

Abstract
Plasmodium falciparum is the parasite responsible for the most severe form of malaria. Its increasing resistance to existing antimalarials represents a major threat to human health and urges the development of new therapeutic strategies to fight malaria. The proteasome is a protease complex essential in all eukaryotes. Accordingly, inhibition of the Plasmodium 20S proteasome is highly toxic for the parasite at all of its infective and developmental stages. Proteasome inhibitors have anti-malaria potential both as curative and transmission blocking agents, but in order to have therapeutic application they must specifically target the Plasmodium proteasome and not its human counterpart. X‐ray crystallography has been widely used to determine structures of yeast and mammalian 20S proteasomes with ligands. However, crystallisation of the Plasmodium proteasome is challenging, as only small quantities of the complex can be directly purified from the parasite. Furthermore, most X-ray structures of proteasome-inhibitor complexes require soaking of crystals with high concentrations of ligand, thus preventing analysis of inhibitor subunit specificity. Instead we chose to determine the Plasmodium falciparum 20S proteasome structure, in the presence of a new rationally designed parasite-specific inhibitor, by high resolution electron cryo‐microscopy and single particle analysis. The resulting map, at a resolution of about 3.6 Å, allows a direct molecular analysis of inhibitor/enzyme interactions. Here we present an overview of this structure, and how it provides valuable information that can be used to assist in the design of improved proteasome inhibitors with the potential to be developed as next-generation anti‐malaria drugs.
Keywords: Proteasome, electron microscopy, cryo-EM, single particle, structure, Plasmodium, malaria, inhibitor, drug design
Graphical abstract
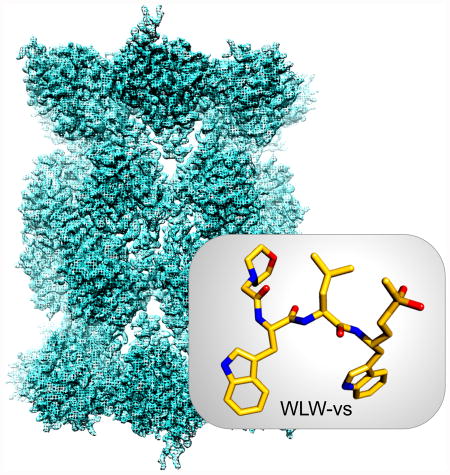
Cryo-EM structure of the Plasmodium falciparum 20S proteasome determined in the presence of WLW-vs (insert), an inhibitor that specifically binds to the parasite proteasome. The cryo‐EM structure reveals the molecular basis of such specificity, contributes to validate the parasite proteasome as a viable target against malaria and guides the development of proteasome inhibitors with potential as new generation antimalarials.
Introduction
Plasmodium falciparum is the parasite responsible for the most severe and deadly form of human malaria, a mosquito transmitted disease that affects millions of people every year particularly in tropical and sub-tropical climates. Metabolically stable analogues of the natural product artemisinin are currently the most effective agents for the treatment of malaria, and their use in combination therapies are recommended for the treatment of P. falciparum infections in areas where there is a widespread resistance to other existing drugs. However, the emergence and recent spread of artemisinin resistant parasites, first identified in Southeast Asia, represent a major threat to human health that jeopardises the current fight to control malaria, and urges the development of new high efficacy antimalarials [1].
The proteasome is a large protease complex essential in all eukaryotes. It comprises a 20S core formed by four hetero-heptameric rings of α and β subunits stacked into a dimeric α7β7β7α7 barrel shaped assembly. The active sites of the proteolytic active subunits, β1, β2 and β5, are located within its inner chamber (figure 1). The organisation of this assembly is well established, with the first structures of 20S proteasomes determined by X-ray crystallography nearly 20 years ago [2, 3]. Apart from its role in general proteostasis, the proteasome is responsible for the highly regulated degradation of proteins, the removal of which coordinates fundamental processes such as cell cycle progression [4]. While the 20S proteasome is a well-established target for cancer therapy [5], its inhibition is being explored as a therapeutic approach for a wider range of conditions including inflammatory disorders [6], viral infections [7] and tuberculosis [8].
Figure 1.

The cryo-EM structure of the Plasmodium falciparum 20S proteasome (EMDB‐3231, PDB 5FMG). (A) Overall view of the 20S proteasome along its two-fold axis, with the location of its α and β hetero-heptameric rings indicated. The back surface of the barrel shaped structure was clipped for clarity. (B) Location of the proteolytic sites within the proteasome inner chamber, where the protein is represented as ribbons and the proteolytic active Thr1 of β1 (green), β2 (magenta) and β5 (orange) are represented as spheres. The front of the structure was clipped for clarity. (C) The same representation as for (B), but viewed along the proteasome long axis, and clipped to show the proteasome inner cavity. The Thr1 of the proteolytic active subunits are colour coded as in (B), but for clarity only those from the β subunit ring proximal to the viewport are labelled.
