

Abstract
Current proteomic approaches include both broad discovery measurements and quantitative targeted analyses. In many cases, discovery measurements are initially used to identify potentially important proteins (e.g. candidate biomarkers) and then targeted studies are employed to quantify a limited number of selected proteins. Both approaches, however, suffer from limitations. Discovery measurements aim to sample the whole proteome but have lower sensitivity, accuracy, and quantitation precision than targeted approaches, whereas targeted measurements are significantly more sensitive but only sample a limited portion of the proteome. Herein, we describe a new approach that performs both discovery and targeted monitoring (DTM) in a single analysis by combining liquid chromatography, ion mobility spectrometry and mass spectrometry (LC-IMS-MS). In DTM, heavy labeled target peptides are spiked into tryptic digests and both the labeled and unlabeled peptides are detected using LC-IMS-MS instrumentation. Compared with the broad LC-MS discovery measurements, DTM yields greater peptide/protein coverage and detects lower abundance species. DTM also achieved detection limits similar to selected reaction monitoring (SRM) indicating its potential for combined high quality discovery and targeted analyses, which is a significant step toward the convergence of discovery and targeted approaches.
The application of both discovery and targeted proteomics approaches is increasingly important across many areas of biological research, such as elucidating molecular mechanism of disease progression and increasing the utility of clinical applications to facilitate early detection and prognostic classification (1, 2). Currently discovery and targeted approaches are performed separately, limiting sample throughput and requiring more material for thorough analyses. In the prevalent discovery-based shotgun approach (3–6), proteins are digested into peptides, separated by liquid chromatography (LC)1, and detected by mass spectrometry (MS), where specific peptides are further selected (typically by abundance) for successive tandem MS/MS for identification. The resulting spectra are then mapped to peptide or protein sequences using highly-evolved database search algorithms with results normally obtained for thousands of unique proteins. However, the large numbers of coeluting peptides and use of MS/MS inevitably limit proteomic measurement effectiveness by at least one of three key metrics: throughput, sensitivity, and proteome coverage. To increase proteome coverage additional fractionation steps and/or extended LC separations are often applied. Although this is advantageous for increasing the comprehensiveness and sensitivity of measurements, the reduced throughput greatly restricts the ability to account for both measurement and biological variability. Additionally, these broad discovery measurements still suffer deficiencies in the detection and precise quantification of low abundance proteins, such as transcription factors, signaling intermediaries or novel protein products of genomic alterations.
The limitations of discovery approaches have driven the development of targeted proteomics approaches such as selected reaction monitoring (SRM, also known as MRM - multiple reaction monitoring) (7–11) and parallel reaction monitoring (PRM) (12, 13). SRM is typically performed with a triple quadrupole mass spectrometer and provides significantly improved reproducibility, sensitivity and quantitation accuracy for targeted species compared with discovery measurements. In SRM, the peptide of interest is selected, fragmented, and typically a few fragment transitions are monitored individually and combined for quantitation (Fig. 1A). Numerous studies have successfully employed SRM measurements for both absolute and relative quantitation of peptide analytes in applications ranging from clinical diagnostics to fundamental studies of biological systems. SRM measurements however are time consuming to develop and can require significant effort to determine the best transitions and measurement parameters for each protein of interest (7–11). These limitations have prompted more interest in PRM where the third quadrupole of a triple quadrupole analyzer is substituted with either a quadrupole time-of-flight (QTOF) or Orbitrap mass analyzer for detection of the precursor and all fragment ions simultaneously and with high mass resolution (Fig. 1B). In addition to reducing the upfront method development time and facilitating automated data analysis, PRM has yielded quantitative dynamic range and linearity that approaches SRM, but often with better selectivity because of the high resolution mass analyzer and greater number of transitions monitored (12). However, challenges remain for both SRM and PRM, including the selection of effective dissociation conditions and especially the limitation on the number of target proteins studied, which essentially removes all other peptides/proteins from possible future data analyses.
Fig. 1.

A schematic representation of the SRM (A), PRM (B), and DTM (C) measurement approaches. In SRM, each product ion transition (fragment) is monitored individually (normally 1 to 5) and then combined for quantitation. In PRM, all product ion transitions and possible product ions are analyzed in concert with high resolution and mass accuracy. DTM allows all product ions to be analyzed concurrently with the heavy and light pairs having the same LC elution times and IMS drift times. High mass accuracy is also possible from the QTOF for ease of peptide identification. The light and heavy pairs are circled in drift cell and on the 2D IMS-MS nested spectrum. Q1 and Q3 refer to the first and third mass-resolving quadrupoles and Q2 to the quadrupole (or cell) where fragmentation is performed.
Ongoing technology developments in targeted and discovery-based approaches continue to provide improved proteomic measurements, but still remain far from a true convergence of the benefits of both. To this end, we have coupled an ion mobility spectrometry (IMS) separations (14, 15) between LC and MS stages (LC-IMS-MS) and explored the potential of this multi-dimensional platform for combining the benefits of both discovery and targeted approaches. The added IMS stage has the attraction of very fast structural separations (milliseconds) and high reproducibility of separation times (<0.5% drift time deviation). We have previously shown that LC-IMS-MS measurements can provide better coverage than other discovery-based LC-MS platforms, increasing the overall measurement peak capacity and thus providing the enhanced selectivity needed to enable more confident identifications (16–18). Utilizing this capability LC-IMS-MS measurements offer great potential for sufficient selectivity, enabling broad and sensitive peptide identifications (16–20). Further, light and heavy labeled isotope pairs have the same LC elution time and IMS drift time, so they can easily be added to samples to both provide more confident identifications as well as for more precise quantification by the QTOF analyzer (21–23). Because the LC-IMS-MS platform is already analyzing highly complex samples (with tens to hundreds of thousands of peptides), the addition of hundreds to thousands of additional heavy labeled peptides does not significantly impact analyses. Herein, we explore the potential of simultaneous proteomic discovery and targeted monitoring (DTM) using the LC-IMS-MS platform (Fig. 1C) in the context of a well characterized patient-derived xenograft (PDX) breast cancer tumor sample as an example of the possible future applications of the platform. We further investigated the potential of the sensitive DTM approach to both broaden discovery efforts as well as improve quantification of targeted proteins of interest by adding heavy labeled peptides into the sample.
EXPERIMENTAL PROCEDURES
Patient-derived Xenograft Tumor Selection and Sample Processing
The PDX tumor sample explored in this works was from the established luminal B (WHIM16) breast cancer subtype raised subcutaneously in an 8-week-old NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ mouse (Jackson Labs, Bar Harbor, Maine) (24). The tumor was harvested by surgical excision at ∼1.5 cm3 with minimal ischemia time because of immediate immersion in a liquid nitrogen bath. Histopathological review classified these PDX tumors as ER positive, PR positive, and HER2 negative. The tumor tissue was then placed in precooled tubes on dry ice and stored at −80 °C until cryopulverized in a Covaris CP02 Cryoprep device. Powdered tissue was decanted to a precooled aluminum weigh dish (VWR #1131–436; VWR Laboratory Supplies, Radnor, PA) and the tissue was thoroughly mixed with a metal spatula precooled in liquid nitrogen before partitioning ∼100 mg aliquots into cryovials (Corning #430487; Corning Inc., Corning, NY). All procedures were carried out on dry ice to ensure thawing of the tissue did not occur.
