

SUMMARY
The transcription factor Foxo3 plays a crucial role in myeloid cell function but its role in lymphoid cells remains poorly defined. Here, we have shown that Foxo3 expression was increased after T cell receptor engagement and played a specific role in the polarization of CD4+ T cells towards pathogenic T helper-1 (Th1) cells producing interferon-γ (IFN-γ) and granulocyte monocyte colony stimulating factor (GM-CSF). Consequently, Foxo3-deficient mice exhibited reduced susceptibility to experimental autoimmune encephalomyelitis. At the molecular level, we identified Eomes as a direct target gene for Foxo3 in CD4+ T cells and we have shown that lentiviral-based overexpression of Eomes in Foxo3-deficient CD4+ T cells restored both IFN-γ and GM-CSF production. Thus, the Foxo3-Eomes pathway is central to achieve the complete specialized gene program required for pathogenic Th1 cell differentiation and development of neuroinflammation.
Introduction
The Foxo (Forkhead Box class O) family of transcription factors (TF) governs processes such as cellular proliferation, apoptosis, energy metabolism, autophagy or stress resistance in response to changes in the abundance of nutrients and growth factors (Eijkelenboom and Burgering, 2013). Foxo proteins can act either as transcriptional activators or repressors upon their high affinity binding to the consensus sequence 5′-GTAAA(T/C)AA-3′, known as the Daf-16 family member-binding element (Obsil and Obsilova, 2010). In addition, Foxo factors can bind and modulate other TF (van der Vos and Coffer, 2010). All of these activities are altered by phosphorylation, acetylation, methylation and ubiquitination, and these post-translational modifications influence Foxo intracellular localization, turnover, transactivation or transcriptional specificity (Zhao et al., 2011).
Foxo TF, through their role in the control of cell cycle progression and apoptosis, were first described as tumor suppressor genes. Nonetheless, numerous studies have revealed that Foxo1 and Foxo3 also play fundamental roles in physiologic and pathologic immune responses (Dejean et al., 2010; Hedrick, 2009; Hedrick et al., 2012; Ouyang and Li, 2010). Because of the similarity between their DNA-binding domains, all Foxo factors can in principle bind to related sequences and therefore should regulate the same target genes. Experiments using mice deficient for a single Foxo isoform however clearly demonstrate that Foxo1 and Foxo3 have independent physiological functions in the immune system, suggesting that Foxo functions could be closely linked to their distinct cell type-specific expression patterns (Dejean et al., 2010; Hedrick, 2009).
Foxo1 is abundantly expressed in lymphoid cells, where it has been shown to regulate many features of lymphocyte homeostasis including survival, homing and differentiation. Indeed, Foxo1 has critical functions in B cell development, homing, class-switch recombination and somatic hypermutation (Amin and Schlissel, 2008; Dengler et al., 2008). Foxo1 also regulates both naive and memory T cell survival and trafficking (Kerdiles et al., 2009; Kim et al., 2013; Ouyang et al., 2009; Ouyang et al., 2010), thymic regulatory T (tTreg) and peripheral regulatory T (pTreg) cell development and function (Kerdiles et al., 2010; Merkenschlager and von Boehmer, 2010; Ouyang et al., 2010; Ouyang et al., 2012), as well as T helper-1 (Th1), Th17 and T follicular helper (Tfh) cell differentiation (Kerdiles et al., 2010; Laine et al., 2015; Merkenschlager and von Boehmer, 2010; Oestreich et al., 2012; Ouyang et al., 2012; Stone et al., 2015). So far, no specific role for Foxo1 has been assigned in immune cells other than lymphocytes.
Foxo3 is the main isoform expressed in the myeloid compartment. Our previous study has shown that Foxo3 is a key suppressor of inflammatory cytokine production by dendritic cells (DC) and macrophages (Dejean et al., 2009). These results are consistent with a non-coding polymorphism in human FOXO3 that limits inflammatory monocyte responses resulting in milder Crohn’s disease and rheumatoid arthritis, but more severe malaria (Lee et al., 2013). The role played by Foxo3 in T cells is less well defined. Using Foxo1−/−Foxo3−/− mice, studies have demonstrated that Foxo1 and Foxo3 cooperatively control the development and function of Foxp3+ Treg cells (Kerdiles et al., 2010; Ouyang et al., 2010). Others have shown that Foxo3 limits the expansion of memory CD8+ T cells during acute or chronic viral infection (Sullivan et al., 2012a; Sullivan et al., 2012b). To date, however, the precise role of Foxo3 in effector CD4+ T cells has not been addressed.
