

Abstract
Each year over 90 million units of blood are transfused worldwide. Our dependence on this blood supply mandates optimized blood management and storage. During storage, red blood cells undergo degenerative processes resulting in altered metabolic characteristics which may make blood less viable for transfusion. However, not all stored blood spoils at the same rate, a difference that has been attributed to variable rates of energy usage and metabolism in red blood cells. Specific metabolite abundances are heritable traits; however, the link between heritability of energy metabolism and red blood cell storage profiles is unclear. Herein we performed a comprehensive metabolomics and proteomics study of red blood cells from 18 mono- and di-zygotic twin pairs to measure heritability and identify correlations with ATP and other molecular indices of energy metabolism. Without using affinity-based hemoglobin depletion, our work afforded the deepest multi-omic characterization of red blood cell membranes to date (1280 membrane proteins and 330 metabolites), with 119 membrane protein and 148 metabolite concentrations found to be over 30% heritable. We demonstrate a high degree of heritability in the concentration of energy metabolism metabolites, especially glycolytic metabolites. In addition to being heritable, proteins and metabolites involved in glycolysis and redox metabolism are highly correlated, suggesting that crucial energy metabolism pathways are inherited en bloc at distinct levels. We conclude that individuals can inherit a phenotype composed of higher or lower concentrations of these proteins together. This can result in vastly different red blood cells storage profiles which may need to be considered to develop precise and individualized storage options. Beyond guiding proper blood storage, this intimate link in heritability between energy and redox metabolism pathways may someday prove useful in determining the predisposition of an individual toward metabolic diseases.
The potency of harvested red blood cells (RBCs)1 depends on their ability to survive and maintain function during storage. RBC viability primarily depends on their ability to resist programmed cell death-related fragmentation and phagocytosis by maintaining proper energetics and avoiding hemolysis, in which they break down into microvesicles and toxic byproducts including iron, heme, hemoglobin, and oxidized lipids. The released iron can feed bacterial infections and free hemoglobin can interfere with nitric oxide signaling (1, 2). A number of small and retrospective studies have suggested that prolonged RBC storage is associated with negative clinical outcomes; however, three larger randomized clinical trials showed no negative effects of longer-stored RBCs (3–6). In short, the viability of stored RBCs is variable and not fully understood, but the accumulation of biophysical and metabolic changes known as storage lesions are linked to the ability to maintain flux through metabolic pathways during storage (7, 8).
Poststorage RBC adenosine triphosphate (ATP) concentration is the single best predictor of RBC in vivo recovery in autologous blood transfusions (9–12). Specifically, high ATP concentrations are correlated with low levels of hemolysis and other storage lesions in RBCs. Interestingly, poststorage ATP levels vary greatly between individuals but are consistent on repeat measure within an individual. This observation suggests that poststorage ATP, and thus stored RBC viability, may be influenced and/or determined by inheritance (13–15). In prior analyses of these samples and in additional studies of mono- and di-zygotic twins, some metabolite concentrations including glucose 6-phosphate, fructose 1,6-bisphosphate, glutathione, and glutathione disulfide were determined to be heritable in stored RBCs (13–16). The metabolite concentrations of ribulose 5-phosphate, sorbitol, and xylulose 5-phosphate are heritable suggesting a genetic control of glucose metabolism (14–16).
Because RBCs eject all organelles, including the nucleus and mitochondria upon maturing, they have no ability to synthesize proteins in response to environmental stimuli. The lack of mitochondria in mature RBCs also leaves these cells unable to rely on oxidative phosphorylation; instead, RBCs are reliant on glycolysis for energy production. These unique metabolic attributes of RBCs provide a highly instructive model for unraveling how genetic regulation of metabolic pathways can impact blood storage viability.
Herein, we describe a multi-omics analysis of genetic and environmental factors dictating RBC variability. Our approach involved an extensive proteomic and metabolomics analysis of RBCs derived from a cohort of 18 mono- and di-zygotic twin-pairs.
The primary challenge of performing proteomic analyses on red blood cells is the wide dynamic range characterized by an abundance of hemoglobin. This was surmounted by focusing our analysis on the membrane fraction of red blood cells. Although other studies have relied on time intensive affinity enrichment, utilizing the membrane fraction granted us the second greatest proteomic depth achieved in red blood cells which allowed us to process a multitude of clinical samples. Furthermore, much of the complexity and diversity in red blood cells is associated with the membrane including many energy metabolism components (17–19).
Despite the simplified composition of mature RBCs, i.e. no nucleus or mitochondria, detection and quantification of the RBC proteome presents a few challenges. First, RBCs must be purified from other blood cells. During this process, typically differential centrifugation, care must be taken to limit contamination, especially from the abundant plasma proteins. The next, and most significant, obstacle is the large dynamic range of protein abundance within the RBC (20, 21). Although the actual dynamic range of the RBC proteome is not yet known, the technical challenges are analogous to measuring the proteome of plasma, which has a dynamic range approaching twelve orders of magnitude (22). Also, similar to the plasma proteome, in which a single protein (albumin) constitutes 55% of the total protein content, hemoglobin comprises 97%, by mass, of the RBC proteome, making protein depletion a necessary consideration (23). Of the remaining 3%, carbonic anhydrase accounts for 1/3, so that the remaining 2% of total protein mass is made up of several thousand different proteins. Identifying these low abundance proteins from the background presented by hemoglobin and carbonic anhydrase, is challenging (24, 25). Several methods attempt to counter this obstacle by use of various types of affinity or ion exchange separation techniques (26–28). Even when employing these methods, most RBC proteome analyses yield detection of less than 1000 proteins, with the exception of one which identified 1,578 (29).
Most of these studies, especially those with the deepest coverage, require extensive protein and/or peptide fractionation which, in turn, yields considerable increases in analysis time—both sample preparation and instrument acquisition. Recent years have ushered in an era of proteomics where advances in peptide separation and mass spectrometer performance has accelerated the rate and depth of proteome analysis (30). We reasoned that application of this technology, combined with straightforward reversed-phase proteome fractionation, could expedite sample preparation and afford reasonably deep RBC proteomic analysis in short order, thus, affording the throughput for quantitative comparison of clinical RBC samples.
Using our method, we show that in RBCs the concentrations of components in crucial energy metabolism pathways are inherited en bloc at distinct levels. This results in different RBC storage phenotypes which can be used to further understanding of changes during storage and develop improved storage guidelines and methods. Furthermore, this rich data set will provide a valuable resource for continuing studies of RBCs and the heritability of disease.
