

Abstract
The diazo group has attributes that complement those of the azido group for applications in chemical biology. Here, we use computational analyses to provide insights into the chemoselectivity of the diazo group in 1,3-dipolar cycloadditions. Dipole distortion energies are responsible for ~80% of the overall energetic barrier for these reactions. Here, we show that diazo compounds, unlike azides, provide an opportunity to decrease that barrier substantially without introducing strain into the dipolarophile. The ensuing rate-enhancement is due to the greater nucleophilic character of a diazo group compared to that of an azido group, which can accommodate decreased distortion energies without pre-distortion. The tuning of distortion energies with substituents in a diazo compound or dipolarophile can enhance reactivity and selectivity in a predictable manner. Notably, these advantages of diazo groups are amplified in water. Our findings provide a theoretical framework that can guide the design and application of both diazo compounds and azides in “orthogonal” contexts, especially for biological investigations.
Graphical Abstract
INTRODUCTION
“If one regards reactions as new only if they have no forerunners, not even singular examples buried in the literature, then 1,3-dipolar additions cannot claim novelty. But if one defines reactions as novel which are for the first time recognized for their generality, scope, and mechanism, the judgment must be different.”
—Rolf Huisgen (1961).1
More than a half-century has passed since this encouraging manifesto. During that time, 1,3-dipolar cycloadditions have elicited extraordinary ingenuity. These reactions are key steps in countless synthetic routes.2 Moreover, 1,3-dipolar cycloadditions have spawned chemoselective reactions for biological contexts, transforming the field of chemical biology.3 Though azido groups have been the most prevalent reactant, their diazo congeners can likewise participate in 1,3-dipolar cycloadditions, and both dipoles survive cellular metabolism.4
The electronic tuning of unstrained dipolarophiles allows for the selective reactivity of diazoacetamides over alkyl azides (Scheme 1A).5 For example, unlike azides, diazoacetamides exhibit site-selective reactivity with a natural amino-acid residue, dehydroalanine (Dha), at rates comparable to those of early strain-promoted azide–alkyne cycloadditions (SPAAC).6 The ability of diazoacetamides to react with electronically activated alkenes facilitates conjugation to the many natural products that contain this moiety (Scheme 1B).
Scheme 1.

(A) Selective reactivity of stabilized diazoacetamides with the activated alkene of dehydroalanine (Dha), a natural amino-acid residue. (B) Exemplary natural products containing activated alkenes.
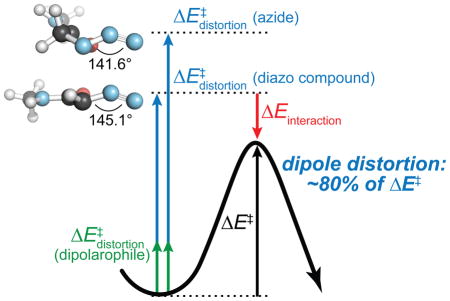
The vast majority of efforts to optimize the kinetics of 1,3-dipolar cycloaddition reactions for biological contexts have focused on dipolarophiles, especially alkynes, that employ either strain6,7 or strain along with electronic effects.8 Far less emphasis has been placed on the dipoles.9,10 This trend contravenes theoretical studies, which suggest that the bulk of the activation barrier (~80%) comes from the energy required to distort the dipole to the geometry of the transition state (TS).11 Owing to the inherent conjugation to its pendant carbon, the diazo group is well-suited for optimization. Such optimization might allow for dual-labeling strategies,4,9b,12 which require orthogonal reactions that remain chemoselective in the presence of biological moieties.3 The small size, high stability, and cycloaddition-reactivity of azido and diazo groups make them attractive chemical reporters for such applications.
Our recent report of diazo-selective reactions with unstrained alkenes in the presence of azides5—a finding largely orthogonal to current optimization strategies—has inspired us to examine the importance of strain and electronic modulation on the rates of 1,3-dipolar cycloadditions of azides and diazo compounds. By using computational methods, including the distortion/interaction analysis, which is also known as the activation strain model,11,13 and Marcus theory,14 we reveal the special importance of electronic effects on the cycloadditions of diazo compounds. We find that the increased nucleophilicity of diazo dipoles leads to unidirectional charge transfer, which is an optimal scenario for stabilizing transition states (TSs) in polar solvents, including water. We conclude that the importance of substituent effects on the cycloadditions of diazo compounds provides a unique opportunity to control reactivity and enable diazo-selective cycloadditions in the presence of azides.
RESULTS AND DISCUSSION
1,3-Cycloadditions of Diazo Compounds and Azides
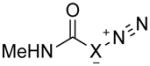
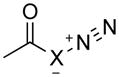
To understand the reactivity of significantly stabilized diazo compounds in contrast to analogous azides while concentrating on the role of substituent effects versus strain-activation, we examined a range of 1,3-cycloadditions between various dipoles. We started with simple dipoles for basic insight (Tables 1 and S1–S4; Figures S1 and S2), and then focused on the stabilizing substituents that allow for experimentally accessible diazo dipoles.
Table 1.
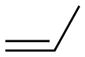
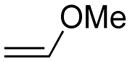
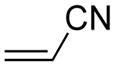
Activation energies for cycloadditions of diazo compounds (X=CH) and azides (X=N) calculated at the M06-2X/6-31+G(2d,p) level of theory. Energies (kcal/mol) include solvation corrections (water) on gas-phase geometries with the IEFPCM model (radii=UFF).
| X |
a |
 b |
 c |
 d |
 e |
 f |
||
|---|---|---|---|---|---|---|---|---|
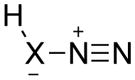
|
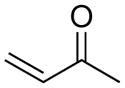
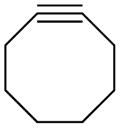
1 | CH | 16.9 | 18.8 | 22.9 | 9.4 | 8.2 | 12.5 |
| 2 | N | 23.3 | 24.3 | 24.3 | 20.4 | 18.7 | 16.7 | |
|
| ||||||||
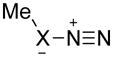
|
3 | CH | 14.5 | 16.3 | 20.4 | 6.7 | 5.6 | 9.7 |
| 4 | N | 21.1 | 22.0 | 23.0 | 19.9 | 15.8 | 14.6 | |
|
| ||||||||
|
5 | CH | 19.5 | 20.8 | 24.9 | 14.2 | 12.3 | 13.9 |
| 6 | N | 23.8 | 24.0 | 23.1 | 23.4 | 20.8 | 16.9 | |
|
| ||||||||
|
7 | CH | 21.4 | 22.7 | 26.5 | 17.1 | 15.1 | 15.8 |
| 8 | N | 22.5 | 22.6 | 21.5 | 22.3 | 19.8 | 15.4 | |
The diazo group has an intrinsic advantage relative to analogous azides: enhanced tunability.15 The total range of activation barriers for cycloadditions with diazo groups spans 21 kcal/mol (3–e TS to 7–c TS), whereas that for azides spans only 10 kcal/mol (2–c TS to 4–f TS) (Table 1). Even with diazoacetamide 5, which is relatively deactivated by conjugation, this range is 13 kcal/mol (Figure 1).
