

Abstract
A 36-year-old woman developed hypokalemic metabolic alkalosis after anti SS-A antibody was found to be positive. Diuretic loading test results were compatible with Gitelman syndrome (GS). The patient had a heterozygous mutation in SLC12A3, which encodes for thiazide-sensitive NaCl cotransporter (NCCT). While the mutation may be responsible for a latent hypofunction of NCCTs, the underlying anti-SSA antibody-associated autoimmunity induced the manifestation of its hypofunction. To the best of our knowledge, this is the first report to demonstrate that anti SS-A antibody-associated autoimmunity may induce GS in a patient with a SLC12A3 heterozygous mutation.
Keywords: Sjögren's syndrome, Gitelman syndrome, SLC12A3 mutation
Introduction
Gitelman syndrome (GS) is an autosomal recessive disease attributed to a mutation in the SLC12A3 gene, which encodes for thiazide-sensitive NaCl cotransporter (NCCT) in the distal renal tubules. The clinical findings of GS are characterized by hypokalemic alkalosis, hypomagnesaemia, hypocalciuria, lower blood pressure, which are attributed to impairment of NCCTs. GS develops with a homozygous mutation or compound heterozygosity in SLC12A3. However, 40% of patients with clinically diagnosed GS have no mutation or a heterozygous mutation in SLC12A3 (1). Fava et al. reported that those with a heterozygous mutation in SLC12A3 had significantly lower blood pressure than those without mutations and suggested that the NCCT function is partially impaired in those with a heterozygous mutation (2). Acquired GS is rare and only six cases have been reported (3-8), four of which were associated with Sjögren's syndrome (SS) (3-6). One case was associated with chronic sialadenitis (7). Therefore, SS may contribute to the development of GS.
SS is a systemic autoimmune disease characterized by lacrimal and salivary gland hyposecretion. It is often accompanied by increased circulating polyclonal immunoglobulins and autoantibodies, and extraglandular complications such as interstitial nephritis, interstitial pneumonia and arthritis are relatively common. The case presented herein had a heterozygous mutation in SLC12A3 and developed GS after anti-SSA antibody was found to be positive. We suspected that our case had an underlying autoimmune response associated with SS, because she also had bilateral parotid swelling, xerostomia with the findings of low grade periductal lymphoid cell infiltration in the salivary gland biopsy and tubulointerstitial inflammation in the renal biopsy.
Case Report
A 36-year-old woman was admitted for muscle cramps, fatigue and a loss of appetite. Four years prior to this admission, subclinical hypokalemia (3.1 mEq/L) and hypochloremia (96 mEq/L) were noted. Serum electrolytes could not be maintained at normal levels without taking potassium supplements. Six months prior to this admission, the patient stopped taking potassium supplements, resulting in muscle cramps in her extremities. One week prior to admission, she developed fatigue and a loss of appetite.
The patient had been healthy except for elevated liver enzyme levels attributed to her excessive alcohol intake. She did not have osteomalacia. Five years prior to the admission, bilateral parotid swelling developed. Testing for SS-A antibody was positive. Schirmer's test, the Saxon text, and chewing gum test were within normal ranges. A salivary gland biopsy revealed two focuses of periductal lymphoid cell infiltration in 24 mm2. However, this finding did not fulfill the diagnostic criterion requiring one or more focuses of periductal lymphoid cell infiltration in 4 mm2. Dry mouth and dry eye subsequently developed. Although the diagnostic criteria of SS were not completely fulfilled, we presumed that she had an underlying autoimmune response associated with SS. The serum IgG4 level was within the normal range (19.2 mg/dL). No organ enlargement except for parotid glands was revealed with a computed tomography (CT) scan. Taken together, we concluded that IgG4-related disease was unlikely, although immunostaining for anti-IgG4 antibodies was not conducted in the salivary glands.
At the time of the development of bilateral parotid swelling, the blood pressure was 106/61 mmHg, and serum levels of potassium (4.9 mEq/L), chloride (109 mEq/L), magnesium (2.3 mEq/L) and creatinine (0.61 mg/dL) were within normal ranges. Regular medical checkups prior to the SS-like autoimmunity development did not reveal any electrolyte abnormalities. It has been well known that interstitial nephritis complicates SS. Although the extent of proteinuria was mild (0.3 g/day), a renal biopsy was performed to clarify the renal involvement. A renal biopsy revealed a small amount of monocyte infiltration around atrophic renal tubules in a localized area (Fig. 1). Concurrently, myelodysplastic syndrome (MDS) was diagnosed because of the increased number of ringed sideroblasts observed by bone marrow aspiration. However, no hematological abnormality was observed in the peripheral blood cells.
Figure 1.