Approximately 100 mg of the frozen, pulverized PDX tissue was homogenized in 600 μl of ice cold lysis buffer (8 m Urea, 0.1% Nonidet P-40, 0.5% Sodium deoxycholate, 10 mm NaF, SIGMA phosphatase inhibitor mixture 2 (Sigma, P 5726; Sigma Aldrich, St. Louis, MO) and SIGMA phosphatase inhibitor mixture 3 (Sigma, P 0044) in 100 mm ammonium bicarbonate, pH 8), shaken in a Thermomixer R for 3 min at 1200 rpm, followed by 3 min sonication in a water bath with ice. The lysate was precleared by centrifugation at 16,500 × g for 5 min at 4 °C and protein concentrations were determined by BCA assay (Pierce; Thermo Fisher Scientific, San Jose, CA). Following the assay, proteins were reduced with 5 mm dithiothreitol for 1 h at 37 °C, and subsequently alkylated with 10 mm iodoacetamide for 1 h at RT in the dark with constant shaking at 1200 rpm in Thermomixer R. The sample was diluted by 1:2 with 50 mm NH4HCO3, 1 mm CaCl2 and digested with sequencing grade modified trypsin protease (Promega, V5113 (Promega, Madison, WI)) at 1:50 enzyme-to-substrate ratio. After 4 h of digestion at 37 °C, the sample was further diluted by 1:4 with the same buffers and another aliquot of trypsin was for further incubated at RT overnight (∼16 h). The digested sample was then acidified with 10% trifluoroacetic acid to ∼pH 2 and transferred into a fresh vial after 20 min at 4000 × g centrifugation in a Sorval centrifuge. The digested sample was desalted using a four-probe positive pressure Gilson GX-274 ASPEC™ system (Gilson Inc., Middleton, WI), first with a Discovery SCX 100 mg/1 ml solid phase extraction tube (Supelco, St. Louis, MO) and second with a Discovery C18 100 mg/1 ml solid phase extraction tube (Supelco). Sample elution from the C18 column used 2 ml 80% ACN and then concentrated down to 200 μl in a SpeedVac SC250 Express (ThermoSavant) before a final BCA to determine the peptide concentration. 800 μg of the sample was used for RP high pH HPLC fractionation. The sample was diluted to a volume of 900 μl with 10 mm ammonium formate buffer (pH 10.0), and resolved on a XBridge C18 250 × 4.6 mm, 5 μm with 4.6 × 20 mm guard column (Waters, Milford, MA) using an Agilent 1200 series HPLC system (Agilent Technologies, Santa Clara, CA) equipped with a quaternary pump, degasser, diode array detector, peltier-cooled autosampler, and fraction collector (set at 4 °C). The mobile phases used were (A) 10 mm Ammonium Formate, pH 10.0 and (B) 10 mm Ammonium Formate, pH 10.0/acetonitrile (10:90). After sample loading and an initial 35 min wash with mobile phase A, the gradient applied was (min:%B); 0:0, 10:5, 70:35, 85:70, 95:70, 105:0, 120:0. Fractions were collected every 65 s from 15 min to 120 min of the gradient, resulting in 96 fractions. The plate was dried in a SpeedVac and each fraction was reconstituted in 100 μl of 50% ACN, 0.1% TFA before being combined into a total of 48 samples. The fractions were then completely dried down again and reconstituted in 50 μl of nanopure water and final peptide concentrations for each fraction were determined by BCA assay. Fractions were then divided into two aliquots for the Q Exactive and IMS-MS instrumental analyses.
Heavy Labeled Peptides
Target heavy labeled peptide standards were purchased from Thermo Fisher Scientific (San Jose, CA) and the last residue of the sequence (Arg or Lys) was modified as either 13C6 and 15N4 or 13C6 and 15N2.
Experimental Design and Statistical Rational: LC-MS and LC-IMS-MS Analyses
Analyses were performed using Thermo Fisher Scientific Q Exactive and Orbitrap Velos instruments, as well as an in-house built IMS-MS instrument that couples a 1-m IMS drift cell with an Agilent 6538 QTOF MS (Agilent Technologies, Santa Clara, CA), providing low ppm mass error and resolution of ∼38,000 (25, 26).
The LC system used for the experiments was custom built using two Agilent 1200 nanoflow pumps and one Agilent 1200 cap pump (Agilent Technologies, Santa Clara, CA), Valco valves (Valco Instruments Co., Houston, TX), and a PAL autosampler (Leap Technologies, Carrboro, NC) (16). Full automation was provided by custom software that allows parallel event coordination and therefore near 100% MS duty cycle through use of two trapping and analytical columns. RP columns were slurry packed in-house with 3 μm Jupiter C18 (Phenomenex, Torrence, CA) into fused silica columns that were 40 cm long × 360 μm o.d. × 75 μm i.d. (Polymicro Technologies Inc., Phoenix, AZ) with a 1-cm sol-gel frit at the end for media retention. Trapping columns were prepared similarly by slurry packing 5-μm Jupiter C18 into fused silica columns that were 4-cm length × 360 μm o.d. × 150 μm i.d. and fritted on both ends. Mobile phases consisted of 0.1% formic acid in water (A) and 0.1% formic acid in acetonitrile (B) and a 100-min gradient profile operated at 300 nL/min flow rate as follows (min:%B); 0:5, 2:8, 20:12, 75:35, 97:60, 100:85. Sample injections of 5 μl were trapped and washed onto the trapping columns at 3 μl/min for 20 min prior to alignment with the analytical columns. Data acquisition lagged the gradient start and end times by 15 min to account for column dead volume allowing for the highest throughput when running in two-column operation mode, so that the columns could to be washed (shortened gradients) and re-generated off-line without any cost to the duty cycle.
The Q Exactive analyses used 2.3 kV applied at the liquid junction for ESI and the MS inlet was maintained at a temperature of 325 °C. Data was collected in data dependent analysis mode and a primary survey scan was performed in the m/z range of 400 to 2000 at a resolution of 70,000 (at m/z 200) and an automatic gain control (AGC) setting of 1 × 106 charges. The top 10 highest intensity ions from the survey scan were selected by a quadrupole mass filter for high energy collision dissociation (HCD) with nitrogen and then mass analyzed by the Orbitrap at a resolution of 17,500. A window of 2 m/z was used for HCD isolation and a normalized collision energy of 30% (a Thermo nomenclature for ion energy for dissociating ions by collision) was applied with an AGC setting of 1 × 105 charges. Mass spectra were recorded for 100 min by repeating this process with a dynamic exclusion of previously selected ions for 30 s. The Velos MS data were collected from 400–2000 m/z at a resolution of 60,000 (AGC: 1 × 106 charges) followed by data dependent ion trap MS/MS spectra (AGC: 1 × 104 charges) of the 10 most abundant ions using a collision energy setting of 35%. A dynamic exclusion time of 60 s was used to discriminate against previously analyzed ions. IMS-MS data were collected from 100–3200 m/z with a cycle time of 3 s/spectra to increase the signal of low abundance species. Ion dissociation (i.e. MS/MS) was not performed in these analyses, and the peptide precursor detected features were compared with an accurate mass and time (AMT) tag databases for identification. We note that MS/MS analysis in both data dependent or independent acquisition mode is feasible with this platform, if desired, but at the cost of decreased spectrum acquisition rate and/or detection sensitivity depending upon experimental details.