Figure 1.

Exemplary transition geometries of diazo- and azido-cycloadditions calculated at the M06-2X/6-31+G(2d,p) level of theory. Energies (kcal/mol) and NBO charges on dipolarophiles (italics) include solvation corrections (water) on gas-phase geometries with the IEFPCM model (radii=UFF).
Diazo compounds prefer to undergo normal electron-demand cycloadditions, in agreement with the increased nucleophilicity of diazo compounds (Table S5).16 Accordingly, we observed an increase in reactivity for diazo groups with dipolarophiles containing electron-withdrawing substituents (d and e).5,15b In contrast, azides show ambiphilic character—benefitting from both donors and acceptors on the dipolarophile. Still, electron-donating substituents on an alkene (e.g., in methyl vinyl ether, c) do not increase reactivity over ethylene (a) until sufficient acceptor substituents are present on the azide (6 and 8). This finding correlates well with earlier studies.9a,9e
The sensitivity to effects on reactivity provided by the strategies of strain-activation versus that of electronic tuning for each type of dipole are apparent. The calculated barriers for cycloadditions of each diazo compound (1, 3, 5, and 7) with electron-deficient methyl vinyl ketone (e) are lower than those for cycloadditions with cyclooctyne (f) (Table 1). In stark contrast, the lowest barriers for cycloadditions of each azide (2, 4, 6, and 8) are observed with cyclooctyne.
Unlike strain, electron-withdrawing substituents on the alkene allowed not only for increased reactivity with diazo compounds, but also increased selectivity over azides. The ΔΔE‡ between diazoacetamide 5 and the analogous azide 6 was 4.3 kcal/mol for the parent ethylene TSs (5–a TS and 6–a TS). Electronic activation increases this difference (ΔΔE‡ = 8.5 kcal/mol for 5–e TS and 6–e TS), whereas strain decreases it (ΔΔE‡ = 3.0 kcal/mol for 5–f TS and 6–f TS). This dichotomy is in agreement with previous findings by Domingo and coworkers, who showed that propargylic-type 1,3-dipoles display zwitterion-type reactivity—concerted reactions proceeding through a polar, asynchronous transition state—and are controlled by the electronic character of reactants.16 To quantify the extent of polarity in the TS, we examine charge transfer (CT) by considering NBO charges on the dipolarophile, where the reciprocal charge resides on the 1,3-dipole (Figure 1).
Decreased Distortion Energies without Pre-distortion
The current strategy for increasing reactivity in uncatalyzed azide–alkyne cycloadditions employs the incorporation of strain or modulation of electronics in cyclic alkynes.3,4,6–8,17 Strain-activation—also termed distortion-acceleration18—has provided reagents for chemical biology, but those reagents have little selectivity between diazo and azido dipoles.5 Although increased reactivity does not necessarily imply an inherent loss of selectivity,19 retaining selectivity while increasing reactivity requires a thorough understanding of those factors that lead to increased reaction rates.
Cycloadditions of vastly different reaction energies often proceed at comparable rates, and thus, cannot be explained via a strict Bell–Evans–Polyani, or Marcus treatment (vide infra).11b,19 As a result, thermodynamic effects on transition state energies and timing is not straightforward. Nevertheless, pre-distortion of the dipolarophile enforces a more reactant-like (i.e., early) TS, disfavoring selectivity. Surprisingly, the approach of pre-distorting starting materials is used commonly to accelerate cycloaddition reactions, even though the dipole displays a much more product-like geometry in the TS,20 whereas the dipolarophile remains relatively linear.11b Based on the Hammond–Leffler postulate,20 the earlier TS of pre-distorted compounds should have a negative effect on selectivity,19 in agreement with calculated activation energies and previous experimental results.4
The selectivity seen in many competing cycloadditions must arise from the alternative strategy of FMO-matching between reactants.9e,21 More specifically, selectivity arises from the smaller distortion energy required by dipoles with more closely matched FMO energies, as revealed with distortion/interaction (or activation strain) analysis—a computational tool utilized to dissect reaction barriers in order to understand their origins.11,13 The energy required to distort isolated reactants to their TS geometries is termed the distortion energy (ΔE‡distortion) and the energy gained when bringing these high energy species together is the interaction energy (ΔE‡interaction). The approach of FMO-matching is superior when the end goal is selective reactivity.19,22
SPAAC relies on decreased distortion energies of the dipolarophile, which is the reactant that contributes only ~20% of the overall barrier,23 whereas ~80% of the distortion energy arises from the 1,3-dipole.11b Diazo-group cycloadditions provide an opportunity to decrease dipole distortion energies but without pre-distortion of the reactants. The distortion energies of simple diazo compounds are, in essence, always lower than those of simple azides because of the higher energy of their highest occupied molecular orbital (HOMO) (Figure S3).11b Although electron-withdrawing substituents on the diazo group have been shown to increase distortion energies as a result of HOMO stabilization,24 analogous azides have not been examined to date. Thus, we sought to reveal factors responsible for the increased reactivity of stabilized diazo compounds over analogous azides.25 We focused on diazoacetamide 5 and the analogous azide 6, as this substitution pattern provides readily accessible compounds but displays attractive substituent effects on reaction kinetics (Figure 2).
Figure 2.

Distortion/interaction analysis for selected diazo- and azido-cycloadditions calculated at the M06-2X/6-31+G(2d,p) level of theory. Energies (kcal/mol) include solvation corrections (water) on gas-phase geometries with the IEFPCM model (radii=UFF).