The initial observation that inhibition of the Plasmodium proteasome is toxic for the parasite [9], and the subsequent demonstration that it has distinct ligand binding preferences to the human complex [10-12], suggest the parasite proteasome as a suitable target for the development of a new class of antimalarials. Furthermore, recent data revealed that inhibition of the Plasmodium proteasome has synergistic activity with front-line artemisinin-based drugs, and that the unfolded protein response (UPR), which is a proteasome mediated stress response pathway, is upregulated in Plasmodium field isolates from Southeast Asia that have a delayed response to those drugs [13]. Thus, inhibitors of the proteasome not only have the potential to kill parasites through disruption of cell cycle and other critical processes, but they may also help to prevent the induction of resistance to artemisinin-based and other antimalarial drugs that induce a UPR response.
When exploring proteasome inhibitors as antimalarials, the ability to obtain a high degree of ligand specificity for the Plasmodium proteasome over the human host complex is paramount to prevent toxicity of any lead molecules. Thus, the characterisation of the parasite 20S proteasome at a molecular level is important to rationalise differences between the parasite and human proteasome ligand binding preferences that can be exploited for drug development efforts [14]. We achieved this goal in a collaborative effort [15], which included determining the structure of the P. falciparum 20S proteasome bound to a new parasite-specific inhibitor by electron cryo‐microscopy (cryo‐EM) and single particle analysis. This structure, at a resolution of about 3.6 Å, when compared with its human counterpart, revealed the molecular basis for the specific ligand binding to the parasite complex. The new structure can further assist in the design and improvement of proteasome inhibitors with credible therapeutic potential against malaria, directly highlighting the impact of the fast evolving field of high resolution cryo-EM on the rational development of therapeutic drugs.
A rational strategy to define the specificity of the P. falciparum active sites
The identification of ligands suitable for drug development frequently relies on brute force high‐throughput screening of the target protein against large libraries of ligands, and the structural characterisation of the subsequent protein-ligand interactions for positive hits usually by X‐ray crystallography. This approach requires the direct determination of the protein structure in the presence of candidate ligands or, if the structure of the protein binding pocket is known, the computational modelling of protein-ligand interactions followed by validation by structure determination. A screening of non-covalent ligands previously showed that the P. falciparum and human proteasomes have distinct ligand binding preferences [11], but without knowledge of the molecular basis for such specificity, the prospects for drug development were limited. In a major step-forward to further characterise these specific ligand binding preferences, we determined cleavage site amino acid frequency profiles of P. falciparum and human proteasomes against a library of diverse peptide substrates [15], and found an unusual preference of the parasite complex for tryptophan residues at the substrate positions P1 and P3 (figure 2). Using this information we prepared peptide-vinyl sulfone prototype inhibitors (WLL-vs, WLW-vs and LLW-vs, figure 2), that act by covalently modifying the Thr1 residue of the proteasome catalytic subunits [16], and tested them in biochemical and cell biological assays to characterise their interactions to each of the individual P. falciparum and human proteasome active sites [15]. We identified WLW-vs as having the highest specificity towards the parasite complex, binding exclusively at its β2 active site. A high resolution structure of the P. falciparum 20S proteasome/inhibitor complex was then required to fully describe the molecular basis for this high degree of specificity.
Figure 2.

The WLW-vs proteasome inhibitor. (A) The WLW peptide was designed based on the proteasome’s optimal substrate binding specificity at positions P1-P3, which are counted upstream of the scissile bond. (B) Structural formula of WLW-vs, with identification of the positions P1-P3.
Determining the structure of the WLW-vs bound P. falciparum 20S proteasome
X-ray crystallography has been extensively used to study the interactions of yeast and mammalian 20S proteasomes with ligands in order to assist drug development [17, 18], but for these studies the protein sample must be amenable to crystallisation. While there are well established protocols for the preparation of highly pure yeast and mammalian 20S proteasomes in quantities and concentrations suitable for crystallography, the preparation of such samples directly from P. falciparum cultures is not practical due to the small yield of proteasomes obtained. On the other hand, the recent revolution in the field of structural cryo-EM has made it a suitable method to determine protein structures at resolutions that used to be achievable only by X-ray crystallography or NMR [19]. Archaeal proteasomes have been used as a test sample for cryo-EM imaging conditions and image processing methods, resulting in structures at resolutions as high as 2.8 Å [20, 21]. However, these are not suitable models to infer detailed ligand selectivity properties of the active sites of eukaryotic proteasomes, which are functionally and structurally more complex.