EXPERIMENTAL PROCEDURES
Twin Subject Enrollment and Sample Collection
This report is a continuation of twin studies reported previously and utilized the same study subjects (14, 16, 31, 32). The study was approved by the Human Subjects office of The University of Iowa Carver College of Medicine. Written informed consent was obtained from all participating subjects. Subjects were qualified for participation by meeting criteria for autologous blood donation according to standard operating procedures of The University of Iowa DeGowin Blood Center. Twin pairs were not required to donate samples at the same time and each individual donated a single blood unit. Standard health history and demographic information were obtained at the time of enrollment and informed consent. Reported height and weight were used to calculate body mass index (BMI). BMI was derived from the formula: BMI = weight (kg)/(height (m))2. Each subject donated one unit of whole blood which were processed according to standard operating procedures into a leukocyte-reduced RBC unit in CP2D/AS-3 extended storage media (Hemonetics Corp, Braintree, MA). During processing, integral leukocyte reduction filters were retained for extraction of DNA.
Sample Preparation
Samples of AS-3 preserved RBC units were prepared from the main unit on each day of sampling. The AS-3 preserved RBCs were sampled by sterile docking of tubing to the RBC unit, back-filling the tubing with RBCs and sectioning into segments. This procedure was performed on the first day after donation (day 0), and every 14 days thereafter until day 56. This resulted in 5 time points at day 0, 14, 28, 42, and 56.
Segments were drained into 5 ml Eppendorf tubes; after mixing an aliquot is removed for complete blood count (CBC) testing using a hematology analyzer (Sysmex XE-2100™ Automated Hematology System, Sysmex Corp, Kobe, Japan). The remaining sample was centrifuged at 500 × g for 5 min, after which the storage media (AS-3) was removed. Samples were further processed and used for measurement of ATP, GSH, and GSSG in RBCs as previously described (14, 16).
Whole venous blood (EDTA, Vacutainer® purple top blood collection tube, 8 ml) collected from participants prior to blood donation was centrifuged at 500 × g for 5 min, followed by removal of the plasma and buffy coat. RBCs were washed twice with cold isotonic saline solution. After washing, a 30 μl aliquot of the packed red blood cells (pRBCs) was removed for complete blood count (CBC) analysis (Sysmex XE-2100™ Automated Hematology System, Sysmex Corp). A 100 μl aliquot of pRBCs was lysed with 900 μl of nanopure water. Samples were thoroughly mixed and stored at −80 °C prior to proteomic and metabolomic analyses.
Zygosity Testing
DNA for zygosity testing was obtained from leukocyte reduction filters by rinsing filters with 15 ml DPBS. The rinse volume was centrifuged at 500 × g for 10 min and the cell pellet was resuspended in 2 ml of DPBS. DNA was extracted from the cell pellet using a nucleic acid extraction instrument (AutoGen QuickGene 610L, AutoGen, Holliston, MA) and kit (Fuji QuickGene DNA Whole Blood Kit, AutoGen).
Genotyping was performed using a previously developed panel of 24 single nucleotide polymorphisms (SNPs) (10). SNP genotyping was performed using PCR assays (TaqMan, Applied Biosystems, Foster City, CA) on a Genotyping System (EP1 SNP, Fluidigm, San Francisco, CA) with a Dynamic Array Integrated Fluidic Circuit (GT48.48, Fluidigm). Monozygotic (MZ) twins were identified by 90% or greater genotype concordance; all other twin pairs were identified as dizygotic (DZ).
Global Metabolomics Profile Analyses
The untargeted metabolic profiling method employed for this analysis combined three independent platforms: ultrahigh performance liquid chromatography/tandem mass spectrometry (UHPLC/MS/MS) optimized for basic species, UHPLC/MS/MS optimized for acidic species, and gas chromatography/mass spectrometry (GC/MS). Samples were analyzed using procedures described in van ′t Erve et al. (14).
Sample Preparation for Proteomic Analysis
Proteolytic Digestion
A 50 μl aliquot of red blood cells lysed in 500 μl DI water was centrifuged at 4 °C for 30 min at 5 G. The supernatant was discarded and the pellet was resuspended in 100 μl lysis buffer (8 m Urea, 100 mm Tris, 10 mm TCEP, 40 mm chloroacetamide). The samples were then diluted with 50 mm Tris pH 7.5 until the pH reached 7.5 (∼ 1 ml). Trypsin digestion was performed overnight at room temperature with trypsin (Promega, Madison, WI) added at a 1:50 (w/w) enzyme to protein ratio with an estimated protein quantity of 500 μg. A second trypsin digestion was performed the following morning at 1:200 (w/w) enzyme to protein ratio for 1 h. Each digest was quenched by the addition of TFA and desalted over tC18 Sep-Pak cartridges (Waters, Milford, MA).
High pH Fraction Collection
Samples were fractionated using high pH reverse phase separation to increase proteomic depth. The solvent system consisted of mobile phase A (20 mm ammonium bicarbonate) and mobile phase B (20 mm ammonium bicarbonate 80% acetonitrile) which was run on an Ultimate 3000 UPLC system (Dionex Sunnyvale, CA) with a reverse phase C18 column. Gradient elution was performed at 400 μl min−1 with the gradient increased from 0 to 6% B over 5 min followed by an increase to 80% B until 24 min and a wash at 100% B for 3 min. Eight fractions were collected from each sample which were subsequently pooled resulting in four total fractions per sample.
nLC-MS/MS Analysis
Samples were analyzed using a LC/MS instrument comprising an Orbitrap Elite hybrid mass spectrometer (Thermo Fisher Scientific). Reverse phase columns were prepared in house using a 75–360 μm inner-outer diameter bare-fused silica capillary with laser pulled tip. The column was packed with 1.7 μm diameter, 130 Å pore size, Bridged Ethylene Hybrid C18 particles (Waters) to a final length of 35 cm. The column was installed on a Dionex Ultimate 3000 UPLC system and heated to 60 °C using an in house designed column heater for all runs (33, 34). Mobile phase buffer A was composed of water, 0.2% formic acid, and 5% DMSO. Mobile phase B was composed of acetonitrile, 0.2% formic acid, and 5% DMSO. 1 μg of sample was injected as determined by quantitative colorimetric peptide assay (Pierce, Rockford, IL). Gradient elution was performed at 300 nL min−1 with the gradient increased linearly from 0 to 60% B over 103 min followed by a linear increase to 100% B until 106 min and a wash at 100% B for 4 min. Survey scans of peptide precursors were collected from 300–1500 Th with an AGC target of 1,000,000 and a resolution of 60,000 followed by data dependent CID MS/MS scans of the 20 most intense peaks in the quadrupole linear ion trap mass analyzer. Precursors with charge states equal to 1 or unassigned were rejected and a 45 s dynamic exclusion was set to expedite identifications.
