

Calcific aortic valve disease (CAVD) is an age-related disorder that causes significant cardiovascular morbidity and mortality, and is directly responsible for ~15,000 patient deaths per year in the USA. Currently, the molecular mechanism by which CAVD initiates is unknown, making discovery of a non-surgical strategy challenging (reviewed in 1). Garg et al. made a seminal discovery in 2005 when they showed NOTCH1 mutations led to heritable CAVD.2 Since then, the heart valve research community has sought a causal mechanism that could connect NOTCH1 dysfunction to idiopathic CAVD. That causal link has been elusive until the recent and impressive work by Hadjj et al., in this issue of Circulation, demonstrated that the promoter region of the long non-coding (lnc) RNA H19 is hypomethylated in patients with CAVD.3 This hypomethylation, in turn, increases H19 expression in the valve interstitial cells (VICs) where it prevents NOTCH1 transcription by seemingly blocking or outcompeting p53’s recruitment to the NOTCH1 promoter. Thus, H19 appears to be the missing link (or lncRNA, if you will) connecting NOTCH1 to idiopathic CAVD.
LncRNAs are transcripts that lack protein translation potential but are master regulators of gene expression and whose own expression dysregulation is associated with a variety of human pathologies. H19 controls gene expression at multiple levels - from chromatin modification to direct protein inhibition - and has been labeled as the multitasking lncRNA prototype.4 H19 expression is controlled via genomic imprinting through methylation of its promoter region, with methylation negatively regulating expression, and is typically expressed at low levels only from the maternal allele after birth.5 In the arterial vasculature, H19 is highly expressed during development but largely absent after birth; however, it reemerges in vascular smooth muscle cells in cases of neointimal hyperplasia6 and atherosclerosis,7 two pathologies that share similar hallmarks with CAVD. This reemerging pattern of H19 in vascular disease is similar to some of the molecular signatures observed during fetal and postnatal development of aortic valves that also reemerge during early CAVD,8 suggesting that CAVD may be an unwelcomed encore presentation of valve development signaling that arises due to the high mechanical demands placed on the aging valve tissue. More recently, H19 has been shown to be upregulated in patients with heart failure and a mouse model of cardiac hypertrophy,9 further supporting its importance in cardiovascular disease pathogenesis. Clarifying the mechanism of H19 methylation will potentially identify new pharmacological targets for slowing or preventing CAVD, which are severely lacking,1 and possibly for treating other cardiovascular pathologies as well.
Perhaps more importantly, the current finding of H19 repressing NOTCH1 should reenergize the CAVD research field. Progress in uncovering the mechanism(s) causing CAVD has been challenging on multiple levels. First, most end-stage human tissue samples are so diseased and calcified that finding an early, molecular signature is nearly impossible. Second, there is discord within the valve research field regarding whether calcification occurs via an osteogenic or a dystrophic process. It is unclear if one is the nidus for the disease since key molecules representing both are present in calcified valves, but individual researchers typically focus their studies on one process of calcification without consideration of the other. Third, there are few animal models of CAVD that have allowed for detailed mechanistic studies probing the etiology, with the notable exception of the Notch1+/− mouse.10, 11 This model has revealed many insights into the heritable form of CAVD, but has heretofore been limited and viewed as disconnected from the idiopathic version of the disease. However, with the newest findings uncovered by Hadjj et al., the past decade of work focused on the molecular mechanism of CAVD in Notch1+/− mice can suddenly be viewed in a much wider context as likely relevant to most or all forms of CAVD.