Identification and quantification of the detected peptide peaks were performed using the AMT tag approach (17, 27, 28). Both Q Exactive and IMS-MS analyses were utilized to populate the database with LC elution times, IMS drift times and accurate mass information for each peptide tag (Fig. 2A). Briefly, Q Exactive MS/MS spectra from the 48 high-pH fractions were searched by MS-GF+, v10072 (6/30/2014) (29), filtered to a false discovery rate (based on MS-GF+ decoy searches) of ≤0.01%, and an observation count ≥2 (the number of MS/MS spectra for a peptide across all data sets). The NCBI's Homo sapiens database (build 37, released December 2, 2011) was combined with NCBI's Mus musculus database (build 37, released December 2, 2011) for a total of 62,433 proteins. The protease searched was trypsin with no limit of missed cleavages, but a maximum peptide length of 50 residues; and fixed alkylation of cysteine (carbamidomethyl) and variable oxidation of methionine modifications were considered. Finally, mass tolerances and parameters for precursor and fragment ions were ± 20 ppm precursor tolerance with a MS-GF+ high resolution and HCD scoring model.
Fig. 2.

The workflow of experiments performed in this manuscript to A, create the AMT tag database for the PDX tumor sample and B, perform discovery-based, DTM and SRM instrumental analyses.
The 48 fractions were then analyzed with the LC-IMS-MS platform to determine IMS drift times for each database feature. Peptides observed in each instrument were compared, and only those agreeing with high mass accuracy (within 2 ppm) and LC elution time (<0.5%) were populated with IMS drift times in the PDX tumor database used in this work (28). The LC-MS and LC-IMS-MS raw data were processed using in-house developed informatics tools (publicly available at https://omics.pnl.gov/software) with algorithms for peak-picking and determining isotopic distributions and charge states for feature definition (30). The detected features were then correlated (i.e. aligned) with the PDX tumor AMT tag database (28). Further downstream data analysis incorporated all possible detected peptides into the VIPER visualization program to associate LC-MS features and the peptide identifications in the AMT tag database (31). VIPER provided an intensity report for all detected features, normalized LC elution times via alignment to the database, and performed feature identification. Although Skyline software (32) was not used in the LC-IMS-MS data analyses in this manuscript, we note that its recent ability to analyze data sets having an IMS dimension will allow its use in future analyses.
SRM Assay Development and Analyses
The SRM analyses used a nanoACQUITY UPLC® system (Waters Corporation) coupled online to a TSQ Vantage triple quadrupole mass spectrometer (Thermo Fisher Scientific). The UPLC® system was equipped with an ACQUITY UPLC BEH 1.7 μm C18 column (100 μm i.d. × 10 cm) and the mobile phases were (A) 0.1% formic acid in water and (B) 0.1% formic acid in 90% acetonitrile. Four microliters of sample were loaded onto the column and separated at a flow rate of 500 nL/min using a 50-min gradient profile as follows (min:%B); 0:0.5, 11:0.5, 13.5:10, 17:15, 38:25, 49:38.5, 50:95. The TSQ Vantage triple quadrupole mass spectrometer was operated with ion spray voltages of 2400 ± 100 V, a capillary offset voltage of 35 V, a skimmer offset voltage of 5 V, and a capillary inlet temperature of 220 °C. Tube lens voltages were obtained from automatic tuning and calibration without further optimization. Both Q1 and Q3 were set at unit resolution of 0.7 FWHM and Q2 gas pressure was optimized at 1.5 mTorr.
Twenty proteotypic peptides were selected for targeted analyses because of their biological activities and concentration levels of interest. For each peptide, six to eight transitions and corresponding optimal collision energy (CE) values were obtained from direct infusion experiments. These SRM transitions were further validated for optimal detection of the target peptides in the PDX sample matrix using LC-SRM analyses. The relative intensity ratios for each given transition were used to assess potential interferences as previously reported (33) and three optimal transitions per peptide were used for the final SRM assay. A reverse calibration curve was built by spiking different concentration of heavy peptides into the same PDX sample matrix (0.125, 0.25, 0.5, 1, 2.5, 5, 5, and 7.5 nm) and compared with light signals for ratio calculation. For endogenous peptide quantification, the sample was prepared with 0.5 nm heavy peptides causing the final peptide matrix concentration to be 0.1875 μg/μl. LC-SRM analyses were performed in triplicate using a scheduled mode with 10 min windows to analyze all peptides and a duty cycle of 1 s.
Skyline software was used to analyze the SRM data (32). The raw data were initially imported into Skyline software for visualization of target peptide chromatograms and to determine which were detected. The best transition for each peptide was used for quantification. Two criteria were used to determine the peak detection and integration: (1) same LC retention time and (2) approximately the same relative SRM peak intensity ratios across multiple transitions between the light peptides and heavy peptide standards. Standard deviation (S.D.) and coefficient of variation (CV) were obtained from three replicates for each sample. All data were manually inspected to ensure correct peak detection and accurate integration. Signal to noise ratios (S/N) were calculated by the peak apex intensity over the highest background noise in a retention time region of ±15s for the target peptides and for surrogate endogenous peptides a S/N of >9 was required for confident quantification. The endogenous level of each peptide in the tissue sample (fmol/μg of total protein) was calculated by the following equation: (fmol/μl concentration of endogenous target peptide calculated based on calibration curve)/(μg/μl digested sample concentration loading on column).
Repository
All data has been submitted to ProteomeXchange (accession PXD004569) and the CPTAC Data Portal.
RESULTS
Discovery-based measurements often use sample fractionation to increase proteome coverage, however, this effectively turns each sample into many samples, significantly lowering analysis throughput and adding challenges for quantitation. Our goal in this work was to increase sample throughput of discovery-based measurements while also achieving high proteomic coverage and providing absolute quantitation for targeted proteins with DTM. DTM was compared with conventional discovery and targeted proteomic approaches using an unfractionated PDX tumor digest from a well-established luminal breast cancer subtype (24) to evaluate its performance. For broad detection, DTM with a LC-IMS-MS platform was assessed against two conventional discovery-based platforms (Orbitrap Velos MS and Q Exactive MS) using the same optimized 100-min LC gradient. For targeted quantitation, DTM was compared to a triple quadrupole performing SRM analyses. These analyses allowed an evaluation of DTMs effectiveness for achieving higher sample throughput, proteomic coverage, and accurate quantitation, while reducing sample size requirements.
AMT Tag Creation and Target Protein Selection
To compare the results for each of the discovery-based platforms, an extensive accurate mass and time (AMT) tag database consisting of highly confident peptide identifications and providing broad protein coverage was created for the PDX tumor sample (27) (Fig. 2A). To construct the AMT tag database, 48 high pH LC peptide fractions were analyzed with the Q Exactive platform to obtain MS/MS-based identifications, and the IMS-MS platform to acquire drift time for each tag. The resulting information from both platforms was then used to provide LC elution times, IMS drift times and accurate masses for the peptides identified from the PDX tumor proteins (28). Q Exactive spectra were searched by MS-GF+ (29) and filtered to a false discovery rate (based on MS-GF+ decoy searches) of ≤ 0.01% for high confidence matching, where only peptides with ≥2 MS/MS spectra across all data sets were used to correlate with those from the IMS-MS instrument. Peptides observed in each instrument were compared and only those within tight mass accuracy (within 2 ppm) and LC elution time (<0.5%) tolerances were populated with IMS drift times and used in the PDX tumor AMT tag database (28). Although creation of this database does take time, it can be applied to all samples having the same characteristics, whereas SRM methods must be redone anytime the peptides of interest change.