We recently used human 20S proteasome samples to demonstrate that cryo‐EM and single particle analysis can be used to determine the structure of eukaryotic proteasomes at resolutions that show the conformation of side-chains of peptide derived ligands bound to the active sites [22]. This analysis also highlighted the significant advantages of using cryo-EM for the study of proteasome-ligand interactions, including using conditions that more closely resemble a physiological environment and preserve the subunit ligand binding specificity patterns. In contrast, analysis by X‐ray crystallography normally requires either protein co-crystallisation, or the soaking of protein crystals with excess of ligand. In either case the protein environment is optimised to facilitate crystallisation, and the exposure to excess ligand under such conditions, together with possible active site access modulation due to crystallographic constraints, can greatly alter the physiological binding selectivity of a molecule. Most importantly, the significantly smaller amounts of purified protein required for cryo-EM methods makes existing preparations of P. falciparum 20S proteasomes adequate for high resolution structure analysis. Using the same strategy as for the cryo-EM analysis of the human proteasome [22], we determined the cryo-EM structure of the Plasmodium falciparum 20S proteasome bound to WLW-vs at a resolution of ∼3.6 Å (Electron Microscopy Data Bank entry EMDB-3231, Protein Data Bank accession number 5FMG, figure 1) [15]. Densities for the ligand were clearly recovered extending from the proteolytic active Thr1 residue of only the β2 subunit (figure 3), thus validating the observed occupancy from the in vitro inhibition studies.
Figure 3.

The conformation of the ligand WLW-vs (represented as grey sticks) covalently bound to the Thr1 (orange sticks) of the Plasmodium proteasome β2 active site. The protein backbone is represented as orange ribbon.
Structural basis for the P. falciparum 20S proteasome ligand binding specificity
Our structure of the P. falciparum 20S proteasome shows that the WLW-vs bound at the β2 subunit active site takes a nearly planar beta secondary structure conformation, analogous to that of other peptide derived inhibitors bound to yeast or mammalian proteasomes [14]. The ligand morpholine, leucine side chain and vinyl sulfone groups are oriented towards the solvent filled inner cavity of the complex, while the tryptophan side chains at positions P1 and P3 face the protein surface (figure 3). Accordingly, our structure shows that the selective binding of WLW-vs arises from the uniquely open Plasmodium β2 ligand binding pocket, which can accommodate tryptophan side chains at positions P1 and P3 while the human complex cannot (figure 4). Furthermore, our structure also shows that this is the only binding site, amongst all of those from the human and parasite proteasomes, compatible with such bulky side chain at the P1 position, providing the basis for the WLW-vs specificity [15].
Figure 4.

Comparison of the ligand accessibility of the Plasmodium falciparum and human proteasome β2 active sites. (A) The tryptophan at the P1 position of the WLW-vs bound to the Plasmodium falciparum β2 active site. (B) The P1 position of the WLW-vs ligand, as in (A), superimposed onto the β2 active site of the cryo-EM structure of the human proteasome (PDB 5A0Q). (C-D) As in (A) and (B), respectively, showing the P3 position of WLW-vs. While the P1 and P3 side chains are well accommodated in the parasite β2 binding pocket (A,C), steric constraints limit accessibility to the human β2 binding pocket (B,D), which is occupied only at higher ligand concentrations where specificity is lost. Equivalent restrictions are observed for the β1 and β5 binding pockets of the parasite and human proteasomes [15].
While WLW-vs is the most specific of the new prototype ligands tested, the exclusive inhibition of the parasite proteasome β2 active site is not sufficiently toxic to kill the parasite [12]. Nevertheless, in vivo assays showed that WLW‐vs successfully synergises with dihydroartemisinin to kill artemisinin-resistant parasites, suggesting a potential therapeutic usage of WLW‐vs derivatives in combination therapy with artemisinin-based compounds. On the other hand, our structure indicates that a tryptophan at the P3 position can be accommodated by both the parasite β2 and β5 sites. The simultaneous inhibition of these two sites results in significantly enhanced toxicity for the parasite, as we demonstrated in vitro and in vivo (in P. falciparum cultures) using the ligand WLL‐vs [15]. Remarkably a single dose of WLL‐vs was sufficient to reduce the parasite to close to undetectable levels in a malaria mouse model, without apparent toxicity to the host. WLL-vs is therefore a suitable foundation for the development of new, highly potent and specific Plasmodium falciparum proteasome inhibitors as antimalarials.