In this study, we show that the expression of Foxo3 was increased in CD4+ T cells following activation and correlated with T cell receptor (TCR) signaling strength. To address the relevance of this up-regulation, we analyzed the impact of Foxo3-deficiency on CD4+ T cell effector functions and found that Foxo3 drives Eomes-dependent differentiation of IFN-γ+ GM-CSF+ pathogenic Th1 cells and that this pathway is needed for the development of central nervous system inflammation.
RESULTS
TCR-triggering leads to increased expression of Foxo3 in CD4+ T cells
In vivo, activated (CD62L−, CD44+) CD4+ T cells were found to exhibit a three-fold increase in Foxo3 expression when compared to naive (CD62L+, CD44−) CD4+ T cells (Figure 1A). We therefore addressed whether CD4+ T cell activation had an impact on the expression of Foxo3. Naive CD4+ T cells were stimulated with plate-bound anti-CD3 mAbs and analyzed for Foxo3 expression. T cell receptor (TCR) triggering resulted in a dose-dependent upregulation of Foxo3 in CD4+ T cells (Figure 1B), with increased expression over time (Figure 1C) whereas CD28-induced costimulation did not influence Foxo3 expression (Figure S1A). A dose-dependent upregulation of Foxo3 was also recorded when OT-II CD4+ T cells were stimulated with antigen presenting cell (APC) loaded with increasing doses of OVA323–339 peptide confirming that TCR-dependent signal intensity regulated Foxo3 expression in activated CD4+ T cells (Figure S1B). To determine key signaling events inducing Foxo3 expression upon stimulation, we next activated CD4+ T cells with anti-CD3 mAbs in the presence of a series of inhibitors that block specific pathways downstream of TCR. We found that inhibition of protein kinase C (PKCs) prevented Foxo3 upregulation whereas inhibition of ERK, p38 or JNK kinase pathways had no effect (Figure S1C). In agreement, stimulation with phorbol 12-myristate 13-acetate (PMA) alone was able to induce Foxo3 expression whereas ionomycin did not (Figure S1D). To dissect the pathway downstream of PKC, we used inhibitors of NF-κB and the NFAT transcription factor and showed that TCR-induced Foxo3 expression was NF-κB dependent (Figure S1E). Taken together, these data suggest that PKCs and NF-κB pathways downstream of TCR positively regulate Foxo3 expression in CD4+ T cells.
Figure 1. Increased Foxo3 expression in CD4+ T cells after TCR engagement.

(A) Foxo3 expression by naive CD62L+, CD44− (white bars) and activated CD62L−, CD44+ (dark grey bars) WT CD4+ T cells (n=7 mice per genotype). (B) Foxo3 expression by naive WT CD4+ T cells stimulated in vitro with the indicated dose of anti-CD3 mAbs (n=4 mice per genotype). (C) Foxo3 expression by naive WT CD4+ T cells stimulated with anti-CD3 mAbs (2μg/mL) for 18, 36 or 72 hours (n=4 mice per genotype). Mean and SEM of the relative MFI of Foxo3 expression was calculated by subtracting the WT MFI from the Foxo3−/− MFI. (D) Immunofluorescence staining of Foxo3 in naive CD4+ T cell from WT or Foxo3−/− mice stimulated in vitro with the indicated dose of anti-CD3 mAbs for 48 hours (Scale bar, 10μm). (E) Immunoblot analysis of Foxo3, PLC-γ and TFIID expression in nuclear and cytoplasmic fractions of naive CD4+ T cells from WT or Foxo3−/− mice stimulated in vitro as in D. Data are representative of three independent experiments. Error bars, SEM.; P values (Mann–Whitney U test). See also Figure S1
Since activation of Foxo3 was correlated with its subcellular localization, immunofluorescence staining and subcellular fractionation combined to Immunoblot analysis were performed. Foxo3 was almost entirely localized in the nucleus of activated CD4+ T cells (Figure 1D, 1E). Altogether, our data show that TCR-dependent signal intensity correlates with Foxo3 expression and nuclear accumulation in activated CD4+ T cells.