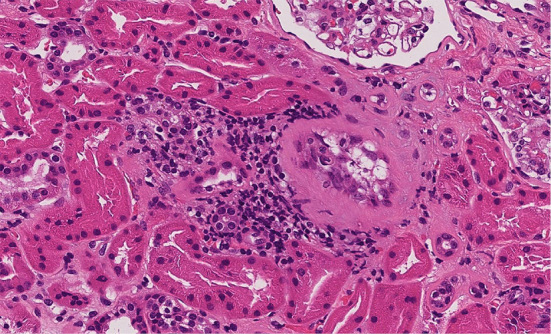
Fifty glomeruli were obtained. One of the glomeruli was sclerosed. Around the sclerosed glomerulus, renal tubular atrophy and interstitial fibrosis were noted. There was focal inflammatory cell infiltration.There was no sign of glomerulonephritis.
On admission, she was normotensive (95/64 mmHg). A physical examination revealed muscle weakness, dry eyes and mouth and bilateral parotid swelling, and most of her teeth had caries. A laboratory examination revealed hypokalemia (2.7 mEq/L), hypochloremia (70 mEq/L), and elevated creatinine (2.46 mg/dL). The plasma renin activity (PRA; 2.6 ng/mL/h), plasma aldosterone concentration (PAC; 112 pg/mL) and PAC/PRA ratio (43) were all within normal ranges. An arterial gas analysis revealed metabolic alkalosis (pH, 7.549; PaCO2, 66.7 mmHg; HCO3-, 56.9 mmol/L). A urine analysis revealed that the urinary pH was 8.5. Both red and white cells were not found in the urine. Urinary protein was 3+, and the protein/creatinine ratio was 0.30 g/gCr. Urinary glucose was negative. N-acetyl-beta-D-glucosaminidase (NAG) was 12.1 IU/g・Cr, and β2-microglobulin was 48,254 μg/g・Cr. A high urinary chloride concentration (27.2 mEq/g・Cr) and low calcium concentration (0.2 mEq/day) were also detected. An electrocardiogram revealed a prolonged QT interval. A chest X-ray revealed no abnormal findings.
Despite hypochloremia, urinary chloride excretion was increased. The transtubular potassium gradient was increased to 4.53, indicating that hypokalemia was due to renal potassium wasting. Primary aldosteronism was unlikely because of the normal PAC/PRA ratio. She had no history of recurrent vomiting, diarrhea, or taking diuretics. After potassium and saline infusion, the clinical symptoms disappeared. The serum levels of creatinine, potassium and urinary levels of NAG and the QT interval were all normalized. Urinary β2-microglobulin decreased to an almost normal level.
GS was suspected because of the findings of relatively low blood pressure, metabolic alkalosis, hypochloremia, and low urinary calcium excretion, although the renin-angiotensin-aldosterone system was normal. Diuretic loading test was conducted to evaluate the activity of NCCTs and furosemide-sensitive Na-K-Cl cotransporters (NKCCs) (Table 1). Furosemide infusion significantly increased the fractional excretion (FE) of sodium (FENa) and chloride (FECl). On the other hand, thiazide infusion increased FENa and FECl to a lesser extent. This indicated a hypofuction of NCCTs and hyperfunction of NKCCs as compensation for the NCCT hypofunction, which was compatible with GS.
Table 1.
Diuretic Loading Test.
Thiazide loading test
| ΔFENa mean±SD | ΔFECl mean±SD | |
|---|---|---|
| GS | 1.16±0.73 | 1.76±1.01 |
| control | 2.52±1.14 | 3.64±1.30 |
| our case | 1.70 | 2.65 |
Furosemide loading test
| ΔFENa mean±SD | ΔFECl mean±SD | |
|---|---|---|
| GS | 22.7±4.6 | 31.6±6.4 |
| control | 17.8±7.3 | 24.5±8.9 |
| our case | 29.3 | 37.7 |
Our protocol followed the protocol employed by Colussi et al (11). They conducted the diuretic loading test to clinically diagnosed patients with GS and healthy controls. The differences between Colussi’s protocol and ours were as follows. In Colussi’s protocol, potassium supplement is discontinued. The furosemide and thiazide loading tests are performed at least 7 days apart. In our case, potassium supplement was continued. The furosemide and thiazide loading tests were performed on consecutive days. The results of the report by Colussi et al. and our case are shown in the Table. ΔFE is the difference between the highest FE after furosemide or thiazide infusion and the mean of the two FE before infusion.
After discharge, the patient continued taking potassium supplements. The serum potassium level normalized, whereas metabolic alkalosis, hypochloremia, and increased urinary chloride excretion persisted. As GS was suspected, a genetic analysis of all exon regions of SLC12A3 using the Sanger sequencing method was conducted. A heterozygous mutation in c.C3052T (p.R1018X) (Fig. 2) was detected, which changes the 1,018th arginine to a stop codon and truncates the C terminal of NCCTs. However, no additional mutation was detected in the exon regions.