Data Analysis
Label free quantification was performed using Maxquant software version 1.5.2.8 (35) and the Andromeda search engine (36). The results were searched against a Homo sapiens database containing 90,482 reviewed proteins plus isoforms downloaded from Uniprot on June 23, 2015. Enzyme specificity was set to fully tryptic with up to two missed cleavages and carbamidomethylation of cysteines as a fixed modification. Oxidation of methionines and protein N-terminal acetylation were set as variable modifications. The match between runs feature was utilized to decrease missing data values within the data set (35). Precursor mass tolerance was 20 ppm and product ions were searched at 0.5 Da tolerances. Peptides were filtered to a 1% FDR and combined to protein groups based on the rules of parsimony, with at least two peptides per protein. Pearson correlations were calculated between each protein and metabolite detected using Perseus software (37–38).
Experimental Design and Statistical Rationale
Five di-zygotic twin pairs and thirteen mono-zygotic twin pairs were used in the study. No biological replicates were available because each individual was only required to donate blood at one time. Proteome and metabolome analyses were performed in randomized order to eliminate systematic biases.
Heritability Calculations
Heritability estimates were calculated for each protein and metabolite concentration measured, and for each measured time point when applicable. The first step to calculating heritability is using the one-way model of intraclass correlation coefficient (ICC) to determine the similarity of a measure in a twin pair: ICC = (MSbetween - MSwithin)/(MSbetween + MSwithin), where MSbetween is the estimate of the mean-square variance between all twin-pairs and MSwithin is the estimate of the mean-square variance within the sets of pairs in that group (13). The ICC is used to compare the variation within specific pairs to that of the population as a whole, and falls on a scale of −1 to +1. Higher positive values indicate that there is less variation within the pairs of subjects than there would be within randomly paired subjects. Positive values approaching 0, as well as negative ICC values, indicate that the variation within pairs of subjects is similar to the variation expected within random pairs. A highly heritable trait between MZ twins would be expected to have an intraclass correlation coefficient near +1. Once ICC values were calculated, heritability was estimated using the method derived by Newman et al., h2 = (ICCMZ - ICCDZ)/(1 - ICCDZ) (39).
RESULTS
To rapidly remove hemoglobin from RBCs, we separated RBCs obtained from whole blood via differential centrifugation. Samples were then centrifuged again, to isolate the membrane fraction, which was kept for further proteomic analysis while the supernatant (containing predominantly hemoglobin) was discarded (40). Although enriching for the membrane fraction will bias our analyses toward the detection of membrane bound proteins, many proteins of interest in red blood cells are associated with the membrane including some glycolytic proteins (18, 19, 41). Following this extraction, proteins were digested with trypsin, and the resultant peptides separated into eight fractionations via high pH reversed-phase liquid chromatography (RPLC). These fractions were recombined, generating four fractions per sample. Each fraction was then analyzed using a 120 min nanoLC-MS/MS method. In total, each sample required eight hours of mass spectrometer analysis, yielding an average of 3678 peptide spectral matches (PSMs), 2357 unique peptides, and 606 proteins per RBC sample.
Once the challenging problem of depleting hemoglobin had been overcome, we turned our attention to clinical RBC samples from 36 individuals including five di-zygotic and 13 mono-zygotic twin pairs. Twins were permitted to donate blood at separate times under separate conditions which serves to strengthen our confidence in heritability calculations. A 50 μl aliquot of washed, lysed RBCs from each patient was analyzed as described above. Across all 36 patients we detected 1280 proteins with an FDR less than 1%. Of these, 105 proteins were detected in all patients. However, of the proteins in our model (CA1, BPGM, and PFK) all were present in at least 27 individuals. No missing value imputation was utilized as this was found to alter our correlation analyses. The large majority (92%) of these proteins were identified with at least two peptides uniquely mapping to their sequence. Our data shows significant overlap with previous results—among our 1280 identified proteins, 941 have previously been observed in RBCs (73%). When performing heritability calculations, protein measurements were required to be present for all five dizygotic twin samples and 10 out of 13 monozygotic samples.
Metabolomic Analysis
As a relative newcomer in the “omics” era, metabolomics lags behind proteomics in the robust quantification of thousands of compounds. Discovery metabolomics aims to identify the entire metabolome present in cells; however, the greatest hurdle is the identification of unknown features. Using such discovery metabolomics assay ∼ 300 unique metabolites were quantified from these same samples.
Briefly, the discovery metabolic profiling method combined three independent platforms: ultrahigh performance liquid chromatography/tandem mass spectrometry (UHPLC/MS/MS) optimized for basic species, UHPLC/MS/MS optimized for acidic species, and gas chromatography/mass spectrometry (GC/MS). This method resulted in the quantification of 328 metabolites including lipids, xenobiotics, dipeptides, and many metabolites from prominent energy pathways (supplemental Table S4). Together, our proteomic and metabolomic data sets comprise the largest multiomic data set of red blood cells.
Correlation Analysis
To identify potentially coregulated proteins and metabolites Pearson correlation analysis and hierarchical clustering were performed between all proteins and metabolites yielding 58,000 correlations either greater than 0.75 or less than −0.75 which corresponds to 5% of the total correlations measured.
These clusters were found to contain unique protein groups containing proteosomal, fatty acid metabolism, or energy metabolism proteins. Of particular interest is a cluster containing numerous proteosomal proteins as well as those involved in glutathione metabolism and glycolysis (Fig. 1). We also observe glycolytic and glutathione metabolism proteins clustering with pyruvate metabolism and carbon fixation. All of these pathways were significantly enriched Kyoto Encyclopedia of Genes and Genomes (KEGG) terms using the online software enrichr (42).
Fig. 1.

Red blood cell proteins and metabolites show clusters of high correlation. Pearson correlation values were calculated between every combination of proteins and metabolites and plotted using Perseus. ™KEGG pathway enrichment of various clusters was measured using enrichr and the negative log of the p value for pathways of interest is reported in the bar charts to the right, where the color of the bars corresponds to the section of the dendrogram where the pathways are enriched. See supplemental information for complete KEGG enrichment results.
Glycolytic protein and metabolite levels were normalized using feature scaling to examine variation within glycolysis. By comparing normalized protein or metabolite levels in glycolysis we note that variation within the glycolytic pathway occurs en bloc at various levels (Fig. 2). The distribution of variation within glycolytic proteins and metabolites indicates that metabolites are more tightly conserved than proteins (Fig. 2C, 2D). Glycolytic proteins and metabolites each cluster together and display a high number of positive correlations supporting our conclusion that variation in glycolysis occurs en bloc.
Fig. 2.