Foxo3 deficiency impairs CD4+ T cell differentiation
To better understand the significance of enhanced Foxo3 expression in effector CD4+ T cells, in vitro experiments were performed in which naive Foxo3−/− or WT CD4+ T cells were stimulated under neutral conditions with increasing concentrations of anti-CD3 mAbs. Under those culture conditions, the frequencies of IFN-γ (Figure 2A, 2B) and GM-CSF (Figure 2C, 2D) secreting cells in Foxo3−/− CD4+ T cells were reduced by half of that observed in WT CD4+ T cells after either 36 or 72 hours of culture whereas survival, proliferation or IL-2, IL-13, IL-4 and TNF production were unaffected (Figure S2A, S2B) and the production of IL-10 and IL-17 was undetectable (data not shown). This decreased frequency of IFN-γ and GM-CSF positive cells was also observed when cells were stimulated with both anti-CD3 and anti-CD28 mAbs, indicating that a co-stimulatory signal was not sufficient to restore cytokine production by Foxo3 deficient cells (Figure S2C). In addition, a delayed and diminished expression of T-bet, the “master regulator” of T helper-1 (Th1) cell differentiation (Szabo et al., 2000), was observed in Foxo3−/− CD4+ T cells upon TCR engagement (Figure 2E, 2F). Decreased IFN-γ production associated with a Foxo3 deficiency was also found under Th1 cell polarizing conditions (Figure 2G) whereas proliferation and survival were not affected (Figure S2E). Moreover, the Foxo3 deficiency not only decreased the frequency of IFN-γ+ cells but also impacted the overall amount of IFN-γ produced on a per-cell basis, as demonstrated by the decreased MFI of IFN-γ expressed by Foxo3−/− CD4+ T cells after either 36 or 72 hours of culture (Figure 2H). The frequency of T-bet expressing cells was equivalent in both Foxo3−/− and WT CD4+ Th1 cells (Figure 2I); however, Foxo3 deficiency was also associated with decreased T-bet MFI in Th1 cells (Figure 2J).
Figure 2. Foxo3 deficiency impaired pathogenic Th1 cell differentiation.
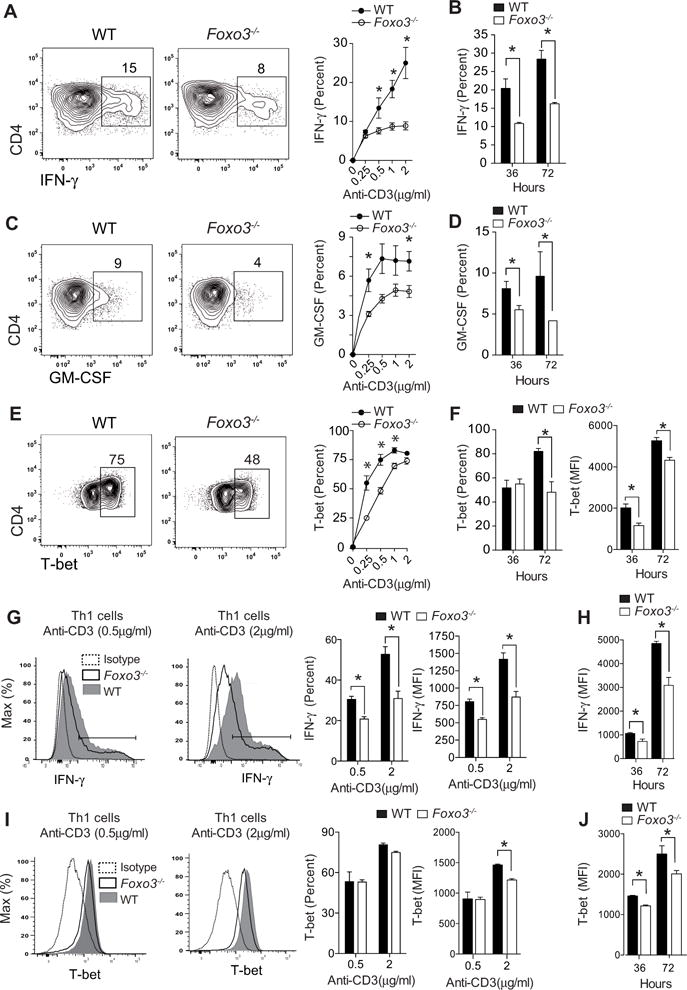
(A) IFN-γ production by WT or Foxo3−/− naive CD4+ T cells stimulated with anti-CD3 (0.5 μg/mL) under non-polarizing condition for 36 hours. Frequency of IFN-γ produced by WT (black circles) or Foxo3−/− (open circles) CD4+ T cells stimulated with anti-CD3 Abs for 36 hours (n=5 mice per genotype) (B) Frequency of IFN-γ production by WT (black bars) or Foxo3−/− (open bars) CD4+ T cells stimulated with anti-CD3 mAbs (2μg/mL) for the indicated time (n=5 mice per genotype). (C) GM-CSF production by WT or Foxo3−/− naive CD4+ T cells stimulated as in A (n=5 mice per genotype). (D) Frequency of GM-CSF production by WT (black bars) or Foxo3−/− (open bars) CD4+ T cells stimulated as in B (n=4 mice per genotype) (E) T-bet expression by WT (black circles) or Foxo3−/− (open circles) CD4+ T cells