Figure 2.
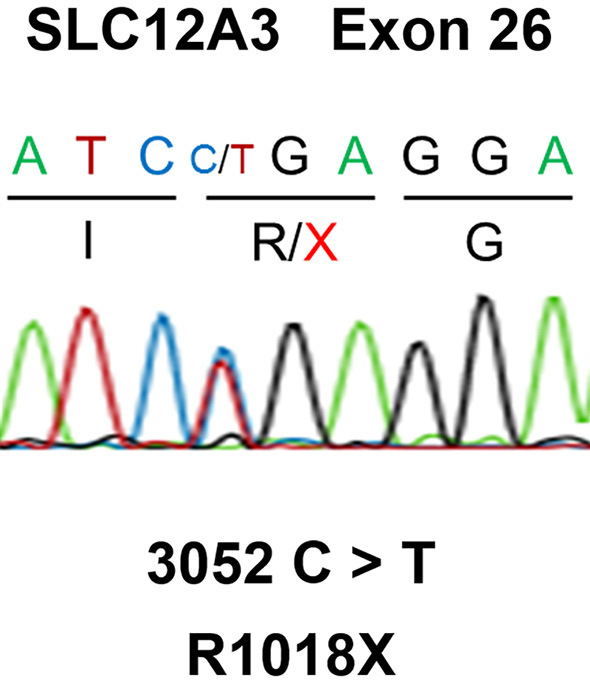
Sanger sequencing of all exon regions of SLC12A3 revealed a heterozygous mutation in c.C3052T, which changes the codon of the 1,018th arginine to a stop codon. No additional mutation was found.
Discussion
We found six cases of acquired GS in the previous literature. Among them, two cases (4,5) underwent a genetic analysis but no mutations in SLC12A3 were found. The heterozygous mutation in SLC12A3 could be responsible for the latent hypofunction of NCCTs. Acid-base imbalance or electrolyte abnormalities were absent before the present patient was found to have an underlying autoimmune response associated with SS. Therefore, we presumed that anti-SSA antibody-associated autoimmunity may have exacerbated the hypofunction of NCCTs, resulting in the development of GS.
The mutation in our case, at the 1,018th arginine in SLC12A3, resulted in the elimination of the C terminal in the NCCT. Berry et al. reported that a patient with a compound heterozygous mutation at the same location developed GS (9), indicating that the truncated NCCTs derived from the mutant allele have an impaired function. No additional mutation was detected in the exon regions. However, the possibility of SLC12A3 compound heterozygosity was not excluded thoroughly in our case, because GS could have developed as a result of mutations in the intronic regions (10).
Several reports have suggested a latent hypofunction of NCCTs induced by heterozygous mutations and the existence of compensation mechanisms (11,12). Colussi et al. reported that furosemide induced a higher FENa and FECl in patients with GS than in controls, indicating that the compensative increase of NaCl reabsorption might have occurred in NKCCs (11). Cruz et al. suggested that patients with heterozygous mutations have a higher salt intake and higher urinary sodium excretion than controls (12). In the present case, an increased response to the furosemide administration was observed. This might reflect the compensative hyperfunction of NKCC. Hypokalemia was not noted before SS was suspected, and no change in dietary habit was observed before the development of GS. In addition to the incomplete function due to the heterozygous mutation in SLC12A3, anti-SSA antibody-associated autoimmunity may have abrogated the compensatory mechanism, resulting in the development of GS.
Among the six cases of acquired GS, four of them were associated with SS (3-6), and one of them was associated with chronic sialadenitis (7) (Table 2). The other case was a kidney recipient from a donor with GS (8). SS is a single comorbidity reported to be associated with acquired GS, suggesting that anti-SSA antibody-associated autoimmunity may have played a role in the development of GS.
Table 2.
Acquired GS Cases.