Relative abundance of glycolytic proteins and glycolytic metabolites is conserved among twin pairs. The average protein (A) and metabolite level (B) in three representative monozygotic twin pairs show variation in glycolytic activity within the population, indicating abundances of these proteins are influenced en bloc. All protein and metabolite levels were normalized to the percentage of maximum protein and metabolite level (see example calculation) using feature scaling. Pyruvate was excluded from the metabolites as it was found to not correlate with the other members. The difference between each protein (C) and metabolite (D) is reported in a histogram. Sequential proteins and metabolites in glycolysis were subtracted to give a scaled difference components of the pathway. The peak is centered at zero for proteins and metabolites indicating that individuals inherit high or low levels of glycolytic compounds together. Glycolytic metabolites appear to be more tightly conserved than protein.
Heritability
Among 18 twin pairs, zygosity testing identified 13 MZ and 5 DZ twin pairs. The means of age, weight, and BMI were not significantly different between MZ and DZ twin groups (Table I). As previously reported, a high degree of estimated heritability for height (96%), weight (97%), and BMI (63%) was observed in this study population (31). The similarity of these results to estimates in a previous report studying 30,111 twin pairs in eight countries supports the validity of the sample population for determination of heritable traits (43).
Table I. Comparison between the mono- and di-zygotic twin populations in this study.
| Trait | Monozygotic (MZ)b | Dizygotic (DZ)b | p valuea |
|---|---|---|---|
| Female pairs | 11 | 2 | |
| Male pairs | 2 | 2 | |
| Male/female pairs | − | 1 | |
| Total pairs | 13 | 5 | |
| Age/Years | 25 ± 7 | 26 ± 9 | 0.7 |
| Weight/kg | 68 ± 14 | 66 ± 8.6 | 0.6 |
| Height/m | 1.68 ± 0.07 | 1.74 ± 0.06 | 0.2 |
| BMI | 24 ± 4.3 | 22 ± 2.7 | 0.11 |
a One way ANOVA DZ versus MZ.
b Mean ± S.E.
Of the proteins and metabolites measured, 119 protein and 148 metabolite concentrations were found to be over 30% heritable, and 73 and 104 were greater than 50% heritable respectively (Fig. 3) (supplemental Table S3). Previous studies using this twin cohort have used 30% heritability as a limit for consideration (14, 14, 31, 32). In particular, we noted that the concentration of the key regulatory enzyme liver type phosphofructokinase (PFK) is 57% heritable as well as the concentration of phosphoglycerate mutase which is 29% heritable. Muscle and platelet PFK isoforms were also detected in our data set, but were not detected in sufficient samples to determine heritability. Additionally, the concentration of bisphosphoglycerate mutase (BPGM), a key enzyme for regulating the oxygen loading capacity of hemoglobin in red blood cells was 50% heritable. No other glycolytic proteins were found to be heritable; however, we were interested to observe that heritable proteins were found at important regulatory steps and branch points in the pathway. The heritability of glycolysis is further supported by high heritability estimates of the metabolites fructose 1,6-bisphosphate, 3-phosphoglycerate, DHAP, 2,3-DPG, phosphoenolpyruvate, and pyruvate. Within glutathione metabolism, GST, GCLC, GPx4, and several hemoglobin subunits were found to be heritable as well as many metabolites including glutamate, cysteinylglycine, GSSG, GSH, and ribose-5-phosphate (Fig. 3).
Fig. 3.

(A) A total of 1,280 proteins and (B) 330 metabolites were detected in red blood cells. Of these, 119 and 148 were found to be over 30% heritable, respectively. To calculate heritability in proteins, measurements were required to be present in all three out of five dizygotic twin pairs and 10 out of 13 dizygotic twin pairs. C, Proteins and metabolites greater than 30% heritable from glycolysis and glutathione metabolism are reported.
High levels of heritability are similarly observed within glutathione metabolism and the pentose phosphate pathway. For example, concentrations of the proteins glutathione peroxidase, glutathione S-transferase, and glutamate cysteine ligase were heritable as well as the metabolite concentrations of ribose-5-phosphate, glutathione, and glutathione disulfide. These heritable metabolite concentrations are in accordance with those detected in previous red blood cell studies (16).
Many of the proteins and metabolites implicated in glutathione metabolism are also correlated and cluster with those discussed previously in glycolysis. This is not surprising, as maintaining redox balance is of key importance to red blood cells and helps preserve sufficient NAD concentration to continue glycolysis. Glycolysis and glutathione metabolism are highly correlated and contain heritable concentrations of proteins and metabolites, implying they are inherited together at varying degrees (Fig. 4).
Fig. 4.

A high number of positive correlations are observed between both proteins and metabolites in the glycolytic and glutathione metabolism pathways. A Pearson correlation greater than 0.5 or less than −0.5 is required to show a connection. Heritability of these pathways can be observed in the shade of the node outline as well as by the gradient outside the network. This figure was created using Cytoscape™ (58).
The best marker for 42 days poststorage ATP concentration in glycolysis and glutathione metabolism is carbonic anhydrase 1 (CA1) which has a −0.56 correlation with ATP. This correlation strengthens with increased time in storage: −0.10 at day 0, −0.22 at day 14, −0.25 at day 28, and −0.56 at day 42. CA1 catalyzes the conversion of CO2 and H2O to produce carbonic acid, which is de-protonated at neutral pH, generating a proton and lowering the pH of stored blood.
The acidification of blood during storage is a well-characterized phenomenon and the resulting decreased pH inhibits PFK. (44) PFK inhibition caused by acidic conditions resulting from CA1 may explain the correlation we observe between higher CA1 concentrations and lower poststorage ATP. The newest blood storage solution, AS-7, buffers blood acidification with the addition of bicarbonate to increase poststorage ATP concentration and in vivo recovery (44–46). Maintaining a high pH during storage is thus imperative for ATP production, and supported by the correlation we have shown between CA1 and poststorage ATP.
Model of Poststorage ATP
An important goal of transfusion medicine is to improve poststorage ATP levels in blood, as RBC ATP concentrations correlate positively with transfused RBC recovery (47, 48). Using our large-scale data set we considered two potential phenotypes of high or low concentrations of ATP at day 42 of storage (Supplemental Fig. 1). The “high” and “low” poststorage ATP phenotypes can be correlated with proteins known to effect ATP concentrations and generate a model to understand poststorage ATP levels. We include five key parameters in this model including PFK, CA1, band 3, BPGM, and pH. Strikingly, concentrations of all protein components of this model were found to be at least 45% heritable. Band 3, BPGM, and CA1 correlate negatively with day 42 poststorage ATP levels (-0.41, −0.39, −0.56) and together may shuttle flux away from glycolysis and ATP production. We also observe positive correlations between pH and ATP early in storage as discussed previously (Fig. 5) which appears to weaken over time. Day 0 ATP correlates positively with pH at day 7, day 14, and day 28 (0.48, 0.80, 0.51) whereas day 42 ATP correlates positively with pH at day 7 and day 14(0.56, 0.57). No positive correlations between pH and ATP are observed at day 42 or 56. However, PFK concentrations correlate positively with pH later in storage at day 42 and 56 (0.45, 0.42) and are associated with increased ATP generation in glycolysis.