Cycloadditions of diazo compounds benefit from decreased distortion energies and increased interaction energies (Table S6; Figures S4 and S5). The total of the distortion energies required to reach the TS of the electronically tuned diazoacetamide–methyl vinyl ketone cycloaddition 5–e TS is significantly less than that for the analogous azido-cycloaddition 6–e TS (23.8 versus 27.4 kcal/mol), and is slightly less than that for the distortion-accelerated methylcarbamoyl azide–cyclooctyne cycloaddition 6–f TS (23.8 versus 24.1 kcal/mol). In addition, the diazoacetamide–methyl vinyl ketone cycloaddition 5–e TS benefits greatly from increased interaction energies relative to both 6–e TS and 6–f TS (−11.4 versus −6.7 and −7.1 kcal/mol, respectively). The large interaction energies are a direct consequence of the high nucleophilicity of the diazo group (Table S5), resulting in highly asynchronous TSs and a high degree of charge transfer in those TSs (Figure 1).26
Distortion/interaction analysis11b,13a reveals further insights into the efficacies of strain-activation and electronic tuning (Figure S6). Again, cycloadditions of diazo compounds are affected more by electronic tuning than are those of the analogous azides. Whereas electronic activation decreases the total of the distortion energies by 2.8 kcal/mol for the diazoacetamide (26.6 versus 23.8 kcal/mol for 5–a TS and 5–e TS), the decrease is only 2.0 kcal/mol for the analogous azide (29.4 versus 27.4 kcal/mol for 6–a TS and 6–e TS). Comparing the same TSs, a much larger increase in interaction energies is observed for the diazoacetamide 5 than for the azide 6 (4.3 versus 1.1 kcal/mol).
The effects of strain-activation are more pronounced for azide 6, despite a lower overall barrier for diazoacetamide 5. Although the diazoacetamide shows a larger decrease in total distortion energies as a result of strain-activation (relative to electronic modulation, ~3 kcal/mol)—4.5 kcal/mol (26.6 versus 22.1 kcal/mol for 5–a TS and 5–f TS)—the decrease is even larger for the analogous azido-cycloadditions—5.3 kcal/mol (29.4 versus 24.1 kcal/mol for 6–a TS and 6–f TS). In addition, the 1.2 kcal/mol increase in interaction energy for diazoacetamide cycloadditions involving strain-activation (5–a TS and 5–f TS) is much smaller than the 4.3 kcal/mol increase as a result of electronic tuning (5–a TS and 5–e TS). In contrast, azide 6 shows a larger increase in interaction energies as a result of strain-activation than as a result of electronic tuning (1.5 versus 1.1 kcal/mol for 6–f TS and 6–e TS, respectively). The distinction stems from the ambiphilic character of the dipole of the azido group, relative to the more nucleophilic dipole of the diazo group, and its benefitting from the decreased gap between the energy of the HOMO and the lowest unoccupied molecular orbital (LUMO) upon alkyne distortion (vide infra).
Electron-withdrawing substituents on diazo compounds 5 and 7 increase the dipole distortion energies relative to diazomethane (1) and diazoethane (3) (Table S6). Yet, little effect is seen on the interaction energies. In comparison, electron-withdrawing substituents on azides 6 and 8 have the same effect on distortion energies, but also decrease interaction energies. With electron-deficient dipoles such as diazo compounds 5 and 7, interaction energies remain largest for the electron-deficient dipolarophile methyl vinyl ketone (e) (−11.4 kcal/mol in 5–e TS). In contrast, azides 6 and 8 show larger interaction energies upon reaction with the electron-rich methyl vinyl ether (c) (−10.6 kcal/mol in 6–c TS), consistent with the ambiphilic nature of azides.27
When strong electron-withdrawing groups are incorporated into the 1,3-dipole, total distortion energies for diazo-cycloadditions become similar to those for the analogous azide (e.g., 7 versus 8) and the interaction energies begin to determine the relative reactivities (Table S6). This trend is expected from the distortion/interaction analysis, as strong intramolecular interactions in stabilized reactants are traded for intermolecular interactions in the transition state, simultaneously increasing both distortion and interaction energies. This trade can be understood by examining the electronic nature of the dipoles of diazo compound 7 and azide 8 (Figure 3). The diazo moiety has a single lone pair of electrons, residing within an unhybridized p-orbital. To react, a diazo group must bend in an alternative mode relative to an azido group, which contains two orthogonal lone pairs. Delocalization of the carbon lone pair of diazopropanone 7 into the carbonyl π* orbital (78.1 kcal/mol) is exchanged for delocalization into the nascent C–C bond. Although much energy is lost upon termination of interactions in the reactant (distortion energy), energy is gained upon bond formation in the TS (interaction energy). The distortion energy of acetyl azide 8 is similar to that of diazopropanone 7, despite a higher degree of bending in the azide (~139° versus ~145°) in its reaction with methyl vinyl ketone (Table S6; Figure S2). The similarity arises from the sacrifice of the weaker nN→σ*C=O hyperconjugative interaction (9.1 kcal/mol) in the azide TS (8–e TS), while maintaining π-conjugation.
Figure 3.

Comparison of bending in diazopropanone (7) and acetyl azide (8) and effects on (hyper)conjugation. The larger decrease in conjugation in diazopropanone leads to similar distortion energies despite less bending. Transition state geometries are for 7–e TS and 8–e TS.
Key Interactions in Cycloadditions of Diazo Compounds
The transient nature of transition states renders their investigation difficult. A number of theoretical methods have been developed to understand the basis for substituent effects on reactivity,14,19,20,28 often examining the nature of the reactants and products of a transformation and describing the TS as a hybrid of these longer-lived species. The effects of reaction thermodynamics on barrier height and timing—in accord with the Hammond–Leffler postulate20 (i.e., early versus late)—are understood by treating each minimum as a parabola and the TS as the point at which the two parabolas cross.14,20,28 Per this analysis, an exothermic reaction results in a crossing point energetically closer to the reactants than in a thermoneutral process.