The pathology of CAVD in Notch1+/− mice is similar to that observed in humans: thickened aortic valve leaflets and varying sized regions of calcification that result in ~three-fold increase in tissue stiffness compared to wild-type mice at one year old.10, 11 Last year, the first VICs from immortalized Notch1+/− animals were successfully isolated and characterized as highly enriched in the inflammatory cell-cell junction protein cadherin-11 (CDH11, also known as OB-cadherin as it was first observed in osteoblasts).11 This enrichment in CDH11 may be a compensatory mechanism adopted by the VICs in order to restore some of the putative cell-cell communication that is lost due to the lack of NOTCH1; however, CDH11 is a robust cell-cell junction that is a primary driver of myofibroblast differentiation12 and a key contributor to inflammatory disease.13 CDH11 overexpression causes CAVD in mice14 and has been found to be upregulated in cases of human idiopathic CAVD (Figure).15 Additionally, Hadjj et al. showed that knockdown of H19 decreased CDH11 by ~30% in human VICs cultured in osteogenic medium (Supplemental Figure 1 in 3). Interestingly, CDH11 transcription was not increased by osteogenic medium, suggesting that H19 expression may directly affect CDH11 through repression of NOTCH1. H19 knockdown also decreased key regulators of osteogenesis BMP2, BGLAP, and RUNX2 (Figure 3 in 3); however, these are not ideal drug targets for CAVD due to their importance in bone homeostasis. Though only a correlate at this point, H19’s repression of NOTCH13 may lead to CDH11 overexpression in cases of CAVD,15 and this is noteworthy because CDH11 is a cell surface receptor on VICs that can likely be targeted as a novel therapy against CAVD (Figure).1
Figure.
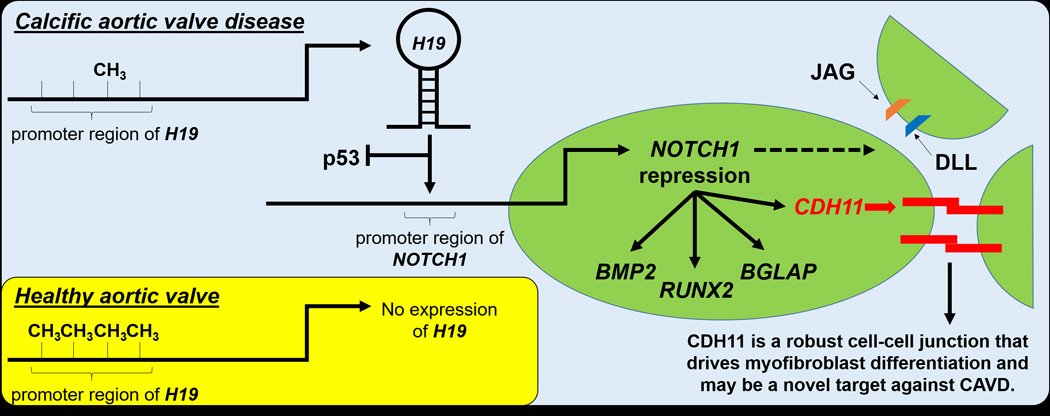
New evidence by Hadjj et al.3 demonstrates that idiopathic calcific aortic valve disease (CAVD) is associated with hypomethylation of the long non-coding (lnc) RNA H19, which prevents p53 from binding to the promoter region of NOTCH1, suppressing transcription. This complements previous data showing that repression of NOTCH1 leads to CAVD, likely through myofibroblast and osteogenic differentiation. CDH11 is particularly of interest as a potential therapeutic as it is an accessible cell surface protein that is enriched in both murine Notch1+/− VICs11 and human CAVD.15
There remain multiple questions to be answered: How is methylation of H19 regulated? Is it genetic or due to environmental factors? Are other genes besides NOTCH1 altered by H19 in CAVD? Regardless, the recent work by Hadjj et al. is the most exciting discovery for heart valve research since Garg et al. found that NOTCH1 haploinsufficiency resulted in heritable CAVD. Further, the renewed focus on NOTCH1, and the accompanying importance of H19, should both energize the heart valve research field and restore hope for clarifying an appropriate target for a pharmacological strategy against idiopathic CAVD.
Acknowledgments
Sources of Funding
W. David Merryman is supported by NIH (R01-HL115103 and R01-HL128715).
Footnotes
Disclosures
None.