Fig. 5.
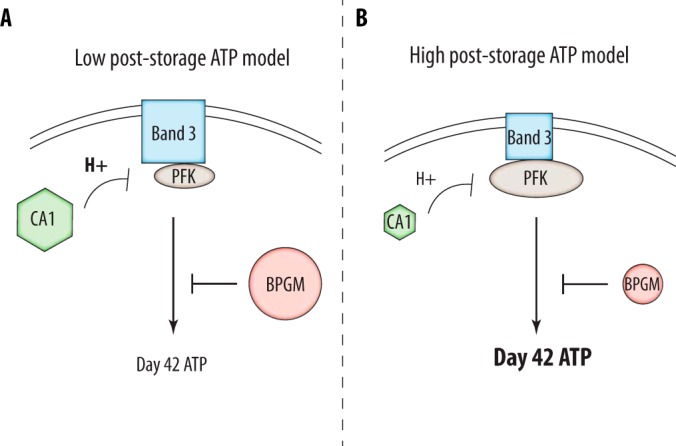
Poststorage ATP levels are determined by several key factors. A, Low ATP levels following 42 days of storage are correlated with high levels of band 3, BPGM, and carbonic anhydrase. Band 3 binds glycolytic proteins decreasing flux through glycolysis whereas BPGM shunts intermediates to the luebering-rapoport pathway away from the generation of ATP. Similarly, high levels of carbonic anhydrase produce acidic conditions and subsequently inhibit PFK. In support of this we observe negative correlations between carbonic anhydrase and pH level during storage. Low poststorage ATP is additionally correlated with low pH. Correlation values of greater than 0.3 were required for consideration. B, The opposite model leads to the generation of high ATP levels following 42 days of storage. The size of the protein in each case is representative of the concentration associated with each phenotype.
DISCUSSION
Our approach to RBC proteome characterization provided an expedient and robust analysis of low abundance RBC membrane proteins, and produced the most thorough analysis of the RBC membrane proteome to-date without the use of affinity based depletion strategies. We expect that further efforts to deplete hemoglobin would result in increased identification of low abundance proteins, but this would also increase processing time per sample. Using a unique data set of twin samples we determined heritability of over 700 proteins and metabolite concentrations.
Within our data set we took particular interest in the interactions between energy and glutathione metabolism. Forty-nine correlations greater than 0.5 are present between the proteins and metabolites of these pathways along with only five negative correlations. The negative correlations included three correlations between pyruvate kinase and other glycolytic proteins, between poststorage ATP and CA1, and between G6P and the delta subunit of hemoglobin. The hemoglobin subunits were included with glutathione metabolism because of their role in oxygen binding and ability to generate superoxide and hydrogen peroxide through hemoglobin (49). Similarly, the anion transport protein band 3 was included with glycolytic proteins and metabolites because of its role binding glycolytic proteins in an oxygen dependent manner (19, 50). We thus propose that glutathione metabolism and glycolysis are highly connected pathways and may be linked by a similar regulatory mechanism.
Because of their role as oxygen carriers and thus the large quantity of oxyhemoglobin, red blood cells have an especially high burden of oxidizing species such as superoxide and hydrogen peroxide. Red blood cells limit the accumulation of these species by maintaining a large pool of reducing equivalents generated from glucose through the pentose phosphate pathway. In the lungs where O2 levels are high, RBCs are exposed to higher levels of oxidative stress necessitating increased flux through the pentose phosphate pathway to generate reducing equivalents and supply the glutathione cycle. Meanwhile, in peripheral tissues with low O2 levels, erythrocytes must pass through narrow capillaries causing distortion from mechanical stress and cation leaks (19). This causes an increased demand for ATP to restore intercellular ion balance. Band 3 specifically binds PFK, GAPDH, and ALDOA in the presence of oxyhemoglobin and diverts flux toward the pentose phosphate pathway to generate NADPH. As we would expect, band 3 correlates negatively with several glycolytic metabolites, including phosphoenolpyruvate and 2,3-DPG (-0.36 and −0.34 respectively). It also positively correlates with several glycolytic proteins, which is compatible with higher concentrations of band 3 being necessary to bind higher levels of glycolytic proteins.
Supporting this model we also observe high levels of correlation between glutathione metabolism and the pentose phosphate pathway. These are intimately linked pathways as the reductive equivalent NADPH generated by the pentose phosphate pathway is necessary to reduce glutathione disulfide generated during removal of hydrogen peroxide. Gluconolactonase is positively correlated with GPx1, GPx4, as well as glutamate cysteine ligase. Also, glutathione is correlated with 6-phosphogluconate, gluconolactonase, and 6-phosphogluconate dehydrogenase demonstrating that the pentose phosphate pathway is essential for the continuation of glutathione metabolism.
Many of the concentrations of proteins and metabolites in these pathways were also found to be heritable. Within glutathione metabolism, glutamate, GPx4, glutamate-cysteine ligase, glutathione, and glutathione disulfide concentrations were all found to be over 45% heritable. Furthermore, in glycolysis, concentrations of the regulatory enzyme PFK as well as BPGM and the metabolites pyruvate, phosphoenolpyruvate, 3-phosphoglycerate, 1,3-bisphosphoglycerate, DHAP, and FDP were all found to be over 50% heritable (Fig. 3). It is also noteworthy that one of the most heritable protein concentrations we observed was carbonic anhydrase, at 85%, which is an important regulator of pH and therefore PFK activity. Based on the strong correlation observed between these pathways, and the high levels of heritably throughout, we infer that glutathione metabolism, pentose phosphate pathway, and glycolysis are coupled pathways that can be inherited en bloc at various levels.
Together our results suggest a model in which inheritance of higher concentrations of band 3 and CA1 reduce flux through the glycolytic pathway by greater binding and inactivation of PFK and by allosteric inhibition of PFK through lower pH. In addition, inheritance of higher concentrations of BPGM may decrease ATP production by competing with PGK for an ATP-producing step in glycolysis. Higher inherited concentrations of PFK may increase flux through the glycolytic pathway. The combined effect of inheritance of these enzyme concentrations accounts both for the en bloc inheritance of glycolytic pathway intermediates, and the heritability of ATP concentration at day 42 and day 28 of storage. The heritable concentrations of many molecules in the pentose phosphate and glutathione pathways may also be predicted by this model.