Based on these principles, Marcus theory is of great utility in the dissection of reaction barriers, correlating kinetic reactivity with thermochemistry, as in the equation:14
| (1) |
The intrinsic reaction barrier (ΔE‡0), which is the reaction barrier for a hypothetical thermoneutral reaction, is useful for understanding electronic effects that govern chemical transformations,29 and the variability of this term overcomes the limitations of simply blending minima to describe the TS.19,30 The last term in the equation can be neglected unless the reaction is highly exothermic (large ΔErxn) or the intrinsic barrier is low (small ΔE‡0). Then, eq. 1 reduces to ΔΔE‡ = ½ ΔErxn, which is the assumption made in the empirical Bell–Evans–Polyani relationship.28
Our focus is on the difference in cycloaddition reactivity between diazo compounds and azides. A general Marcus relationship was observed previously for the cycloadditions of simple dipoles with the simplest dipolarophile, ethylene.11b Our study examines a broad range of cycloadditions, thereby providing the opportunity to illuminate subtle electronic differences and their importance throughout the reaction coordinate. For example, a plot of ΔE‡ versus ΔErxn for all alkene cycloadditions from Table 1 reveals little correlation (R2 = 0.27, Figure S7). Separating the diazo and azido dipoles gives even lower correlation (R2 = 0.23 and 0.0003, respectively, Figure S7). Interestingly, separate plots of electron-deficient alkenes (d and e) and of electron-neutral/rich alkenes (a, b, and c) show a high correlation between ΔE‡ and ΔErxn (Figure 4) especially for the cycloadditions of diazo compounds (R2 = 0.98 and 0.92, respectively). Notably, the differences in intrinsic barriers between the two sets of reactions for diazo-cycloadditions are much larger (≥10 kcal/mol) than for azido-cycloadditions (4–5 kcal/mol) (Table S7). The greater importance of substituent effects, especially in the TS, for cycloadditions of diazo compounds provides the opportunity for increased reactivity and selectivity (for further analysis of substituent effects utilizing global reactivity indices, see: Table S5 and Figures S8 and S9).
Figure 4.

Activation energies versus reaction energies separated by dipoles and activated and non-activated dipolarophiles for cycloadditions from Table 1.31
Implications for Design of Orthogonal Cycloadditions
Our comparison of azides and diazo compounds in 1,3-dipolar cycloadditions unveils important principles for endowing reactivity and selectivity (Figure 5). As azides are relatively ambiphilic, strain-activation decreases the activation barriers not only by pre-distorting towards TS geometries, but also by increasing the ambiphilicity of the alkyne by decreasing the HOMO–LUMO gap. As a result, when strain alone is used to increase reactivity, as with bicyclo[6.1.0]non-4-yne (BCN), cycloaddition of an alkyl azide actually outcompetes that of the diazoacetamide.4 On the other hand, even the stabilized diazoacetamide 5 is a relatively nucleophilic dipole. Hence, increased electrophilicity of reacting partners—attained by incorporating electron-withdrawing substituents—becomes more important than in cycloadditions with azido congeners.5,32
Figure 5.

Alternative principles govern the cycloadditions of azides and diazo compounds. Alkyne distortion increases ambiphilicity relative to 2-butyne, which is an optimal scenario for the ambiphilic azide dipole (left). Increasing electrophilicity primes the alkyne for increased reactivity with the more nucleophilic diazo compound (right). Orbital energies are given relative to the HOMO of 2-butyne for the ground state (GS) and transition state (TS) geometries, as indicated. aEnergies given for the LUMO+1 (see: Figures S3 and S5).
To evaluate the degree of selectivity provided by each principle, we sought to compare the efficacy of strain-activation and electronic tuning amongst model dipoles resembling those used previously as chemical reporters (Figures 6 and S10).4 A general decrease in barriers for both diazoacetamide 5 and azide 9 is observed with increasing strain (Figure 6A) or with increased electron-withdrawing capability (Figure 6B). When activated by strain, the reactivity of both dipoles is highly sensitive to increased strain in the cycloalkyne. Still, the ΔΔE‡ values between the azide and diazo group are within ~0.6 kcal/mol for the reactions with cyclooctyne, 2-butyne, and cyclononyne (f–h), providing little selectivity. Selectivity is predicted in highly strained systems like cycloheptyne or cyclohexyne, but this outcome arises from the lowering of the LUMO energy by alkyne bending (Figure S6).8e Overall, our computational data and previous experimental results5 demonstrate that strain-activation alone is not able to generate selective reactivity for a diazoacetamide versus an analogous azide.
Figure 6.

Comparison of activation via strain (A) or electron-withdrawing substituents (B) for 1,3-dipolar cycloadditions of diazoacetamide 5 (filled circles) and azide 9 (open circles). Strain energies were calculated from the isodesmic equation in ref. 33. Activation energies were calculated at the M06-2X/6-31+G(2d,p) level of theory. Energies (kcal/mol) include solvation corrections (water) on gas-phase geometries with the IEFPCM model (radii=UFF).
Diazo-cycloadditions show a greater dependency on the LUMO energy of the dipolarophile than do azido-cycloadditions (Figure 6B). Simply changing the dipolarophile from ethylene (a) to nitroethylene (n) increases the relative cycloaddition rate from 7 to >103. Despite the deactivating and stabilizing effects of conjugation, the diazo HOMO remains higher in energy than does that of the azide (Figures S3 and S6). In addition, the calculated barrier of (12.3 kcal/mol) for the cycloaddition of diazoacetamide 5 with methyl vinyl ketone (e) is lower than that (14.4 kcal/mol) for the cycloaddition of azide 9 with cyclooctyne (f). Again, the difference is in accord with the increased reactivity of diazo groups over alkyl azides,5 allowing for reactions with unstrained alkenes that are able to surpass first-generation SPAAC.6
Solvent Effects on Cycloadditions
The higher-energy HOMO of stabilized diazo compounds as compared to similar azides results in both decreased distortion energies and increased interaction energies, allowing for the chemoselectivity of diazo groups with matched dipolarophiles. Importantly, these interactions are largely unidirectional—from the diazo compound to the dipolarophile—leading to a highly asynchronous TS. The result is a significant degree of charge transfer in the TS, a feature that can be exploited in polar solvents.4 Reactions insensitive to (and even accelerated by) water are of particular interest for green chemistry34 and an absolute requirement for chemical biology.35
The early notion that cycloaddition rates have little solvent-dependence36 was abandoned long ago.37 Experimental38 and theoretical39 reports show that not only aggregation, but also TS stabilization via hydrogen-bonding, are responsible for enhanced rates in water. In previous reports, we observed larger rate increases in aqueous conditions for diazo- than azido-cycloadditions.4,5,15b Here, we examined solvent effects on a series of electron-deficient alkenes examined experimentally5 in comparison to solvent effects with a strained dipolarophile, cyclooctyne.