| reference | our case | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|
| age | 36 | 46 | 38 | 32 | 62 | 39 |
| sex | F | F | F | F | F | F |
| underlying disease | anti SS-A antibody associated autoimmunity | SS | SS | SS, Systemic Sclerosis, Hashimoto disease | SS | chronic sialadenitis |
| analysis of SLC12A3 | heterozygous mutation | not performed | no mutations | no mutations | not performed | not performed |
| renal biopsy | slight tubulointerstitium inflammation | interstitial nephritis | interstitial nephritis | slight interstitial abnormality | not performed | not performed |
| response to thiazide response to furosemide | blunted response increased response | not performed | blunted response increased response | not performed | not performed | not performed |
| potassium | ||||||
| prednisolone, | and magnesium | |||||
| potassium and | potassium and | supplementation, | ||||
| magnesium | magnesium | levothyroxine, eplerenone, | prednisolone, | prednisolone, | ||
| potassium | supplementation, | supplementation, | hydroxychloroquine, | potassium | potassium | |
| treatment | supplementation | spironolactone | spironolactone | methotrexate | supplementation | supplementation, |
The link between the autoimmune response and NCCT hypofunction remains unclear. Kim et al. suggested the presence of anti-NCCT antibody in the patient's serum. They reported a patient with interstitial nephritis associated with SS who later developed GS (3). The patient underwent immunosuppressive therapy, and electrolyte abnormality and proteinuria disappeared. An immunohistochemical study of the distal convoluted tubules demonstrated the absence of the NCCT expression, indicating that an autoantibody against NCCT might have existed in the serum of the patient.
Immunosuppressive therapy may have normalized the electrolyte abnormality in a similar manner to the case reported by Kim et al. (3). However, this therapy was not conducted in the present study because there were no findings of active tubulointerstitial nephritis, and our case was managed with potassium supplements alone.
We searched for renal complications of MDS and found only several cases of glomerulonephritis with MDS. Moreover, there have been no reports of renal tubular diseases associated with MDS. Therefore, we concluded that the development of GS was not associated with MDS.
This is the first report to demonstrate that anti-SSA antibody-associated autoimmunity may contribute to the development of GS in a patient with a SLC12A3 heterozygous mutation.
The authors state that they have no Conflict of Interest (COI).
Acknowledgement
We thank Dr. Kiyotaka Nagahama for offering the kidney biopsy results and for providing valuable advice.
References
- 1. Nakhoul F, Nakhoul N, Dorman E, Berger L, Skorecki K, Magen D. Gitelman's syndrome: a pathophysiological and clinical update. Endocrine 41: 53-57, 2012. [DOI] [PubMed] [Google Scholar]
- 2. Fava C, Montagnana M, Rosberg L, et al. . Subjects heterozygous for genetic loss of function of the thiazide-sensitive cotransporter have reduced blood pressure. Hum Mol Genet 17: 413-418, 2008. [DOI] [PubMed] [Google Scholar]
- 3. Kim YK, Song HC, Kim WY, et al. . Acquired Gitelman syndrome in a patient with primary Sjögren syndrome. Am J Kidney Dis 52: 1163-1167, 2008. [DOI] [PubMed] [Google Scholar]
- 4. Chen YC, Yang WC, Yang AH, Lin SH, Li HY, Lin CC. Primary Sjögren's syndrome associated with Gitelman's syndrome presenting with muscular paralysis. Am J Kidney Dis 42: 586-590, 2003. [DOI] [PubMed] [Google Scholar]
- 5. Hinschberger O, Martzolff L, Ioannou G, Baumann D, Jaeger F, Kieffer P. Acquired Gitelman syndrome associated with Sjögren's syndrome and scleroderma. Rev Med interne 32: e96-e98, 2011(in French, Abstract in English). [DOI] [PubMed] [Google Scholar]
- 6. Schwarz C, Barisani T, Bauer E, Druml W. A woman with red eyes and hypokalemia: a case of acquired Gitelman syndrome. Wien Klin Wochenschr 118: 239-242, 2006. [DOI] [PubMed] [Google Scholar]
- 7. Casatta L, Ferraccioli GF, Bartoli E. Hypokalaemic alkalosis, acquired Gitelman's and Bartter's syndrome in chronic sialoadenitis. Br J Rheumatol 36: 1125-1128, 1997. [DOI] [PubMed] [Google Scholar]
- 8. Bansal R, Ranga VK. Acquired Gitelman's syndrome: an oxymoron? Int Urol Nephrol 43: 233-236, 2011. [DOI] [PubMed] [Google Scholar]
- 9. Berry MR, Robinson C, Karet Frankl FE. Unexpected clinical sequelae of Gitelman syndrome: hypertension in adulthood is common and females have higher potassium requirements. Nephrol Dial Transplant 28: 1533-1542, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lo YF, Nozu K, Iijima K, et al. . Recurrent deep intronic mutations in the SLC12A3 gene responsible for Gitelman's syndrome. Clin J Am Soc Nephrol 6: 630-639, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Colussi G, Bettinelli A, Tedeschi S, et al. . A thiazide test for the diagnosis of renal tubular hypokalemic disorders. Clin J Am Soc Nephrol 2: 454-460, 2007. [DOI] [PubMed] [Google Scholar]
- 12. Cruz DN, Simon DB, Nelson-Williams C, et al. . Mutations in the Na-Cl cotransporter reduce blood pressure in humans. Hypertension 37: 1458-1464, 2001. [DOI] [PubMed] [Google Scholar]