References
- 1.Hutcheson JD, Aikawa E, Merryman WD. Potential drug targets for calcific aortic valve disease. Nature reviews Cardiology. 2014;11:218–231. doi: 10.1038/nrcardio.2014.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Garg V, Muth AN, Ransom JF, Schluterman MK, Barnes R, King IN, Grossfeld PD, Srivastava D. Mutations in NOTCH1 cause aortic valve disease. Nature. 2005;437:270–274. doi: 10.1038/nature03940. [DOI] [PubMed] [Google Scholar]
- 3.Hadjj F, Boulanger M-C, Guay S-P, Gaudreault N, Amellah S, Mkannez G, Bouchareb R, Marchand JT, Nsaibia MJ, Guauque-Olarte S, Pibarot P, Bouchard L, Bosse Y, Mathieu P. Altered DNA methylation of long non-coding RNA H19 in calcific aortic valve disease promotes mineralization by silencing NOTCH1. Circulation. 2016;134:xx–xx. doi: 10.1161/CIRCULATIONAHA.116.023116. [DOI] [PubMed] [Google Scholar]
- 4.Angrand PO, Vennin C, Le Bourhis X, Adriaenssens E. The role of long non-coding RNAs in genome formatting and expression. Front Genet. 2015;6:165. doi: 10.3389/fgene.2015.00165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feil R, Walter J, Allen ND, Reik W. Developmental control of allelic methylation in the imprinted mouse Igf2 and H19 genes. Development. 1994;120:2933–2943. doi: 10.1242/dev.120.10.2933. [DOI] [PubMed] [Google Scholar]
- 6.Kim DK, Zhang L, Dzau VJ, Pratt RE. H19, a developmentally regulated gene, is reexpressed in rat vascular smooth muscle cells after injury. J Clin Invest. 1994;93:355–360. doi: 10.1172/JCI116967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Han DK, Khaing ZZ, Pollock RA, Haudenschild CC, Liau G. H19, a marker of developmental transition, is reexpressed in human atherosclerotic plaques and is regulated by the insulin family of growth factors in cultured rabbit smooth muscle cells. J Clin Invest. 1996;97:1276–1285. doi: 10.1172/JCI118543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aikawa E, Whittaker P, Farber M, Mendelson K, Padera RF, Aikawa M, Schoen FJ. Human semilunar cardiac valve remodeling by activated cells from fetus to adult: implications for postnatal adaptation, pathology, and tissue engineering. Circulation. 2006;113:1344–1352. doi: 10.1161/CIRCULATIONAHA.105.591768. [DOI] [PubMed] [Google Scholar]
- 9.Greco S, Zaccagnini G, Perfetti A, Fuschi P, Valaperta R, Voellenkle C, Castelvecchio S, Gaetano C, Finato N, Beltrami AP, Menicanti L, Martelli F. Long noncoding RNA dysregulation in ischemic heart failure. J Transl Med. 2016;14:183. doi: 10.1186/s12967-016-0926-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nigam V, Srivastava D. Notch1 represses osteogenic pathways in aortic valve cells. J Mol Cell Cardiol. 2009;47:828–834. doi: 10.1016/j.yjmcc.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen J, Ryzhova LM, Sewell-Loftin MK, Brown CB, Huppert SS, Baldwin HS, Merryman WD. Notch1 Mutation Leads to Valvular Calcification Through Enhanced Myofibroblast Mechanotransduction. Arterioscler Thromb Vasc Biol. 2015;35:1597–1605. doi: 10.1161/ATVBAHA.114.305095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hinz B, Pittet P, Smith-Clerc J, Chaponnier C, Meister JJ. Myofibroblast development is characterized by specific cell-cell adherens junctions. Mol Biol Cell. 2004;15:4310–4320. doi: 10.1091/mbc.E04-05-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chang SK, Noss EH, Chen M, Gu Z, Townsend K, Grenha R, Leon L, Lee SY, Lee DM, Brenner MB. Cadherin-11 regulates fibroblast inflammation. Proc Natl Acad Sci U S A. 2011;108:8402–8407. doi: 10.1073/pnas.1019437108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sung DC, Bowen CJ, Vaidya KA, Zhou J, Chapurin N, Recknagel A, Zhou B, Chen J, Kotlikoff M, Butcher JT. Cadherin-11 Overexpression Induces Extracellular Matrix Remodeling and Calcification in Mature Aortic Valves. Arterioscler Thromb Vasc Biol. 2016;36:1627–1637. doi: 10.1161/ATVBAHA.116.307812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hutcheson JD, Chen J, Sewell-Loftin MK, Ryzhova LM, Fisher CI, Su YR, Merryman WD. Cadherin-11 regulates cell-cell tension necessary for calcific nodule formation by valvular myofibroblasts. Arterioscler Thromb Vasc Biol. 2013;33:114–120. doi: 10.1161/ATVBAHA.112.300278. [DOI] [PMC free article] [PubMed] [Google Scholar]