Solvation optimizations with the IEFPCM model give similar activation energies for cycloadditions in acetonitrile and water (Figure S11). As the IEFPCM solvation model does not include explicit hydrogen-bonding, its corrections should be considered to be only a first approximation of solvation effects,40 especially when explicit hydrogen-bonding can lead to a large stabilization of the TS.39c
Effects of hydrogen-bonding on reaction barriers were examined by using explicit water molecules (Figure S11). To simplify calculations and provide a simple method to predict the extent of solvent effects, we focused our attention on the extent of charge transfer in the transition state. Upon calculating NBO charges on reacting fragments in the TS, we found a much larger degree of charge transfer in cycloadditions of diazoacetamide 5 in comparison to those of azide 9, especially with methyl vinyl ketone (e) (Figure 7).21b This dichotomy is indicative of a more polar TS for the reaction with the diazo compound, and thus larger rate enhancements due to polar solvents. This distinction is noteworthy, as the diazo group is conjugated to the amide, whereas the azido group is not.
Figure 7.

Electrostatic potential maps calculated at the B3LYP/6-31G(d) level of theory on M06-2X/6-31+G(2d,p) geometries. NBO charges (italics) were calculated at the M06-2X/6-31+G(2d,p) level of theory and include solvation corrections (water) on gas-phase geometries with the IEFPCM model (radii=UFF).
The charge transfer in cycloadditions with strained aliphatic dipolarophiles is typically less than that with electron-deficient alkenes. In contrast, we recently reported on diazoacetamide–aza-dibenzocyclooctyne (DIBAC) cycloadditions, which display a transfer of 0.14 e− from the diazo compound, but only a transfer of 0.08 e− from the azide.4,5 Recently reported inverse electron-demand cycloadditions of azides with BCN show a similar magnitude of charge transfer in the TS, but these cycloadditions transfer electrons from the electron-rich BCN to electron-poor azides (0.22–0.26 e−).9e
Conclusions
Diazo compounds have numerous advantages as dipoles in cycloadditions. First and foremost is the intrinsic tunability of their dipole to facilitate reaction with a particular dipolarophile. Others have stated that the “primary importance in the design and synthesis of reactive cycloalkynes for copper-free click chemistry is ring strain, followed by electronic activation and steric effects.”41,42 In contrast, we believe that a strategy based on the proper pairing of electronics in reacting partners is a more powerful means to achieve high and selective reactivity in 1,3-dipolar cycloadditions.19
The greater nucleophilic character of diazo groups over that of azido groups leads to more rapid cycloadditions. This nucleophilicity accommodates decreased distortion energies without the need for pre-distortion of dipolarophiles. In addition to decreased distortion energies, tuned cycloadditions of diazo compounds also display increased interaction energies. Thus, the tuning of FMOs allows for increased selectivity and reactivity for diazo compounds. In contrast, the strategy of strain-activation (or distortion-acceleration18) decreases total distortion energies to a larger extent, but at the expense of a smaller increase in interaction energies. Accordingly, the prominence of substituent effects in 1,3-dipolar cycloadditions of diazo compounds allows for the development of chemoselective transformations in the presence of azides and ensuing dual-labeling strategies. In this regard, the diazo group is poised for application in chemical biology to meet the rising demand for “orthogonal–bioorthogonal” reactions in bioconjugation and chemical reporter strategies.3
EXPERIMENTAL SECTION
Computational Details
Optimizations were performed with Gaussian 09 software43 at the M06-2X level of theory44 and the 6-31+G(2d,p) basis set. M06-2X has been shown to describe trends in reactivity accurately for cycloadditions.45 Optimizations were performed in the gas-phase, followed by single-point solvation corrections. An IEFPCM dielectric continuum solvent model for water with UFF radii was employed. Frequency calculations were performed to confirm stationary points as minima or first-order saddle points. All ΔE and ΔE‡ values include zero-point corrections.
Alternative mechanisms for 1,3-dipolar cycloadditions are sometimes possible (e.g., stepwise or biradical).16b,46 We do not expect these mechanisms to be operational in our cycloaddtions, due to seminal work,47 small solvent effects in previous studies,5 and previous calculations suggesting that a diradical mechanism is unlikely in reactions of propargylic 1,3-dipoles.11b,48 Nevertheless, we explored alternative possibilities and confirmed that a concerted mechanism is the most energetically favored in cases where a stepwise TS was located (Figure S12).
Supplementary Material
Acknowledgments
This work was supported by Grant R01 GM044783 (NIH). Computational resources were supported in part by Grant CHE-0840494 (NSF). We are grateful to R. W. Newberry, I. C. Tanrikulu, and C. L. Jenkins (University of Wisconsin–Madison) for contributive discussions.
Footnotes
The authors declare no competing financial interest.