Our model has implications in the management of blood storage as it confirms that energy metabolism and ATP concentrations are heritable traits. In the future, we can imagine blood donors being tested once for levels of key heritable markers to determine a blood storage profile and optimum storage period. Based on our results, some donors actually show an increase in ATP concentrations early in storage suggesting this blood could theoretically be stored longer than individuals that show a continual decrease in ATP. Individuals that have increasing ATP during storage also show correlations with higher PFK and lower BPGM. This could prevent potential ATP loss by the diversion of 1,3-bisphosphoglyverate to produce 2,3-bisphosphoglycerate. Furthermore, individuals that have decreasing ATP during storage have lower PFK and higher BPGM concentrations. These correlations are suggestive of heritable markers which could someday be used to predict poststorage ATP levels in blood donors.
The negative correlation identified between CA1 and poststorage ATP is significant in that it provides one reasonable explanation for ATP decreases that occur in storage. RBC units are stored in gas permeable bags allowing CO2 to diffuse into the bag causing acidification and inhibiting PFK. This will decrease the natural rate of energy metabolism and subsequently ATP production. Atmospheric conditions have been shown capable of regulating RBC metabolism in storage previously in the case of oxygen saturation (50). Our results suggest that the addition of CA1 inhibitors to stored blood, or the selection of donors known to have low inherited levels of CA1 are potential facile methods to increase the quality and lifetime of stored blood. The importance of pH modulation in blood storage was observed to be of key importance in the development of the newest blood storage solution, additive solution-7, which was primarily improved by increased buffering capacity (45).
RBC blood bank storage is not an activity that occurs in nature, so the strong genetic components suggests that it intersects with a deeper problem in evolutionary biology such as the tradeoff involved in oxygen-based energetics with the risks of oxygen-induced biochemical damage. Keeping RBC glycolytic flux high is known to be advantageous in RBC storage and function and probably represents one of the poles of the deeper tradeoffs in cellular or whole body energetics. The implications for RBC storage are that it is possible to both identify markers to identify individual blood donors with better blood storage or to support identified aspects of metabolism in all donors that make all cells store better.
The heritability we observed in many pathways within erythrocytes may have ramifications in metabolic disease. Diseases such as Alzheimer's disease and cancer are known to involve aberrations of energy metabolism (51–56). Current disease models suggest that a low glycolytic capacity may confer a risk of Alzheimer's disease but protect against cancer. Because we have determined many components of glycolysis to be heritable in erythrocytes, we hypothesize that other cell types are similarly affected, and individuals who inherit low levels of glycolytic proteins and metabolites may be more prone to developing Alzheimer's disease later in life. Similarly, people who inherit high levels of glycolysis may be inclined to develop cancer. Inverse comorbidity has been documented between these diseases, supporting our hypothesis that inheritance of energy metabolism along a spectrum may contribute to the incidence of cancer or Alzheimer's disease (57).
Supplemental Information
All raw files and annotated spectra for single peptide protein identifications from these experiments are available on Chorus (Project ID 1114). Annotated spectra of proteins identified by a single peptide can be viewed on MS Viewer with the key jow7i89mmv.
Supplementary Material
Acknowledgments
We thank Allison Momany, and Dee A. Even, Jessica Nichol, and Jamie L'Heureux (The University of Iowa) for their technical expertise on twin studies and zygosity testing; the Widness lab (The University of Iowa) and the Sysmex Corp. (Kobe, Japan) for the use of the XE-2100 and XT-2000 automated hematology analyzers (P01 HL46925); the staff of The University of Iowa DeGowin Blood Center in recruiting subjects and obtaining the blood samples; and the ESR Facility for invaluable assistance. We also thank Brett A. Wagner, Sean M. Martin, C. Michael Knudson, Robyn Blendowski, Gary R. Buettner, Kelli K. Ryckman, Benjamin W. Darbro, Jeffrey C. Murray, and Emery Bresnick for acquisition of data and thoughtful conversation.
Footnotes
Author contributions: E.M.W., T.J.v., T.J.R., and J.J.C. designed research; E.M.W. and T.J.v. performed research; E.M.W. and T.J.v. analyzed data; E.M.W., T.J.v., N.M.R., and J.R.H. wrote the paper.
* This work was supported by the National Center for Advancing Translational Sciences, through Grant 2UL1TR000442, and National Institutes of Health Grants P41 GM108538, P42 ES013661. TJvE thanks The University of Iowa Graduate College for support. NMR gratefully acknowledges support from a National Science Foundation Graduate Research Fellowship (DGE-1256259).
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- RBC
- red blood cells 2,3-DPG, 2,3-disphosphoglycerate
- 3PG
- 3-phosphoglycerate
- 6PGD
- 6-phosphogluconate dehydrogenase
- ADP
- adenosine diphosphate
- ALDOA
- fructose bisphosphate aldolase
- AMP
- adenosine monophosphate
- ATP
- adenosine triphosphate
- BPGM
- bisphosphoglycerate mutase
- CA1
- carbonic anhydrase
- DHAP
- dihydroxyacetone phosphate
- ENO1
- Alpha-enolase
- ESI
- electrospray ionization
- FDP
- fructose diphosphate
- GAPDH
- glyceraldehyde 3-phosphate dehydrogenase
- G6P
- glucose 6 phosphate
- GCLC
- glutamate-cysteine ligase
- GPx1
- glutathione peroxidase 1
- GPx4
- glutathione peroxidase 4
- GPI
- glucose phosphate isomerase
- GSH
- glutathione reduced
- GSSG
- glutathione oxidized
- GST
- glutathione S transferase
- LDH
- lactate dehydrogenase
- MS
- mass spectrometry
- MS/MS
- tandem mass spectrometry
- nLC-MS/MS
- nanoflow liquid chromatography-tandem mass spectrometry
- PEP
- phosphoenolpyruvate
- PFK
- phosphofructokinase
- PGAM1
- phosphoglycerate mutase
- PGK1
- phosphoglycerate kinase
- PKLR
- pyruvate kinase
- PGM2
- phosphoglucomutase-2.