MS/MS spectral count information from the fraction data sets was then used to select a subset of 10 target proteins ranging in abundance and biological activity for analysis in the targeted studies (Table I). Two moderately abundant proteins, DJ-1 and calmodulin, were selected first because they had >500 spectral counts in the fractions and should be easily observed in all analyses. Next, four lower concentration proteins (∼100 spectral counts) were chosen based on their function. Membrane-associated progesterone receptor component 1, membrane-associated progesterone receptor component 2, and epidermal growth factor receptor isoform a precursor, were chosen as receptors in breast cancer tumor cells and kynureninase isoform b is overexpressed in the luminal subtype. The final four proteins (transmembrane and TPR repeat-containing protein 3, MAP7 domain-containing protein 1, parafibromin, and T-complex protein 11-like protein 1) were selected because they are known to be low abundance proteins in this sample and expected to be difficult to detect in the unfractionated parent sample. Spectral counts from the fractionated studies were again used to select two target peptides for each of the moderately and low abundance proteins. Each peptide chosen for the moderately abundant proteins had >20 spectral counts each whereas those selected for the lowest concentrations had ∼4 spectral counts each. The 20 heavy labeled peptides corresponding to the light peptides were then purchased from Thermo Fisher Scientific.
Table I. SRM and DTM concentrations (fmol/μg) and CVs for target peptides.
| Peptide | Protein name | Gene symbol | SRM Conca | SRM CVa | DTM Concb | DTM CVb | DTM/SRM Avg Concc | DTM/SRM CVc |
|---|---|---|---|---|---|---|---|---|
| R.TTILQSTGK.N | Parafibromin | CDC73 | 0.36 | 0.19 | 0.96 | 0.19 | 0.66 | 0.64 |
| R.IEDILEVIEK.E | Kynureninase isoform b | KYNU | 0.76 | 0.03 | 1.49 | 0.09 | 1.12 | 0.46 |
| R.SAPLEIGLQR.S | Parafibromin | CDC73 | 0.83 | 0.09 | 2.15 | 0.11 | 1.49 | 0.63 |
| R.LSASTASELSPK.S | MAP7 domain-containing protein 1 | MAP7D1 | 2.04 | 0.16 | 1.28 | 0.22 | 1.66 | 0.32 |
| R.EIFSVLDLMK.V | T-complex protein 11-like protein 1 | TCP11L1 | 1.28 | 0.02 | 2.66 | 0.08 | 1.97 | 0.50 |
| K.TYLEEELDKWAK.I | Kynureninase isoform b | KYNU | 4.34 | 0.11 | 2.09 | 0.12 | 3.21 | 0.49 |
| R.GLATFCLDKDALR.D | Membrane-associated progesterone receptor component 2 | PGRMC2 | 2.70 | 0.13 | 4.44 | 0.10 | 3.57 | 0.35 |
| R.TPETLLPFAEAEAFLKK.A | MAP7 domain-containing protein 1 | MAP7D1 | 4.56 | 0.03 | 2.75 | 0.19 | 3.65 | 0.35 |
| K.ETLLSFLLPGHTR.L | T-complex protein 11-like protein 1 | TCP11L1 | 2.65 | 0.03 | 6.47 | 0.08 | 4.56 | 0.59 |
| R.CLLETLALAPHEEYIQR.H | Transmembrane and TPR repeat-containing protein 3 | TMTC3 | 4.05 | 0.04 | 9.61 | 0.11 | 6.83 | 0.58 |
| R.DFTPAELR.R | Membrane-associated progesterone receptor component 1 | PGRMC1 | 13.05 | 0.03 | 10.23 | 0.11 | 11.64 | 0.17 |
| K.TIQEVAGYVLIALNTVER.I | Epidermal growth factor receptor isoform a precursor | EGFR | 12.82 | 0.36 | 10.49 | 0.10 | 11.66 | 0.14 |
| K.ALPILEELLR.Y | Transmembrane and TPR repeat-containing protein 3 | TMTC3 | 10.31 | 0.09 | 16.40 | 0.07 | 13.36 | 0.32 |
| R.GLGAGAGAGEESPATSLPR.M | Membrane-associated progesterone receptor component 2 | PGRMC2 | 12.25 | 0.16 | 17.91 | 0.06 | 15.08 | 0.27 |
| R.VFDKDGNGYISAAELR.H | Calmodulin | CALM2 | 22.14 | 0.04 | 29.39 | 0.07 | 25.76 | 0.20 |
| K.FYGPEGPYGVFAGR.D | Membrane-associated progesterone receptor component 1 | PGRMC1 | 24.44 | 0.08 | 32.01 | 0.06 | 28.22 | 0.19 |
| R.DVVICPDASLEDAKK.E | Protein DJ-1 | PARK7 | 30.83 | 0.17 | 32.64 | 0.06 | 31.73 | 0.04 |
| K.LTQLGTFEDHFLSLQR.M | Epidermal growth factor receptor isoform a precursor | EGRF | NA | NA | 32.42 | 0.06 | 32.42 | NA |
| K.VTVAGLAGKDPVQCSR.D | Protein DJ-1 | PARK7 | 40.87 | 0.08 | 43.06 | 0.07 | 41.97 | 0.04 |
| K.EAFSLFDKDGDGTITTK.E | Calmodulin | CALM2 | 113.46 | 0.06 | 126.28 | 0.03 | 119.87 | 0.08 |
The concentration (fmol/μg) and CV values observed for each peptide by aSRM and bDTM analyses.
c The average concentration and error for both analyses were also computed to understand differences.
Prior to spiking the heavy labeled peptides into the tumor sample, the peptides were mixed together in an aqueous solution and analyzed with the LC-IMS-MS platform using the same conditions as would be applied to the PDX sample. This provided specific LC elution times and IMS drift times (and CCS values) for the heavy labeled target peptides, as well as confirmation of their purity, expected charge states, and monoisotopic masses. Because both light and heavy labeled peptides have the same LC elution time and IMS drift times (21–23), this analysis also shows where the endogenous species will occur in the complex proteome digests (and potentially reveals other species that may be difficult to distinguish). Upon completion, the heavy labeled peptides were then spiked into the PDX sample digest at four different concentrations (2, 10, 20 and 40 fmol/μg), which were selected to mimic the range of endogenous peptides in the sample.