Supporting Information. Tables S1–S7, Figures S1–S12, and Cartesian coordinates, energies, and imaginary frequencies (for TSs) of optimized structures. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Huisgen R. Proc Chem Soc. 1961:357–396. [Google Scholar]
- 2.(a) Pechmann vH. Ber Dtsch Chem Ges. 1898;31:2950–2951. [Google Scholar]; (b) Cowell GW, Ledwith A. Q Rev Chem Soc. 1970;24:119–167. [Google Scholar]; (c) Maas G. In Organic Synthesis, Reactions and Mechanisms. Springer; 1987. pp. 75–253. [Google Scholar]; (d) Synthesis of Heterocycles via Cycloadditions. Springer; 2008. [Google Scholar]; (e) Regitz M. Diazo Compounds: Properties and Synthesis. Elsevier; 2012. [Google Scholar]; (f) Sears JE, Boger DL. Acc Chem Res. 2016;49:241–251. doi: 10.1021/acs.accounts.5b00510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Lang K, Chin JW. ACS Chem Biol. 2014;9:16–20. doi: 10.1021/cb4009292. [DOI] [PubMed] [Google Scholar]; (b) Patterson DM, Nazarova LA, Prescher JA. ACS Chem Biol. 2014;9:592–605. doi: 10.1021/cb400828a. [DOI] [PubMed] [Google Scholar]; (c) Patterson DM, Prescher JA. Curr Opin Chem Biol. 2015;28:141–149. doi: 10.1016/j.cbpa.2015.07.006. [DOI] [PubMed] [Google Scholar]
- 4.Andersen KA, Aronoff MR, McGrath NA, Raines RT. J Am Chem Soc. 2015;137:2412–2415. doi: 10.1021/ja5095815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aronoff MR, Gold B, Raines RT. Org Lett. 2016;18:1538–1541. doi: 10.1021/acs.orglett.6b00278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Agard NJ, Prescher JA, Bertozzi CR. J Am Chem Soc. 2004;126:15046–15047. doi: 10.1021/ja044996f. [DOI] [PubMed] [Google Scholar]
- 7.(a) Ning X, Guo J, Wolfert MA, Boons GJ. Angew Chem, Int Ed. 2008;47:2253–2255. doi: 10.1002/anie.200705456. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Poloukhtine AA, Mbua NE, Wolfert MA, Boons GJ, Popik VV. J Am Chem Soc. 2009;131:15769–15776. doi: 10.1021/ja9054096. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Jewett JC, Sletten EM, Bertozzi CR. J Am Chem Soc. 2010;132:3688–3690. doi: 10.1021/ja100014q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Dommerholt J, Schmidt S, Temming R, Hendriks LJA, Rutjes FPJT, van Hest JCM, Lefeber DJ, Friedl P, van Delft FL. Angew Chem, Int Ed. 2010;49:9422–9425. doi: 10.1002/anie.201003761. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Varga BR, Kállay M, Hegyi K, Béni S, Kele P. Chem Eur J. 2012;18:822–828. doi: 10.1002/chem.201102329. [DOI] [PubMed] [Google Scholar]
- 8.(a) Baskin JM, Prescher JA, Laughlin ST, Agard NJ, Chang PV, Miller IA, Lo A, Codelli JA, Bertozzi CR. Proc Natl Acad Sci USA. 2007;104:16793–16797. doi: 10.1073/pnas.0707090104. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chenoweth K, Chenoweth D, Goddard WA., III Org Biomol Chem. 2009;7:5255–5258. doi: 10.1039/b911482c. [DOI] [PubMed] [Google Scholar]; (c) Schoenebeck F, Ess DH, Jones GO, Houk KN. J Am Chem Soc. 2009;131:8121–8133. doi: 10.1021/ja9003624. [DOI] [PubMed] [Google Scholar]; (d) Debets MF, van Berkel SS, Schoffelen S, Rutjes FPJT, van Hest JCM, van Delft FL. Chem Commun. 2010;46:97–99. doi: 10.1039/b917797c. [DOI] [PubMed] [Google Scholar]; (e) Gold B, Dudley GB, Alabugin IV. J Am Chem Soc. 2013;135:1558–1569. doi: 10.1021/ja3114196. [DOI] [PubMed] [Google Scholar]; (f) Garcia-Hartjes J, Dommerholt J, Wennekes T, van Delft FL, Zuilhof H. Eur J Org Chem. 2013;2013:3712–3720. [Google Scholar]; (g) Ni R, Mitsuda N, Kashiwagi T, Igawa K, Tomooka K. Angew Chem, Int Ed. 2015;54:1190–1194. doi: 10.1002/anie.201409910. [DOI] [PubMed] [Google Scholar]
- 9.(a) Jones GO, Houk KN. J Org Chem. 2008;73:1333–1342. doi: 10.1021/jo702295d. [DOI] [PubMed] [Google Scholar]; (b) Sanders BC, Friscourt F, Ledin PA, Mbua NE, Arumugam S, Guo J, Boltje TJ, Popik VV, Boons GJ. J Am Chem Soc. 2010;133:949–957. doi: 10.1021/ja1081519. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Yoshida S, Shiraishi A, Kanno K, Matsushita T, Johmoto K, Uekusa H, Hosoya T. Sci Rep. 2011;1:82. doi: 10.1038/srep00082. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Zimmerman ES, Heibeck TH, Gill A, Li X, Murray CJ, Madlansacay MR, Tran C, Uter NT, Yin G, Rivers PJ, Yam AY, Wang WD, Steiner AR, Bajad SU, Penta K, Yang W, Hallam TJ, Thanos CD, Sato AK. Bioconjugate Chem. 2014;25:351–361. doi: 10.1021/bc400490z. [DOI] [PubMed] [Google Scholar]; (e) Dommerholt J, van Rooijen O, Borrmann A, Guerra CF, Bickelhaupt FM, van Delft FL. Nat Commun. 2014;5:5378. doi: 10.1038/ncomms6378. [DOI] [PubMed] [Google Scholar]; (f) Xie S, Lopez SA, Ramström O, Yan M, Houk KN. J Am Chem Soc. 2015;137:2958–2966. doi: 10.1021/ja511457g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.There are notable alternative reactions, exemplified as follows. Tetrazine–trans-cyclooctene: Blackman ML, Royzen M, Fox JM. J Am Chem Soc. 2008;130:13518–13519. doi: 10.1021/ja8053805.Darko A, Wallace S, Dmitrenko O, Machovina MM, Mehl RA, Chin JW, Fox JM. Chem Sci. 2014;5:3770–3776. doi: 10.1039/C4SC01348D.Selvaraj R, Giglio B, Liu S, Wang H, Wang M, Yuan H, Chintala SR, Yap LP, Conti PS, Fox JM, Li Z. Bioconjugate Chem. 2015;26:435–442. doi: 10.1021/acs.bioconjchem.5b00089.. Tetrazine–cyclopropene: Yang J, Šečkutė J, Cole CM, Devaraj NK. Angew Chem, Int Ed. 2012;51:7476–7479. doi: 10.1002/anie.201202122.Patterson DM, Nazarova LA, Xie B, Kamber DN, Prescher JA. J Am Chem Soc. 2012;134:18638–18643. doi: 10.1021/ja3060436.Kamber DN, Nazarova LA, Liang Y, Lopez SA, Patterson DM, Shih HW, Houk KN, Prescher JA. J Am Chem Soc. 2013;135:13680–13683. doi: 10.1021/ja407737d.Yang J, Liang Y, Šečkutė J, Houk KN, Devaraj NK. Chem Eur J. 2014;20:3365–3375. doi: 10.1002/chem.201304225.Sachdeva A, Wang K, Elliott T, Chin JW. J Am Chem Soc. 2014;136:7785–7788. doi: 10.1021/ja4129789.. Triazine–cyclopropene: Kamber DN, Liang Y, Blizzard RJ, Liu F, Mehl RA, Houk KN, Prescher JA. J Am Chem Soc. 2015;137:8388–8391. doi: 10.1021/jacs.5b05100.Nitrile imine–cyclopropene: Wang Y, Song W, Hu WJ, Lin Q. Angew Chem, Int Ed. 2009;48:5330–5333. doi: 10.1002/anie.200901220.