REFERENCES
- 1. Hod E. A., and Spitalnik S. L. (2012) Stored red blood cell transfusions: Iron, inflammation, immunity, and infection. Transfus. Clin. Biol. 19, 84–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alexander J. T., El-Ali A. M., Newman J. L., Karatela S., Predmore B. L., Lefer D. J., Sutliff R. L., and Roback J. D. (2013) Red blood cells stored for increasing periods produce progressive impairments in nitric oxide–mediated vasodilation. Transfusion 53, 2619–2628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Offner P. J., Moore E. E., Biffl W. L., Johnson J. L., and Silliman C. C. (2002) INcreased rate of infection associated with transfusion of old blood after severe injury. Arch. Surg. 137, 711–717 [DOI] [PubMed] [Google Scholar]
- 4. Sparrow R. L. (2015) Red Blood Cell Storage Duration and Trauma. Transfus. Med. Rev. 29, 120–126 [DOI] [PubMed] [Google Scholar]
- 5. Zallen G., Offner P. J., Moore E. E., Blackwell J., Ciesla D. J., Gabriel J., Denny C., and Silliman C. C. (1999) Age of transfused blood is an independent risk factor for postinjury multiple organ failure. Am. J. Surg. 178, 570–572 [DOI] [PubMed] [Google Scholar]
- 6. Mary Keller Jean Raymond Wayne LaMorte Frederick Millham and Erwin Hirsch. (2002) Effects of age of transfused blood on length of stay in trauma patients: a preliminary report. J. Trauma Inj. Infect. Crit. Care 53, 1023–1025 [DOI] [PubMed] [Google Scholar]
- 7. Hess J. R. (2010) Red cell changes during storage. Transfus. Apher. Sci. 43, 51–59 [DOI] [PubMed] [Google Scholar]
- 8. Hess J. R., and for the Biomedical Excellence for Safer Transfusion (BEST) Collaborative. (2012) Scientific problems in the regulation of red blood cell products. Transfusion 52, 1827–1835 [DOI] [PubMed] [Google Scholar]
- 9. Reid T. J., Babcock J. G., Derse-Anthony C. P., Hill H. R., Lippert L. E., and and Hess J. R. (1999) The viability of autologous human red cells stored in additive solution 5 and exposed to 25°C for 24 hours. Transfusion 39, 991–997 [DOI] [PubMed] [Google Scholar]
- 10. Hess J. R. (2014) Measures of stored red blood cell quality. Vox Sang. 107, 1–9 [DOI] [PubMed] [Google Scholar]
- 11. Luten M., Roerdinkholder-Stoelwinder B., Schaap N. P. M., De Grip W. J., Bos H. J., Bosman G. J. C. G. M. (2008) Survival of red blood cells after transfusion: a comparison between red cells concentrates of different storage periods. Transfusion 48, 1478–1485 [DOI] [PubMed] [Google Scholar]
- 12. Nakao K., Wada T., Kamiyama T., Nakao M., and Nagano K. (1962) A direct relationship between adenosine triphosphate-level and in vivo viability of erythrocytes. Nature 194, 877–878 [DOI] [PubMed] [Google Scholar]
- 13. Gilroy T. E., Brewer G. J., and Sing C. F. (1980) Genetic control of glycolysis in human erythrocytes. Genetics 94, 719–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. van ′t Erve T. J., Wagner B. A., Martin S. M., Knudson C. M., Blendowski R., Keaton M., Holt T., Hess J. R., Buettner G. R., Ryckman K. K., Darbro B. W., Murray J. C., and Raife T. J. (2014) The heritability of metabolite concentrations in stored human red blood cells. Transfusion 54, 2055–2063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chakraborty S., and Chaudhuri A. B. D. (2001) Heritability of Some Important Parameters of the Antioxidant Defense System Like Glucose-6-Phosphate Dehydrogenase, Catalase, Glutathione Peroxidase and Lipid Peroxidation in Red Blood Cells by Twin Study. 1, 1–4 [Google Scholar]
- 16. van ′t Erve T. J., Doskey C. M., Wagner B. A., Hess J. R., Darbro B. W., Ryckman K. K., Murray J. C., Raife T. J., and and Buettner G. R. (2014) Heritability of glutathione and related metabolites in stored red blood cells. Free Radic. Biol. Med. 76, 107–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mohandas N., and Gallagher P. G. (2008) Red cell membrane: past, present, and future. Blood 112, 3939–3948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Almizraq R., Tchir J. D. R., Holovati J. L., and Acker J. P. (2013) Storage of red blood cells affects membrane composition, microvesiculation, and in vitro quality. Transfusion 53, 2258–2267 [DOI] [PubMed] [Google Scholar]
- 19. Lewis I. A., Campanella M. E., Markley J. L., and Low P. S. (2009) Role of band 3 in regulating metabolic flux of red blood cells. Proc. Natl. Acad. Sci. U.S.A. 106, 18515–18520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pasini E. M., Kirkegaard M., Salerno D., Mortensen P., Mann M., and Thomas A. W. (2008) Deep coverage mouse red blood cell proteome a first comparison with the human red blood cell. Mol. Cell. Proteomics 7, 1317–1330 [DOI] [PubMed] [Google Scholar]
- 21. Pasini E. M., Lutz H. U., Mann M., and Thomas A. W. (2010) Red blood cell (RBC) membrane proteomics — Part II: Comparative proteomics and RBC patho-physiology. J. Proteomics 73, 421–435 [DOI] [PubMed] [Google Scholar]
- 22. Anderson N. L., and Anderson N. G. (2002) The human plasma proteome: history, character, and diagnostic prospects. Mol. Cell. Proteomics MCP 1, 845–867 [DOI] [PubMed] [Google Scholar]
- 23. Barasa B., and Slijper M. (2014) Challenges for red blood cell biomarker discovery through proteomics. Biochim. Biophys. Acta 1844, 1003–1010 [DOI] [PubMed] [Google Scholar]
- 24. Zubarev R. A. (2013) The challenge of the proteome dynamic range and its implications for in-depth proteomics. Proteomics 13, 723–726 [DOI] [PubMed] [Google Scholar]
- 25. Goodman S. R., Daescu O., Kakhniashvili D. G., and Zivanic M. (2013) The proteomics and interactomics of human erythrocytes. Exp. Biol. Med. 238, 509–518 [DOI] [PubMed] [Google Scholar]
- 26. Ringrose J. H., van Solinge W. W., Mohammed S., O'Flaherty M. C., van Wijk R., Heck A. J. R., and Slijper M. (2008) Highly efficient depletion strategy for the two most abundant erythrocyte soluble proteins improves proteome coverage dramatically. J. Proteome Res. 7, 3060–3063 [DOI] [PubMed] [Google Scholar]
- 27. D'Amici G. M., Rinalducci S., and Zolla L. (2012) Depletion of hemoglobin and carbonic anhydrase from erythrocyte cytosolic samples by preparative clear native electrophoresis. Nat. Protoc. 7, 36–44 [DOI] [PubMed] [Google Scholar]
- 28. Walpurgis K., Kohler M., Thomas A., Wenzel F., Geyer H., Schänzer W., and Thevis M. (2012) Validated hemoglobin-depletion approach for red blood cell lysate proteome analysis by means of 2D PAGE and Orbitrap MS. Electrophoresis 33, 2537–2545 [DOI] [PubMed] [Google Scholar]