DTM with LC-IMS-MS Compared with LC-MS and LC-MS/MS Approaches
Triplicate analyses of the unfractionated PDX peptides with 2 fmol/μg spiked heavy peptides were obtained for the Orbitrap Velos, Q Exactive, and IMS-MS using the same 100-min LC separation conditions (Fig. 2B). Each data set was then aligned to the PDX tissue AMT tag database as described (27, 34), providing identifications for 29,551 human and mouse peptides (Fig. 3A and supplemental Table S1) across all platforms. Of these, 9832 peptides were detected by all platforms, and 2511 peptides were uniquely detected in the Q Exactive data, 2012 in the Velos data, and 8055 in the DTM data, respectively. The 8055 peptides uniquely detected with DTM exhibited a significantly lower average relative abundance (peak area) compared with the 9832 peptides detected across all platforms (Fig. 3B), indicating higher sensitivity than other discovery platforms. Consistent with this observation, none of the heavy peptides spiked at 2 fmol/μg were observed by the Orbitrap Velos or Q Exactive platforms, but all were observed with DTM. Previously, this increased sensitivity has been correlated with the extra dimension added by the IMS separation in conjunction with the constraints on charge capacity for Orbitrap-based platforms. The automated gain control restricts the accumulation times in congested TIC areas or where high abundance species are present, limiting the time needed for low abundance species to collect enough to be detected (16–18). This phenomenon results in a lower level of detection with such platforms, which we refer to as “ion suppression” (as opposed to “ionization suppression,” which occurs because of the presence of high concentrations of competing species during ESI). We note that when using MS/MS instead of the AMT tag approach, the Q Exactive had 19,096 identifications with <1% FDR (similar to the AMT tag approach with the extremely tight database), whereas the Velos only had 11,105 identifications, primarily attributed to its slower MS/MS speed.
Fig. 3.
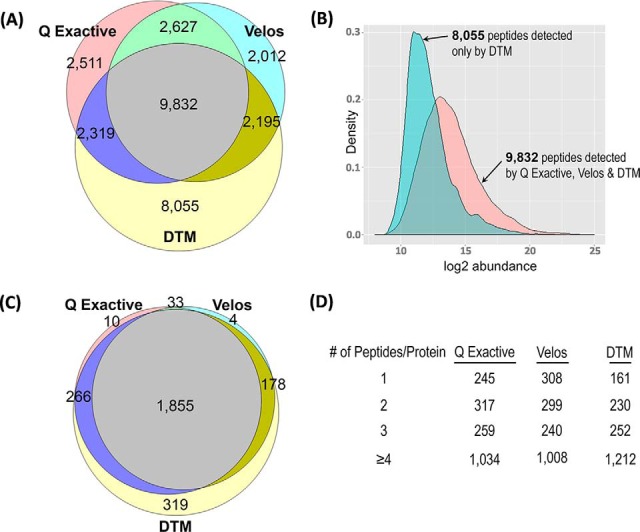
A comparison of the peptides and proteins detected with the discovery-based measurements. A, The peptide identifications for each of the three discovery platforms illustrating the overlap and uniqueness of each. B, The abundance of the 8055 unique DTM peptide identifications was found to be less than those for the 9832 peptides detected by all platforms, and primarily attributed to more effective detection of low abundance species. C, The protein overlap and unique identifications for each discovery based platform. D, The number of peptides for each of the 1855 proteins observed in all platforms. The sequence coverage for the DTM approach was higher than the others with more proteins having ≥4 peptides/protein.
To explore the protein overlap from the different platforms, the peptides were “rolled up” into protein (or protein group) identifications (17) as illustrated in Fig. 3C (supplemental Table S1). In the case of a single peptide matching multiple proteins (typically protein isoforms), a representative protein was chosen, therefore, each reported peptide matches back to a single nonredundant protein. Fig. 3C depicts the 2665 nonredundant human proteins identified by at least 2 peptides in one of the three platforms, illustrating that 1855 proteins were common to all three datatypes and 319 proteins were uniquely detected with the DTM approach. The 1855 common proteins where further explored in Fig. 3D to understand protein coverage for each platform. DTM displayed the best average protein sequence coverage with both the highest number of proteins having ≥4 peptide identifications and the least number of proteins with ≤2 peptide identifications. Both the higher sensitivity and sequence coverage illustrated the utility of DTM in discovery measurements.
DTM Compared with SRM Results
To evaluate the simultaneous use of DTM for targeted measurements, the 20 endogenous PDX peptides in Table I were quantitatively measured with both SRM and DTM. As noted above for the DTM measurements, the 20 heavy peptides were initially mixed together and analyzed with the LC-IMS-MS platform to determine their LC elution times, IMS drift times and observed charge states. Because light and heavy peptides have the same elution and drift times, the pairs can typically be readily discerned in complex biological matrices. The heavy peptides were then spiked into the PDX sample at concentrations of 2, 10, 20, and 40 fmol/μg, and a linear response was observed for the triplicate DTM heavy peptide measurements (Fig. 4A). Even though the PDX sample is unfractionated, all twenty endogenous peptides were confidently detected. Additionally, the nontargeted peptides identified using DTM with the 2 fmol/μg spiked heavy peptides were used to compare the Q Exactive and Orbitrap Velos discovery-based platforms (above discussion) and we note that similar numbers of nontarget peptides were observed using the three other higher spiking levels. Thus, there was no trade off observed using DTM for targeted detection. The concentration for each target peptide was determined based on their light versus heavy peak area ratios and specific spiking amounts (Table I and Fig. 4B). The unfractionated separation with DTM and light and heavy peptide pairs with different endogenous peptide concentrations are shown in Figs. 5A, 5B, and 5C. The same LC elution time and IMS drift time were observed for each pair (Figs. 5B and 5C) and the measurements had high mass accuracy. The multiple dimensions provided separation from most all peptides in the solution. However, in certain cases, peptides with similar or identical masses to the heavy or light precursor coeluted, making identifications difficult, and highlighting the potential utility of more sophisticated feature definition software. Alternatively, these cases also illustrate the utility of having even higher IMS resolving power than presently achieved for improved selectivity and separation, in order to avoid false positives or degraded quantitation.
Fig. 4.

A comparison of the peptides monitored for absolute quantitation in the targeted measurements. A, The DTM log/log linear response curves of four different spiking levels (2, 10, 20 and 40 fmol/ug) for each heavy peptide. Only 10 heavy peptides are shown for clarity and error bars are given for each point. B, The DTM versus SRM calculated concentrations for each of the 20 target peptides and the error bars associated with each measurement, and showing a good linear correlation between the measurements.
Fig. 5.

An example of the DTM measurements possible when analyzing complex samples with a LC-IMS-MS platform. A, The LC and extracted IMS-MS nested spectra (summed for 1-s) illustrating the complexity of the PDX tissue sample. The XICs and IMS-MS nested spectra for target peptides for the B, kynureninase and C, transmembrane proteins. The XICs were extracted using both mass and drift time to eliminate any contributions from interfering peaks. Because the kynureninase peptide was observed at a low concentration its IMS-MS nested spectra is illustrated with the 2 fmol/μg heavy spiking level while the transmembrane peptide was detected at a higher concentration so the 20 fmol/μg spiking level is shown. The IMS-MS nested spectra for the DTM measurements with no heavy spiking are shown to the right to illustrate the lack of interference in the heavy region because of the IMS separation.