- 11.(a) Ess DH, Houk KN. J Am Chem Soc. 2007;129:10646–10647. doi: 10.1021/ja0734086. [DOI] [PubMed] [Google Scholar]; (b) Ess DH, Houk KN. J Am Chem Soc. 2008;130:10187–10198. doi: 10.1021/ja800009z. [DOI] [PubMed] [Google Scholar]
- 12.Stockmann H, Neves AA, Day HA, Stairs S, Brindle KM, Leeper FJ. Chem Commun. 2011;47:7203–7205. doi: 10.1039/c1cc12161h.Karver MR, Weissleder R, Hilderbrand SA. Angew Chem, Int Ed. 2012;51:920–922. doi: 10.1002/anie.201104389.Plass T, Milles S, Koehler C, Szymański J, Mueller R, Wießler M, Schultz C, Lemke EA. Angew Chem, Int Ed. 2012;51:4166–4170. doi: 10.1002/anie.201108231.Willems LI, Li N, Florea BI, Ruben M, van der Marel GA, Overkleeft HS. Angew Chem, Int Ed. 2012;51:4431–4434. doi: 10.1002/anie.201200923.Patterson DM, Nazarova LA, Xie B, Kamber DN, Prescher JA. J Am Chem Soc. 2012;134:18638–18643. doi: 10.1021/ja3060436.Cole CM, Yang J, Šečkutė J, Devaraj NK. ChemBioChem. 2013;14:205–208. doi: 10.1002/cbic.201200719.Kamber DN, Nazarova LA, Liang Y, Lopez SA, Patterson DM, Shih HW, Houk KN, Prescher JA. J Am Chem Soc. 2013;135:13680–13683. doi: 10.1021/ja407737d.Sachdeva A, Wang K, Elliott T, Chin JW. J Am Chem Soc. 2014;136:7785–7788. doi: 10.1021/ja4129789.
- 13.For a tutorial review of the distortion/interaction model (which is also known as the activation strain model), see: Fernández I, Bickelhaupt FM. Chem Soc Rev. 2014;43:4953–4967. doi: 10.1039/c4cs00055b.Wolters LP, Bickelhaupt FM. WIREs Comput Mol Sci. 2015;5:324–343. doi: 10.1002/wcms.1221.
- 14.(a) Marcus RA. J Chem Phys. 1956;24:966–978. [Google Scholar]; (b) Marcus RA. Annu Rev Phys Chem. 1964;15:155–196. [Google Scholar]; (c) Marcus RA. J Phys Chem. 1968;72:891–899. [Google Scholar]; (d) Koeppl GW, Kresge AJ. J Chem Soc, Chem Commun. 1973:371–373. [Google Scholar]
- 15.(a) Bihlmaier W, Huisgen R, Reissig HU, Vass S. Tetrahedron Lett. 1979;20:2621–2624. [Google Scholar]; (b) McGrath NA, Raines RT. Chem Sci. 2012;3:3237–3240. doi: 10.1039/C2SC20806G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Domingo LR, Chamorro E, Pérez P. J Org Chem. 2008;73:4615–4625. doi: 10.1021/jo800572a. [DOI] [PubMed] [Google Scholar]; (b) Domingo LR, Emamian SR. Tetrahedron. 2014;70:1267–1273. [Google Scholar]
- 17.(a) Gold B, Shevchenko NE, Bonus N, Dudley GB, Alabugin IV. J Org Chem. 2012;77:75–89. doi: 10.1021/jo201434w. [DOI] [PubMed] [Google Scholar]; (b) Gold B, Batsomboon P, Dudley GB, Alabugin IV. J Org Chem. 2014;79:6221–6232. doi: 10.1021/jo500958n. [DOI] [PubMed] [Google Scholar]
- 18.Lopez SA, Houk KN. J Org Chem. 2013;78:1778–1783. doi: 10.1021/jo301267b. [DOI] [PubMed] [Google Scholar]
- 19.Mayr H, Ofial AR. Angew Chem, Int Ed. 2006;45:1844–1854. doi: 10.1002/anie.200503273. [DOI] [PubMed] [Google Scholar]
- 20.(a) Leffler JE. Science. 1953;117:340–341. doi: 10.1126/science.117.3039.340. [DOI] [PubMed] [Google Scholar]; (b) Hammond GS. J Am Chem Soc. 1955;77:334–338. [Google Scholar]
- 21.(a) Sustmann R. Tetrahedron Lett. 1971;12:2717–2720. [Google Scholar]; (b) Epiotis ND. J Am Chem Soc. 1972;94:1924–1934. [Google Scholar]; (c) Houk KN. J Am Chem Soc. 1972;94:8953–8955. [Google Scholar]; (d) Houk KN, Sims J, Duke RE, Strozier RW, George JK. J Am Chem Soc. 1973;95:7287–7301. [Google Scholar]; (e) Houk KN, Sims J, Watts CR, Luskus LJ. J Am Chem Soc. 1973;95:7301–7315. [Google Scholar]; (f) MacKenzie DA, Pezacki JP. Can J Chem. 2014;92:337–340. [Google Scholar]
- 22.Atthough the FMO and distortion/interaction analysis is a powerful tool for predicting the energetics of type I and III cycloadditions, type II cycloadditions are more recalcitrant to this approach. A recent study has elucidated factors responsible for high reactivity of dipoles displaying pseudodiradical character in the ground state—a set largely comprised of allylic-type rather than propargylic-type dipoles. Here, negligible dipole distortion is required to obtain favorable electronic character for highly synchronous bond formation. In cases where pseudodiradical character is small—as in dipoles currently under investigation—the predictive power of the pseudodiradical character is less reliable and reactivity is understood via electronic differences provided by the FMO approach. For additional information, see: ref. 16b and references therein.
- 23.Distortion of dipolarophiles also decreases the gap in FMO energies between reacting partners, thereby decreasing dipole distortion indirectly by favoring an early TS Gordon CG, Mackey JL, Jewett JC, Sletten EM, Houk KN, Bertozzi CR. J Am Chem Soc. 2012;134:9199–9208. doi: 10.1021/ja3000936.
- 24.In addition, the present study uses the M06-2X level of theory, which has proven to be better suited for asynchronous cycloadditions. Linder M, Brinck T. Phys Chem Chem Phys. 2013;15:5108–5114. doi: 10.1039/c3cp44319a.
- 25.Interaction energies differentiate reactivity when distortion energies are nearly constant (ref. 10).