stimulated as in A (n=5 mice per genotype). (F) Frequency and MFI of T-bet expression by WT (black bars) or Foxo3−/− (open bars) CD4+ T cells stimulated as in B (n=5 mice per genotype). (G) Frequency and MFI of IFN-γ+ expression by WT (black bars) or Foxo3−/− (open bars) CD4+ T cells stimulated with anti-CD3 mAbs in Th1 cell polarizing conditions for 36 hours (n=5 mice per genotype) or (H) stimulated with 2μg/mL of anti-CD3 mAbs in Th1 cell polarizing conditions for 36 or 72 hours (n=5 mice per genotype). (I) Frequency and MFI of T-bet expression by naive CD4+ T from WT (black bars) or Foxo3-deficient mice (open bars) stimulated as in G (n=5 mice per genotype) or (J) as in H. Data are representative of three independent experiments. Error bars, SEM.; P values (Mann–Whitney U test). See also Figure S2 and S3
We next assessed the ability of Foxo3−/− CD4+ T cell to differentiate into different Th cell lineages when stimulated in polarizing conditions. We showed that Foxo3 deficiency did not impact Th2, Th17 or Foxp3 Treg cell differentiation (Figure S3A). In particular, Foxo3−/− CD4+ T cells were fully able to differentiate into Foxp3+ pTreg cells induced by transforming growth factor (TGF-β) signaling (Figure S3B) or suboptimal TCR activation (Figure S3C) (Li et al., 2013a). Moreover, we showed that tTreg cells from Foxo3−/− mice were as suppressive as WT tTreg cells (Figure S3D). Collectively, these results show that Foxo3 promotes TCR-induced production of IFN-γ and GM-CSF and has no notable impact on Th2, Th17 or Treg cell differentiation.
Foxo3 is required for TCR-induced Eomes expression by CD4+ T cells
To understand the molecular mechanisms whereby Foxo3 controls CD4+ T cell differentiation, unbiased analysis of genes differentially expressed in Foxo3-deficient vs. Foxo3-sufficient CD4+ T cells was achieved using both resting and activated CD4+ T cells obtained following 12 or 24 hours of stimulation with anti-CD3 mAbs. When comparing unstimulated WT and Foxo3−/− CD4+ T cells, only 5 transcripts showed greater than 2-fold change, suggesting that Foxo3 plays minimal role in resting CD4+ T cells (Figure S4A). This number increased upon TCR engagement suggesting that Foxo3 is mainly active following TCR stimulation (FDR≤ 0.05) (Figure 3A and S4B). Three main networks were impacted by Foxo3 deletion among which the “IFN-γ and IFN- γ response” was the most dysregulated pathway (Figure 3B). The second network was enriched for metabolic functional categories, confirming the role of Foxo3 in the regulation of cellular metabolism (Figure S4C). The third identified cluster was enriched in genes involved in “immune cell trafficking” suggesting that Foxo3 might have a role in T cell migration and homing (Figure S4D)
Figure 3. Foxo3 is required for Eomes expression in CD4+ T cells.