For the SRM analyses, the three best transitions were selected for each peptide and a reverse calibration curve was built by spiking different concentrations of the heavy peptides into the PDX sample (Fig. 2B). To determine the endogenous peptide values, 0.5 nm of the heavy peptides were spiked into the PDX sample with a tissue concentration of 0.185 μg/μl. The resulting values were then matched back to the calibration curve for concentration determination (Table I). Nineteen of the 20 peptides could be quantified with SRM, whereas one had an interfering endogenous signal possibly because of the shorter LC gradient. When the endogenous concentrations of the SRM and DTM results were compared (Fig. 4B), a linear relationship was observed for the measurements (coefficient of determination (r2) ≥ 0.88) indicating a high degree of correlation between the instrumental platforms. Four peptides had slightly less agreement than the others, as shown in Table I, an observation that we attribute to the use of features that incorporate some signal contributions from nontarget peptides. The instrument-to-instrument CVs showed the best agreement at higher concentrations as expected and interestingly, the CVs for the DTM measurements dropped with increasing concentrations, whereas those for SRM did not drop as drastically possibly because of partial interferences in certain transitions (Fig. 4B), consistent with the above speculation. Most importantly, the high sensitivity provided by the DTM measurements was confirmed by agreement with the SRM results, thus showing the utility of DTM for targeted measurements while extending the information provided with the addition of broad discovery analyses.
Benefits of Additional Coverage from DTM Analyses
To illustrate the benefit of simultaneous discovery and targeted measurements, we further analyzed the proteins observed only in the DTM analyses and compared them to the Human Protein Atlas (HPA) breast cancer stains (supplemental Table S1). The HPA contains protein expression data derived from antibody-based protein profiling using immunohistochemistry where the data is presented as pathology-based annotation of protein expression levels denoted as low, medium and high (35). Of the 319 proteins uniquely observed in the DTM analyses, 49 ranked as “high” in at least 25% of HPA breast cancer stains. Twenty five of these unique proteins further ranked as high in at least 50% of HPA breast cancer stains and when investigated were found to localize to distinct subcellular regions and have diverse functional roles in cancer progression including transport, transcription and transcription regulation, host-virus interaction, DNA replication and repair, catabolic and anabolic metabolism, and tissue remodeling (Fig. 6). Of significant interest was the detection of MEPCE, FAP and APLP2, which in addition to ranking high in at least 50% of HPA breast cancer stains, have been positively correlated with invasive and metastatic potential. RNA methyltransferase (MEPCE - 7SK snRNA methylphosphate capping enzyme) has been found to be overexpressed in highly tumorigenic breast cancer stem cells (36, 37), prolyl endopeptidase FAP (FAP) is a cell surface glycoprotein serine protease that participates in malignant cell invasiveness by degrading the extracellular matrix (38) and amyloid-like protein 2 (APLP2) has higher expression within invasive breast cancer because it promotes rearrangement of the actin cytoskeleton by increasing cortical actin (39, 40). Detection of these proteins illustrates the power of DTM analyses for finding new information in a high throughput unfractionated approach while simultaneously targeting specific proteins. DTM also provides the option to target these proteins in future studies by purchasing heavy labeled peptides for each and adding them to different sample types for their quantification while also discovering other novel proteins. This capability further illustrates the potential of DTM in targeting and quantitating thousands of peptides (which is difficult to do with SRM currently), while performing discovery-based analyses to learn more about novel protein changes.
Fig. 6.

A schematic illustrating the distinct sub-cellular localization of the 25 unique proteins found by DTM and ranked as 'high' in at least 50% of HPA breast cancer stains. The diverse functional roles necessary for cancer progression including transport, transcription and transcription regulation, host-virus interaction, DNA replication and repair, catabolic and anabolic metabolism, and tissue remodeling are color coded for each and gene symbol and description are given by the table on the left.
DISCUSSION
In this manuscript we report a new DTM approach for the simultaneous acquisition of discovery and targeted measurements based upon a LC-IMS-MS platform. When compared with other discovery-based platforms, the high sensitivity DTM measurements were capable of detecting lower concentration peptides while also providing absolute quantification of specific targeted endogenous peptides. Comparison to SRM analyses illustrated that DTM results required less upfront method development, proved highly reproducible quantitation values, and correlated well with detected SRM concentrations. Moreover, by combining the targeted and discovery analyses, important biological information was attained that would normally be discarded by SRM alone. These high throughput, high sensitivity and quantitative capabilities make DTM extremely attractive for large scale applications, such as patient screening, where thousands of peptides could be spiked into biological samples to obtain information on their abundances in a variety of control and disease conditions. Because this type of targeting is difficult for current SRM platforms, DTM could be an attractive future clinical approach combining high quality discovery and targeted analyses, which is a significant step toward the convergence of these methods. DTM also has other advantages and capabilities that were not fully explored in this work. First, the presence of labeled peptides provides a basis for their use as “anchors” for more precise quantitation of the other detected peptides. Second, the QTOF in the LC-IMS-MS platform could be utilized in either a data independent or dependent acquisition mode for the acquisition of corresponding MS/MS information. However, using the 3-dimensional AMT tag databases developed with MS/MS spectra, allows less reliance on MS/MS information acquired for each sample and becomes increasingly effective as the IMS and LC peak capacities, as well as the MS accuracies increase. Third, DTM could be applied to fractionated samples to achieve even greater depth of coverage than with unfractionated samples (as used in this work), especially because thousands of heavy labeled peptides could be spiked into the samples prior to fractionation for extended quantitation. Finally, we note that future DTM measurements have even greater potential because the dynamic range of IMS-QTOF MS platform results in the detection of many more features than other platforms, and improved separation stages are expected to increase the feature numbers even higher. In this regard, we are presently pursuing much higher IMS separation power using long path length ion mobility separations in Structures for Lossless Ion Manipulations (41, 42) and the initial results look very promising for enhancing future DTM measurements.
Supplementary Material
Acknowledgments
We thank Nathan Johnson for assistance in preparing the figures. This work was performed in the W. R. Wiley Environmental Molecular Sciences Laboratory (EMSL), a DOE national scientific user facility at the Pacific Northwest National Laboratory (PNNL). PNNL is operated by Battelle for the DOE under contract DE-AC05–76RL0 1830.
Footnotes
Author contributions: K.E.B., T.S., W.Q., T.L., E.S.B., and R.D.S. designed research; K.E.B., S.N., C.P.C., D.J.O., Y.M.I., M.A.G., T.R.C., A.K.S., R.J.M., S.O.P., and E.S.B. performed research; D.J.O., Y.M.I., R.J.M., and E.S.B. contributed new reagents or analytic tools; K.E.B., S.N., C.P.C., M.E.M., T.L., and E.S.B. analyzed data; K.E.B., S.N., M.A.G., T.R.C., A.K.S., T.S., W.Q., T.L., E.S.B., and R.D.S. wrote the paper.
* Portions of this research were supported by grants from NCI/Leidos, National Institute of General Medical Sciences (P41 GM103493), the National Institute of Environmental Health Sciences of the NIH (R01 ES022190) and the National Institute of Allergy and Infectious Diseases (U19 AI106772). This research utilized capabilities developed by the Pan-omics program (funded by the U.S. Department of Energy Office of Biological and Environmental Research Genome Sciences Program) and by the National Institute of Allergy and Infectious Diseases under grant U19 AI106772. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- LC
- liquid chromatography
- IMS
- ion mobility spectrometry
- DTM
- discovery and targeted monitoring
- AMT
- accurate mass and time
- MS/MS
- tandem MS
- SRM
- selected reaction monitoring
- MRM
- multiple reaction monitoring
- PRM
- parallel reaction monitoring
- QTOF
- quadrupole time-of-flight
- PDX
- patient-derived xenograft
- WHIM16
- highly characterized, HER2, ER+, PR+ PDX model of breast cancer
- ER
- estrogen-receptor
- PR
- progesterone-receptor
- HER2
- human epidermal growth factor receptor 2
- CCS
- collision cross sections
- SID
- standard isotope dilution
- HPA
- human protein atlas
- MEPCE
- 7SK snRNA methylphosphate capping enzyme
- FAP
- prolyl endopeptidase FAP
- APLP2
- amyloid-like protein 2.