- 26.(a) Domingo LR, Sáez JA. Org Biomol Chem. 2009;7:3576–3583. doi: 10.1039/b909611f. [DOI] [PubMed] [Google Scholar]; (b) Domingo LR, Aurell MJ, Pérez P, Sáez JA. RSC Adv. 2012;2:1334–1342. [Google Scholar]
- 27.Diazo compounds have also been shown to display “type II” behavior, though with stronger donors than a methoxy group (ref. 15a).
- 28.(a) Bell RP. Proc Roy Soc London. 1936;A154:414–429. [Google Scholar]; (b) Evans MG, Polanyi M. Trans Faraday Soc. 1938;34:11–24. [Google Scholar]
- 29.(a) Yoo HY, Houk KN. J Am Chem Soc. 1997;119:2877–2884. [Google Scholar]; (b) Aviyente V, Yoo HY, Houk KN. J Org Chem. 1997;62:6121–6128. [Google Scholar]; (c) Aviyente V, Houk KN. J Phys Chem A. 2001;105:383–391. [Google Scholar]; (d) Würthwein EU, Lang G, Schappele LH, Mayr H. J Am Chem Soc. 2002;124:4084–4092. doi: 10.1021/ja0121540. [DOI] [PubMed] [Google Scholar]; (e) Alabugin IV, Manoharan M, Breiner B, Lewis FD. J Am Chem Soc. 2003;125:9329–9342. doi: 10.1021/ja035729x. [DOI] [PubMed] [Google Scholar]; (f) Mayr H, Breugst M, Ofial AR. Angew Chem, Int Ed. 2011;50:6470–6505. doi: 10.1002/anie.201007100. [DOI] [PubMed] [Google Scholar]; (g) Alabugin IV, Gilmore K, Manoharan M. J Am Chem Soc. 2011;133:12608–12623. doi: 10.1021/ja203191f. [DOI] [PubMed] [Google Scholar]; (h) Mondal S, Gold B, Mohamed RK, Alabugin IV. Chem Eur J. 2014;20:8664–8669. doi: 10.1002/chem.201402843. [DOI] [PubMed] [Google Scholar]; (i) Mayr H, Ofial AR. Acc Chem Res. 2016;49:952–965. doi: 10.1021/acs.accounts.6b00071. [DOI] [PubMed] [Google Scholar]
- 30.Pross A, Shaik SS. J Am Chem Soc. 1982;104:1129–1130. [Google Scholar]
- 31.Cyclooctyne is excluded to avoid complications arising from the formation of aromatic versus nonaromatic products.
- 32.Aronoff MR, Gold B, Raines RT. Tetrahedron Lett. 2016;57:2347–2350. doi: 10.1016/j.tetlet.2016.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bach RD. J Am Chem Soc. 2009;131:5233–5243. doi: 10.1021/ja8094137. [DOI] [PubMed] [Google Scholar]
- 34.Anastas PT, Warner JC. Green Chemistry: Theory and Practice. Oxford University Press; New York: 1998. [Google Scholar]
- 35.Kolb HC, Finn MG, Sharpless KB. Angew Chem, Int Ed. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 36.(a) Sauer J, Sustmann R. Angew Chem, Int Ed. 1980;19:779–807. [Google Scholar]; (b) Huisgen R. Pure Appl Chem. 1980;52:2283–2302. [Google Scholar]
- 37.Breslow R. Acc Chem Res. 1991;24:159–164. [Google Scholar]
- 38.(a) Meijer A, Otto S, Engberts JBFN. J Org Chem. 1998;63:8989–8994. [Google Scholar]; (b) Narayan S, Muldoon J, Finn MG, Fokin VV, Kolb HC, Sharpless KB. Angew Chem, Int Ed. 2005;44:3275–3279. doi: 10.1002/anie.200462883. [DOI] [PubMed] [Google Scholar]
- 39.(a) Chandrasekhar J, Shariffskul S, Jorgensen WL. J Phys Chem B. 2002;106:8078–8085. [Google Scholar]; (b) Ess DH, Jones GO, Houk KN. Adv Synth Catal. 2006;348:2337–2361. [Google Scholar]; (c) Jung Y, Marcus RA. J Am Chem Soc. 2007;129:5492–5502. doi: 10.1021/ja068120f. [DOI] [PubMed] [Google Scholar]
- 40.(a) Miertuš S, Scrocco E, Tomasi J. Chem Phys. 1981;55:117–129. [Google Scholar]; (b) Tomasi J, Mennucci B, Cancès E. J Mol Struct. 1999;464:211–226. [Google Scholar]; (c) Tomasi J, Mennucci B, Cammi R. Chem Rev. 2005;105:2999–3094. doi: 10.1021/cr9904009. [DOI] [PubMed] [Google Scholar]
- 41.Sletten EM, de Almeida G, Bertozzi CR. Org Lett. 2014;16:1634–1637. doi: 10.1021/ol500260d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Our focus was on alkenes, as a result of their readily tunable electronics and abundance in natural products (Scheme 1).
- 43.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark MJ, Heyd J, Brothers EN, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell AP, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam NJ, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09. Gaussian, Inc; Wallingford, CT, USA: 2009. [Google Scholar]
- 44.Zhao Y, Truhlar D. Theoret Chem Acc. 2008;120:215–241. [Google Scholar]
- 45.Lan Y, Zou L, Cao Y, Houk KN. J Phys Chem A. 2011;115:13906–13920. doi: 10.1021/jp207563h. [DOI] [PubMed] [Google Scholar]
- 46.(a) Huisgen R, Mloston G, Langhals E. J Am Chem Soc. 1986;108:6401–6402. [Google Scholar]; (b) Haberhauer G, Gleiter R, Woitschetzki S. J Org Chem. 2015;80:12321–12332. doi: 10.1021/acs.joc.5b02230. [DOI] [PubMed] [Google Scholar]; (c) Domingo LR, Ríos-Gutiérrez M, Pérez P. Tetrahedron. 2016;72:1524–1532. [Google Scholar]
- 47.Huisgen R, Reissig H-U, Huber H, Voss S. Tetrahedron Lett. 1979;20:2987–2990. [Google Scholar]
- 48.(a) Braida B, Walter C, Engels B, Hiberty PC. J Am Chem Soc. 2012;132:7631–7637. doi: 10.1021/ja100512d. [DOI] [PubMed] [Google Scholar]; (b) Domingo LR, Emamian SR. Tetrahedron. 2014;70:1267–1273. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.