Final remarks
While the proteasome is conserved amongst all eukaryotes, we showed that sufficient structural differences exist in complexes from different species to allow their specific targeting. While our study focused on the Plasmodium falciparum proteasome as an antimalarial target, it strongly suggests that a similar strategy may be applicable against other disease causing protozoan parasites, including Trypanosoma, Leishmania, Toxoplasma and Entamoeba. In all these cases, high resolution cryo-EM is likely to be the method of choice to obtain the structural information required to guide drug design. Finally, cryo-EM does not yet provide the high-throughput structural data attainable by X-ray crystallography, which when using favourable proteins allows a fast, direct screening of their binding with candidate ligands. However, our results emphasise that structures determined by cryo-EM can provide information highly suitable for drug development, with the significant advantage of preserving ligand specificity. Furthermore, ongoing developments in cryo-EM instrumentation and data analysis are expected to yield consistently higher resolution structures, at a higher throughput. Such prospects, together with the already applicable advantages of using cryo-EM in the structural analysis of protein-ligand interactions, as we have shown, makes it foreseeable that in the near future cryo-EM may become the primary method of choice for such studies.
Acknowledgments
This work was support by Medical Research Council grant MC_UP_1201/5 to P.C.A.dF., the National Institutes of Health grants R01AI078947 and R01EB05011 to M.B. and a NSS‐PhD scholarship from the Agency for Science, Technology and Research (A*STAR) Singapore to H.L..
Abbreviations
- Cryo-EM
electron cryo-microscopy
- P. falciparum
Plasmodium falciparum
Footnotes
Conflicts of Interest. The authors declare no conflicts of interest
Author contributions: All authors contributed for the writing of this review.
References
- 1.Ashley EA, Dhorda M, Fairhurst RM, Amaratunga C, Lim P, Suon S, Sreng S, Anderson JM, Mao S, Sam B, Sopha C, Chuor CM, Nguon C, Sovannaroth S, Pukrittayakamee S, Jittamala P, Chotivanich K, Chutasmit K, Suchatsoonthorn C, Runcharoen R, Hien TT, Thuy-Nhien NT, Thanh NV, Phu NH, Htut Y, Han KT, Aye KH, Mokuolu OA, Olaosebikan RR, Folaranmi OO, Mayxay M, Khanthavong M, Hongvanthong B, Newton PN, Onyamboko MA, Fanello CI, Tshefu AK, Mishra N, Valecha N, Phyo AP, Nosten F, Yi P, Tripura R, Borrmann S, Bashraheil M, Peshu J, Faiz MA, Ghose A, Hossain MA, Samad R, Rahman MR, Hasan MM, Islam A, Miotto O, Amato R, MacInnis B, Stalker J, Kwiatkowski DP, Bozdech Z, Jeeyapant A, Cheah PY, Sakulthaew T, Chalk J, Intharabut B, Silamut K, Lee SJ, Vihokhern B, Kunasol C, Imwong M, Tarning J, Taylor WJ, Yeung S, Woodrow CJ, Flegg JA, Das D, Smith J, Venkatesan M, Plowe CV, Stepniewska K, Guerin PJ, Dondorp AM, Day NP, White NJ. Spread of artemisinin resistance in Plasmodium falciparum malaria. The New England journal of medicine. 2014;371:411–23. doi: 10.1056/NEJMoa1314981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lowe J, Stock D, Jap B, Zwickl P, Baumeister W, Huber R. Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 A resolution. Science (New York, NY) 1995;268:533–9. doi: 10.1126/science.7725097. [DOI] [PubMed] [Google Scholar]
- 3.Groll M, Ditzel L, Lowe J, Stock D, Bochtler M, Bartunik HD, Huber R. Structure of 20S proteasome from yeast at 2.4A resolution. Nature. 1997;386:463–471. doi: 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]
- 4.Ciechanover A. Intracellular protein degradation: from a vague idea through the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Bioorganic & medicinal chemistry. 2013;21:3400–10. doi: 10.1016/j.bmc.2013.01.056. [DOI] [PubMed] [Google Scholar]
- 5.Kisselev AF, van der Linden WA, Overkleeft HS. Proteasome inhibitors: an expanding army attacking a unique target. Chemistry & biology. 2012;19:99–115. doi: 10.1016/j.chembiol.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huber EM, Groll M. Inhibitors for the immuno- and constitutive proteasome: current and future trends in drug development. Angewandte Chemie (International ed in English) 2012;51:8708–20. doi: 10.1002/anie.201201616. [DOI] [PubMed] [Google Scholar]