REFERENCES
- 1. Végvári Á., and Marko-Varga G. (2010) Clinical protein science and bioanalytical mass spectrometry with an emphasis on lung cancer. Chem. Rev. 110, 3278–3298 [DOI] [PubMed] [Google Scholar]
- 2. Xiao G. G., Recker R. R., and Deng H. W. (2008) Recent advances in proteomics and cancer biomarker discovery. Clin. Med. Oncol. 2, 63–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wu C. C., and MacCoss M. J. (2002) Shotgun proteomics: tools for the analysis of complex biological systems. Curr. Opin. Mol. Ther. 4, 242–250 [PubMed] [Google Scholar]
- 4. Meissner F., and Mann M. (2014) Quantitative shotgun proteomics: considerations for a high-quality workflow in immunology. Nat. Immunol. 15, 112–117 [DOI] [PubMed] [Google Scholar]
- 5. Domon B., and Aebersold R. (2010) Options and considerations when selecting a quantitative proteomics strategy. Nat. Biotechnol. 28, 710–721 [DOI] [PubMed] [Google Scholar]
- 6. Baker E. S., Liu T., Petyuk V. A., Burnum-Johnson K. E., Ibrahim Y. M., Anderson G. A., and Smith R. D. (2012) Mass spectrometry for translational proteomics: progress and clinical implications. Genome Med. 4, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gupta M. K., Jung J. W., Uhm S. J., Lee H., Lee H. T., and Kim K. P. (2009) Combining selected reaction monitoring with discovery proteomics in limited biological samples. Proteomics 9, 4834–4836 [DOI] [PubMed] [Google Scholar]
- 8. Yang X., and Lazar I. M. (2009) MRM screening/biomarker discovery with linear ion trap MS: a library of human cancer-specific peptides. BMC Cancer 9, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schmidt A., Gehlenborg N., Bodenmiller B., Mueller L. N., Campbell D., Mueller M., Aebersold R., and Domon B. (2008) An integrated, directed mass spectrometric approach for in-depth characterization of complex peptide mixtures. Mol. Cell. Proteomics 7, 2138–2150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lange V., Picotti P., Domon B., and Aebersold R. (2008) Selected reaction monitoring for quantitative proteomics: a tutorial. Mol Syst. Biol. 4, 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Addona T. A., Abbatiello S. E., S.chilling B., Skates S. J., Mani D. R., Bunk D. M., Spiegelman C. H., Zimmerman L. J., Ham A. J., Keshishian H., Hall S. C., Allen S., Blackman R. K., Borchers C. H., Buck C., Cardasis H. L., Cusack M. P., Dodder N. G., Gibson B. W., Held J. M., Hiltke T., Jackson A., Johansen E. B., Kinsinger C. R., Li J., Mesri M., Neubert T. A., Niles R. K., Pulsipher T. C., Ransohoff D., Rodriguez H., Rudnick P. A., Smith D., Tabb D. L., Tegeler T. J., Variyath A. M., Vega-Montoto L. J., Wahlander A., Waldemarson S., Wang M., Whiteaker J. R., Zhao L., Anderson N. L., Fisher S. J., Liebler D. C., Paulovich A. G., Regnier F. E., Tempst P., and Carr S. A. (2009) Multi-site assessment of the precision and reproducibility of multiple reaction monitoring-based measurements of proteins in plasma. Nat. Biotechnol. 27, 633–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Peterson A. C., Russell J. D., Bailey D. J., Westphall M. S., and Coon J. J. (2012) Parallel reaction monitoring for high resolution and high mass accuracy quantitative, targeted proteomics. Mol. Cell. Proteomics 11, 1475–1488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rauniyar N. (2015) Parallel reaction monitoring: a targeted experiment performed using high resolution and high mass accuracy mass spectrometry. Int. J. Mol. Sci. 16, 28566–28581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mason E., and McDaniel E. (1988) Transport properites of ions in gases. Wiley: New York [Google Scholar]
- 15. McLean J. A., Ruotolo B. T., Gillig K. J., and Russell D. H. (2005) Ion mobility-mass spectrometry: a new paradigm for proteomics. Int. J. Mass Spectrom. 240, 301–315 [Google Scholar]
- 16. Baker E. S., Livesay E. A., Orton D. J., Moore R. J., Danielson W. F. 3rd, Prior D. C., Ibrahim Y. M., LaMarche B. L., Mayampurath A. M., Schepmoes A. A., Hopkins D. F., Tang K., Smith R. D., and Belov M. E. (2010) An LC-IMS-MS platform providing increased dynamic range for high-throughput proteomic studies. J. Proteome Res. 9, 997–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Baker E. S., Burnum-Johnson K. E., Jacobs J. M., Diamond D. L., Brown R. N., Ibrahim Y. M., Orton D. J., Piehowski P. D., Purdy D. E., Moore R. J., Danielson W. F. 3rd, Monroe M. E., Crowell K. L., Slysz G. W., Gritsenko M. A., Sandoval J. D., Lamarche B. L., Matzke M. M., Webb-Robertson B. J., Simons B. C., McMahon B. J., Bhattacharya R., Perkins J. D., Carithers R. L. Jr., Strom S., Self S. G., Katze M. G., Anderson G. A., and Smith R. D. (2014) Advancing the high throughput identification of liver fibrosis protein signatures using multiplexed ion mobility spectrometry. Mol. Cell. Proteomics 13, 1119–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Baker E. S., Burnum-Johnson K. E., Ibrahim Y. M., Orton D. J., Monroe M. E., Kelly R. T., Moore R. J., Zhang X., Theberge R., Costello C. E., and Smith R. D. (2015) Enhancing bottom-up and top-down proteomic measurements with ion mobility separations. Proteomics 15, 2766–2776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu X., Plasencia M., Ragg S., Valentine S. J., and Clemmer D. E. (2004) Development of high throughput dispersive LC-ion mobility-TOFMS techniques for analysing the human plasma proteome. Brief Funct. Genomic Proteomic 3, 177–186 [DOI] [PubMed] [Google Scholar]
- 20. Valentine S. J., Plasencia M. D., Liu X., Krishnan M., Naylor S., Udseth H. R., Smith R. D., and Clemmer D. E. (2006) Toward plasma proteome profiling with ion mobility-mass spectrometry. J. Proteome Res. 5, 2977–2984 [DOI] [PubMed] [Google Scholar]
- 21. Sturm R. M., Lietz C. B., and Li L. (2014) Improved isobaric tandem mass tag quantification by ion mobility mass spectrometry. Rapid Commun. Mass Spectrom. 28, 1051–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Myung S., Lee Y. J., Moon M. H., Taraszka J., Sowell R., Koeniger S., Hilderbrand A. E., Valentine S. J., Cherbas L., Cherbas P., Kaufmann T. C., Miller D. F., Mechref Y., Novotny M. V., Ewing M. A., Sporleder C. R., and Clemmer D. E. (2003) Development of high-sensitivity ion trap ion mobility spectrometry time-of-flight techniques: a high-throughput nano-LC-IMS-TOF separation of peptides arising from a Drosophila protein extract. Anal. Chem. 75, 5137–5145 [DOI] [PubMed] [Google Scholar]
