as Triton X-100 and SDS.
If DNA contamination is suspected, RT-minus PCR amplification reactions can be performed. If there are amplifications, genomic DNA could be contaminated during immunoprecipitation. In this case, increased amount of DNase can be utilized with longer incubation time.
This method can be used to map RBPs binding to mRNAs. Some attention should be paid to the fact that mRNAs may be extensively engaged with polyribosomes and hence the coding region may not be available to RBPs and PCR amplification of certain regions may be challenging.
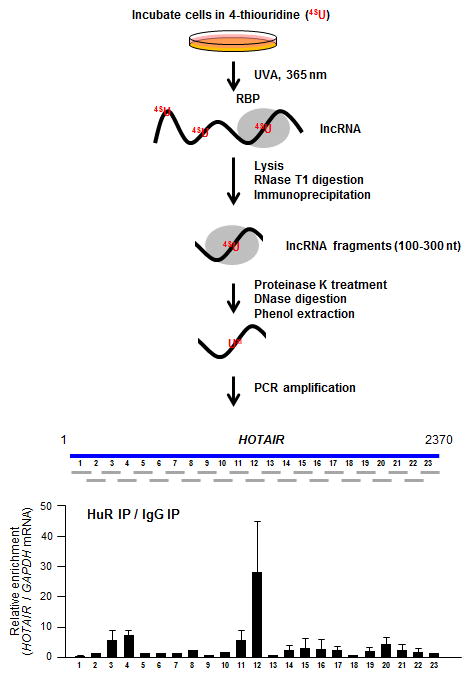
Figure 1.
Acknowledgments
JHY and MG were supported by the National Institute on Aging Intramural Research Program, National Institutes of Health.
References
- 1.Berger SL. Histone modifications in transcriptional regulation. Curr Opin Genet Dev. 2002;12:142–148. doi: 10.1016/s0959-437x(02)00279-4. [DOI] [PubMed] [Google Scholar]
- 2.Glisovic T, Bachorik JL, Yong J, Dreyfuss G. RNA-binding proteins and post-transcriptional gene regulation. FEBS Lett. 2008;582:1977–1986. doi: 10.1016/j.febslet.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet. 2008;9:102–114. doi: 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- 4.Yoon JH, Abdelmohsen K, Gorospe M. Posttranscriptional gene regulation by long noncoding RNA. J Mol Biol. 2013;425:3723–3730. doi: 10.1016/j.jmb.2012.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoon JH, Abdelmohsen K, Gorospe M. Functional interactions among microRNAs and long noncoding RNAs. Semin Cell Dev Biol. 2014;34:9–14. doi: 10.1016/j.semcdb.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81:145–166. doi: 10.1146/annurev-biochem-051410-092902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Riley KJ, Steitz JA. The “Observer Effect” in genome-wide surveys of protein-RNA interactions. Mol Cell. 2013;49:601–604. doi: 10.1016/j.molcel.2013.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lin RJ. RNA -protein interaction protocols. Methods Mol Biol. 2008;488:v–vii. doi: 10.1007/978-1-60327-475-3. [DOI] [PubMed] [Google Scholar]
- 9.Tenenbaum SA, Carson CC, Lager PJ, Keene JD. Identifying mRNA subsets in messenger ribonucleoprotein complexes by using cDNA arrays. Proc Natl Acad Sci U S A. 2000;97:14085–14090. doi: 10.1073/pnas.97.26.14085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mukherjee N, Corcoran DL, Nusbaum JD, Reid DW, Georgiev S, Hafner M, Ascano M, Jr, Tuschl T, Ohler U, Keene JD. Integrative regulatory mapping indicates that the RNA-binding protein HuR couples pre-mRNA processing and mRNA stability. Mol Cell. 2011;43:327–339. doi: 10.1016/j.molcel.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ule J, et al. CLIP identifies Nova-regulated RNA networks in the brain. Science. 2003;302:1212–1215. doi: 10.1126/science.1090095. [DOI] [PubMed] [Google Scholar]
- 12.Licatalosi DD, et al. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature. 2009;456:464–469. doi: 10.1038/nature07488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chi SW1, Zang JB, Mele A, Darnell RB. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature. 2009;460:479–486. doi: 10.1038/nature08170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hafner M, et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010;141:129–141. doi: 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.König J, et al. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat Struct Mol Biol. 2010;17:909–915. doi: 10.1038/nsmb.1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kudla G, et al. Cross-linking, ligation, and sequencing of hybrids reveals RNA-RNA interactions in yeast. Proc Natl Acad Sci U S A. 2011;108:10010–10015. doi: 10.1073/pnas.1017386108. [DOI] [PMC free article] [PubMed] [Google Scholar]