- 29. Roux-Dalvai F., Peredo A. G. de, Simó C., Guerrier L., Bouyssié D., Zanella A., Citterio A., Burlet-Schiltz O., Boschetti E., Righetti P. G., and Monsarrat B. (2008) Extensive analysis of the cytoplasmic proteome of human erythrocytes using the peptide ligand library technology and advanced mass spectrometry. Mol. Cell. Proteomics 7, 2254–2269 [DOI] [PubMed] [Google Scholar]
- 30. Riley N. M., Hebert A. S., and Coon J. J. (2016) Proteomics moves into the fast lane. Cell Syst. 2, 142–143 [DOI] [PubMed] [Google Scholar]
- 31. van ′t Erve T. J., Wagner B. A., Ryckman K. K., Raife T. J., and and Buettner G. R. (2013) The concentration of glutathione in human erythrocytes is a heritable trait. Free Radic. Biol. Med. 65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Van ′t Erve T. J., Wagner B. A., Martin S. M., Knudson C. M., Blendowski R., Keaton M., Holt T., Hess J. R., Buettner G. R., Ryckman K. K., Darbro B. W., Murray J. C., and and Raife T. J. (2015) The heritability of hemolysis in stored human red blood cells. Transfusion 55, 1178–1185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hebert A. S., Richards A. L., Bailey D. J., Ulbrich A., Coughlin E. E., Westphall M. S., and Coon J. J. (2014) The one hour yeast proteome. Mol. Cell. Proteomics MCP 13, 339–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Richards A. L., Hebert A. S., Ulbrich A., Bailey D. J., Coughlin E. E., Westphall M. S., and Coon J. J. (2015) One-hour proteome analysis in yeast. Nat. Protoc. 10, 701–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cox J., and Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 36. Cox J., Neuhauser N., Michalski A., Scheltema R. A., Olsen J. V., and Mann M. (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 [DOI] [PubMed] [Google Scholar]
- 37. Cox J., and Mann M. (2012) 1D and 2D annotation enrichment: a statistical method integrating quantitative proteomics with complementary high-throughput data. BMC Bioinformatics 13, S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tyanova S., Temu T., Sinitcyn P., Carlson A., Hein M. Y., Geiger T., Mann M., and Cox J. (2016) The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods advance online publication [DOI] [PubMed] [Google Scholar]
- 39. Roberts J. A. F. (1938) Twins: a study of heredity and environment. Eugen. Rev. 30, 61–62 [Google Scholar]
- 40. Pesciotta E. N., Sriswasdi S., Tang H.-Y., Mason P. J., Bessler M., and Speicher D. W. (2012) A label-free proteome analysis strategy for identifying quantitative changes in erythrocyte membranes induced by red cell disorders. J. Proteomics 76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Campanella M. E., Chu H., and Low P. S. (2005) Assembly and regulation of a glycolytic enzyme complex on the human erythrocyte membrane. Proc. Natl. Acad. Sci. U.S.A. 102, 2402–2407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen E. Y., Tan C. M., Kou Y., Duan Q., Wang Z., Meirelles G. V., Clark N. R., and Ma'ayan A. (2013) Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Silventoinen K., Sammalisto S., Perola M., Boomsma D. I., Cornes B. K., Davis C., Dunkel L., De Lange M., Harris J. R., Hjelmborg J. V. B., Luciano M., Martin N. G., Mortensen J., Nisticò L., Pedersen N. L., Skytthe A., Spector T. D., Stazi M. A., Willemsen G., and Kaprio J. (2003) Heritability of adult body height: a comparative study of twin cohorts in eight countries. Twin Res. Off. J. Int. Soc. Twin Stud. 6, 399–408 [DOI] [PubMed] [Google Scholar]
- 44. Hess J. R., Rugg N., Knapp A. D., Gormas J. F., Hill H. R., Oliver C. K., Lippert L. E., and Greenwalt T. J. (2001) The role of electrolytes and pH in RBC ASs. Transfusion 41, 1045–1051 [DOI] [PubMed] [Google Scholar]
- 45. Cancelas J. A., Dumont L. J., Maes L. A., Rugg N., Herschel L., Whitley P. H., Szczepiokowski Z. M., Siegel A. H., Hess J. R., and Zia M. (2015) Additive solution-7 reduces the red blood cell cold storage lesion. Transfusion 55, 491–498 [DOI] [PubMed] [Google Scholar]
- 46. Hess J. R., Rugg N., Joines A. D., Gormas J. F., Pratt P. G., Silberstein E. B., and and Greenwalt T. J. (2006) Buffering and dilution in red blood cell storage. Transfusion 46, 50–54 [DOI] [PubMed] [Google Scholar]
- 47. Dern R. J., and John JWiorkowski. (1969) Studies on the preservation of human blood. IV. The hereditary component of pre- and poststorage erythrocyte adenosine triphosphate levels. J. Lab. Clin. Med. 73, 1019–1029 [PubMed] [Google Scholar]
- 48. Hess J. R. (2006) An update on solutions for red cell storage. Vox Sang. 91, 13–19 [DOI] [PubMed] [Google Scholar]
- 49. Johnson R. M., Goyette G. Jr., Ravindranath Y., and Ho Y.-S. (2005) Hemoglobin autoxidation and regulation of endogenous H2O2 levels in erythrocytes. Free Radic. Biol. Med. 39, 1407–1417 [DOI] [PubMed] [Google Scholar]
- 50. Harrison M. L., Rathinavelu P., Arese P., Geahlen R. L., and Low P. S. (1991) Role of band 3 tyrosine phosphorylation in the regulation of erythrocyte glycolysis. J. Biol. Chem. 266, 4106–4111 [PubMed] [Google Scholar]
- 51. Reitz C. (2015) Genetic diagnosis and prognosis of Alzheimer's disease: challenges and opportunities. Expert Rev. Mol. Diagn. 15, 339–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Demetrius L. A., and Simon D. K. (2012) An inverse-Warburg effect and the origin of Alzheimer's disease. Biogerontology 13, 583–594 [DOI] [PubMed] [Google Scholar]
- 53. Demetrius L. A., Magistretti P. J., and Pellerin L. (2015) Alzheimer's disease: the amyloid hypothesis and the Inverse Warburg effect. Front. Physiol. 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hsu P. P., and Sabatini D. M. (2008) Cancer cell metabolism: Warburg and beyond. Cell 134, 703–707 [DOI] [PubMed] [Google Scholar]
- 55. Palsson-McDermott E. M., and O'Neill L. A. J. (2013) The Warburg effect then and now: From cancer to inflammatory diseases. BioEssays 35, 965–973 [DOI] [PubMed] [Google Scholar]
- 56. Demetrius L. A., and Driver J. (2013) Alzheimer's as a metabolic disease. Biogerontology 14, 641–649 [DOI] [PubMed] [Google Scholar]
- 57. Driver J. A. (2014) Inverse association between cancer and neurodegenerative disease: review of the epidemiologic and biological evidence. Biogerontology 15, 547–557 [DOI] [PubMed] [Google Scholar]
- 58. Shannon P., Markiel A., Ozier O., Baliga N. S., Wang J. T., Ramage D., Amin N., Schwikowski B., and Ideker T. (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.