- 23. Merkley E. D., Baker E. S., Crowell K. L., Orton D. J., Taverner T., Ansong C., Ibrahim Y. M., Burnet M. C., Cort J. R., Anderson G. A., Smith R. D., and Adkins J. N. (2013) Mixed-isotope labeling with LC-IMS-MS for characterization of protein-protein interactions by chemical cross-linking. J.Am. Soc. Mass Spectrom. 24, 444–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li S., Shen D., Shao J., Crowder R., Liu W., Prat A., He X., Liu S., Hoog J., Lu C., Ding L., Griffith O. L., Miller C., Larson D., Fulton R. S., Harrison M., Mooney T., McMichael J. F., Luo J., Tao Y., Goncalves R., Schlosberg C., Hiken J. F., Saied L., Sanchez C., Giuntoli T., Bumb C., Cooper C., Kitchens R. T., Lin A., Phommaly C., Davies S. R., Zhang J., Kavuri M. S., McEachern D., Dong Y. Y., Ma C., Pluard T., Naughton M., Bose R., Suresh R., McDowell R., Michel L., Aft R., Gillanders W., DeSchryver K., Wilson R. K., Wang S., Mills G. B., Gonzalez-Angulo A., Edwards J. R., Maher C., Perou C. M., Mardis E. R., and Ellis M. J. (2013) Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep. 4, 1116–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Baker E. S., Clowers B. H., Li F., Tang K., Tolmachev A. V., Prior D. C., Belov M. E., and Smith R. D. (2007) Ion mobility spectrometry-mass spectrometry performance using electrodynamic ion funnels and elevated drift gas pressures. J. Am. Soc. Mass Spectrom. 18, 1176–1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ibrahim Y. M., Baker E. S., Danielson W. F. 3rd, Norheim R. V., Prior D. C., Anderson G. A., Belov M. E., and Smith R. D. (2015) Development of a new ion mobility (quadrupole) time-of-flight mass spectrometer. Int. J. Mass Spectrom. 377, 655–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zimmer J. S., Monroe M. E., Qian W. J., and Smith R. D. (2006) Advances in proteomics data analysis and display using an accurate mass and time tag approach. Mass Spectrom. Rev. 25, 450–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Burnum K. E., Hirota Y., Baker E. S., Yoshie M., Ibrahim Y. M., Monroe M. E., Anderson G. A., Smith R. D., Daikoku T., and Dey S. K. (2012) Uterine deletion of Trp53 compromises antioxidant responses in the mouse decidua. Endocrinology 153, 4568–4579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kim S., Gupta N., and Pevzner P. A. (2008) Spectral probabilities and generating functions of tandem mass spectra: a strike against decoy databases. J. Proteome Res. 7, 3354–3363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jaitly N., Mayampurath A., Littlefield K., Adkins J. N., Anderson G. A., and Smith R. D. (2009) Decon2LS: An open-source software package for automated processing and visualization of high resolution mass spectrometry data. BMC Bioinformatics 10, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Monroe M. E., Tolic N., Jaitly N., Shaw J. L., Adkins J. N., and Smith R. D., VIPER: an advanced software package to support high-throughput LC-MS peptide identification. Bioinformatics 23, 2021–2023 [DOI] [PubMed] [Google Scholar]
- 32. MacLean B., Tomazela D. M., Shulman N., Chambers M., Finney G. L., Frewen B., Kern R., Tabb D. L., Liebler D. C., and MacCoss M. J. (2010) Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Abbatiello S. E., Mani D. R., Keshishian H., and Carr S. A. (2002) Automated detection of inaccurate and imprecise transitions in peptide quantification by multiple reaction monitoring mass spectrometry. Clin. Chem. 56, 291–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Smith R. D., Anderson G. A., Lipton M. S., Pasa-Tolic L., Shen Y., Conrads T. P., Veenstra T. D., and Udseth H. R. (2002) An accurate mass tag strategy for quantitative and high-throughput proteome measurements. Proteomics 2, 513–523 [DOI] [PubMed] [Google Scholar]
- 35. Ponten F., Jirstrom K., and Uhlen M. (2008) The Human Protein Atlas–a tool for pathology. J. Pathol. 216, 387–393 [DOI] [PubMed] [Google Scholar]
- 36. Liu R., Wang X., Chen G. Y., Dalerba P., Gurney A., Hoey T., Sherlock G., Lewicki J., Shedden K., and Clarke M. F. (2007) The prognostic role of a gene signature from tumorigenic breast-cancer cells. New Engl. J.Med. 356, 217–226 [DOI] [PubMed] [Google Scholar]
- 37. Xhemalce B., Robson S. C., and Kouzarides T. (2012) Human RNA methyltransferase BCDIN3D regulates microRNA processing. Cell 151, 278–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Waumans Y., Baerts L., Kehoe K., Lambeir A. M., and De Meester I. (2015) The dipeptidyl peptidase family, prolyl oligopeptidase, and prolyl carboxypeptidase in the immune system and inflammatory disease, including atherosclerosis. Front. Immunol. 6, 387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Abba M. C., Drake J. A., Hawkins K. A., Hu Y., Sun H., Notcovich C., Gaddis S., Sahin A., Baggerly K., and Aldaz C. M. (2004) Transcriptomic changes in human breast cancer progression as determined by serial analysis of gene expression. Breast Cancer Res. 6 , R499–R513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pandey P., Rachagani S., Das S., Seshacharyulu P., Sheinin Y., Naslavsky N., Pan Z., Smith B. L., Peters H. L., Radhakrishnan P., McKenna N. R., Giridharan S. S. P., Haridas D., Kaur S., Hollingsworth M. A., MacDonald R. G., Meza J. L., Caplan S., Batra S. K., and Solheim J. C. (2015) Amyloid precursor-like protein 2 (APLP2) affects the actin cytoskeleton and increases pancreatic cancer growth and metastasis. Oncotarget 6, 2064–2075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Deng L., Ibrahim Y. M., Hamid A. M., Garimella S. V., Webb I. K., Zheng X., Prost S. A., Sandoval J. A., Norheim R. V., Anderson G. A., Tolmachev A. V., Baker E. S., and Smith R. D. (2016) Ultra-high resolution ion mobility separations utilizing traveling waves in a 13 m serpentine path length structures for lossless ion manipulations module. Anal. Chem. 88, 8957–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Deng L., Ibrahim Y. M., Baker E. S., Aly N. A., Hamid A. M., Zhang X., Zheng X., Garimella S. V. B., Webb I. K., Prost S. A., Sandoval J. A., Norheim R. V., Anderson G. A., Tolmachev A. V., and Smith R. D. (2016) Ion mobility separations of isomers based upon long path length structures for lossless ion manipulations combined with mass spectrometry. ChemistrySelect 1, 2396–2399 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.