- 7.Luo H. Interplay between the virus and the ubiquitin-proteasome system: molecular mechanism of viral pathogenesis. Current opinion in virology. 2015;17:1–10. doi: 10.1016/j.coviro.2015.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lin G, Li D, de Carvalho LP, Deng H, Tao H, Vogt G, Wu K, Schneider J, Chidawanyika T, Warren JD, Li H, Nathan C. Inhibitors selective for mycobacterial versus human proteasomes. Nature. 2009;461:621–6. doi: 10.1038/nature08357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gantt SM, Myung JM, Briones MR, Li WD, Corey EJ, Omura S, Nussenzweig V, Sinnis P. Proteasome inhibitors block development of Plasmodium spp. Antimicrobial agents and chemotherapy. 1998;42:2731–8. doi: 10.1128/aac.42.10.2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li H, Ponder EL, Verdoes M, Asbjornsdottir KH, Deu E, Edgington LE, Lee JT, Kirk CJ, Demo SD, Williamson KC, Bogyo M. Validation of the proteasome as a therapeutic target in Plasmodium using an epoxyketone inhibitor with parasite-specific toxicity. Chemistry & biology. 2012;19:1535–45. doi: 10.1016/j.chembiol.2012.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li H, Tsu C, Blackburn C, Li G, Hales P, Dick L, Bogyo M. Identification of potent and selective non-covalent inhibitors of the Plasmodium falciparum proteasome. Journal of the American Chemical Society. 2014;136:13562–5. doi: 10.1021/ja507692y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li H, van der Linden WA, Verdoes M, Florea BI, McAllister FE, Govindaswamy K, Elias JE, Bhanot P, Overkleeft HS, Bogyo M. Assessing subunit dependency of the Plasmodium proteasome using small molecule inhibitors and active site probes. ACS chemical biology. 2014;9:1869–76. doi: 10.1021/cb5001263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dogovski C, Xie SC, Burgio G, Bridgford J, Mok S, McCaw JM, Chotivanich K, Kenny S, Gnädig N, Straimer J, Bozdech Z, Fidock DA, Simpson JA, Dondorp AM, Foote S, Klonis N, Tilley L. Targeting the Cell Stress Response of Plasmodium falciparum to Overcome Artemisinin Resistance. PLoS Biol. 2015;13:e1002132. doi: 10.1371/journal.pbio.1002132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Le Chapelain C, Groll M. Rational Design of Proteasome Inhibitors as Antimalarial Drugs. Angewandte Chemie (International ed in English) 2016;55:6370–2. doi: 10.1002/anie.201602519. [DOI] [PubMed] [Google Scholar]
- 15.Li H, O'Donoghue AJ, van der Linden WA, Xie SC, Yoo E, Foe IT, Tilley L, Craik CS, da Fonseca PC, Bogyo M. Structure- and function-based design of Plasmodium-selective proteasome inhibitors. Nature. 2016;530:233–6. doi: 10.1038/nature16936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bogyo M, McMaster JS, Gaczynska M, Tortorella D, Goldberg AL, Ploegh H. Covalent modification of the active site threonine of proteasomal beta subunits and the Escherichia coli homolog HslV by a new class of inhibitors. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:6629–34. doi: 10.1073/pnas.94.13.6629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Borissenko L, Groll M. 20S proteasome and its inhibitors: crystallographic knowledge for drug development. Chemical reviews. 2007;107:687–717. doi: 10.1021/cr0502504. [DOI] [PubMed] [Google Scholar]
- 18.Beck P, Dubiella C, Groll M. Covalent and non-covalent reversible proteasome inhibition. Biological chemistry. 2012;393:1101–20. doi: 10.1515/hsz-2012-0212. [DOI] [PubMed] [Google Scholar]
- 19.Kuhlbrandt W. The resolution revolution. Science (New York, NY) 2014;343:1443–4. doi: 10.1126/science.1251652. [DOI] [PubMed] [Google Scholar]
- 20.Campbell MG, Veesler D, Cheng A, Potter CS, Carragher B. 2.8 A resolution reconstruction of the Thermoplasma acidophilum 20S proteasome using cryo-electron microscopy. eLife. 2015;4:e06380. doi: 10.7554/eLife.06380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grant T, Grigorieff N. Measuring the optimal exposure for single particle cryo-EM using a 2.6 A reconstruction of rotavirus VP6. eLife. 2015;4:e06980. doi: 10.7554/eLife.06980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.da Fonseca PC, Morris EP. Cryo-EM reveals the conformation of a substrate analogue in the human 20S proteasome core. Nature communications. 2015;6:7573. doi: 10.1038/ncomms8573. [DOI] [PMC free article] [PubMed] [Google Scholar]