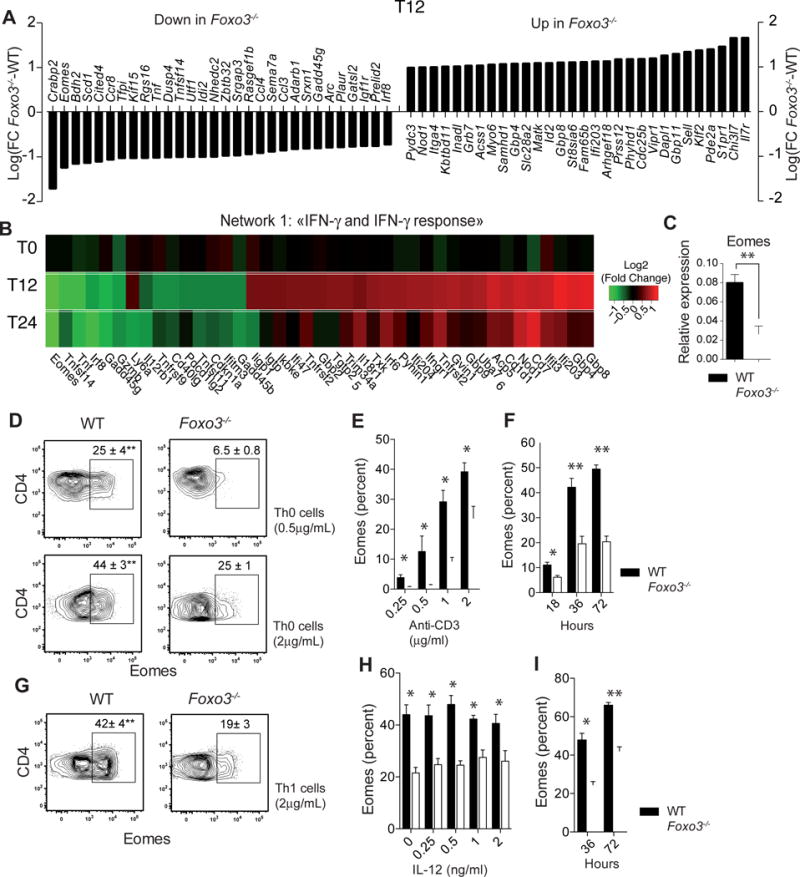
(A) Gene expression microarray experiments comparing WT (n=4) versus Foxo3−/− (n=4) CD4+ T cells after 12h of stimulation in neutral condition with 2 μg/mL of anti-CD3 mAbs. Data are expressed as Log2(Fold Change Foxo3−/−-WT) of the top 30 most significantly regulated genes (FDR ≤ 0.05 and fold change >2 or <2). (B) Gene expression fold changes (Log2(FC Foxo3−/−-WT) of the top most significantly regulated (FDR ≤ 0.05 and fold change > 1.5) genes within the “IFN-γ and IFN-γ response” pathway shown as a Heatmap of over-(red) or under-(green) expressed genes in naive Foxo3−/− CD4+ T cells unstimulated (T0) or stimulated with anti-CD3 mAbs for 12 (T12) or 24 hours (T24) (C) WT (black bars) or Foxo3−/− (open bars) naive CD4+ T cells were stimulated under non-polarizing conditions for 18 hours with 2 μg/mL anti-CD3 mAbs and the mRNA expression of Eomes gene was measured by quantitative real-time RT-PCR (n=4 mice per genotype). (D) Intracellular staining of Eomes expressed by WT or Foxo3−/− naive CD4+ T cells stimulated with 0.5 or 2 μg/mL anti-CD3 in Th0 cell polarizing condition. (E) Frequency of Eomes+ CD4+ T cells in WT (black bars) of Foxo3−/− (open bars) naive CD4+ T cells stimulated under neutral polarizing condition with indicated doses of anti-CD3 mAbs or (F) with 2 μg/mL of anti-CD3 mAbs for the indicated time (n=4 mice per genotype) (G) Eomes expressed by naive CD4+ T cells from WT (black bars) or Foxo3−/− mice (open bars) stimulated with 2 μg/mL of anti-CD3 mAbs under Th1 cell polarizing condition (n=4 mice per genotype) (H) Frequency of Eomes+ CD4+ T cells in WT (black bars) or Foxo3−/− (open bars) naive CD4+ T stimulated with 2 μg/mL of anti-CD3 mAbs and IL-12 (n=4 mice per genotype) for 36 hours or (I) with 2 μg/mL of anti-CD3 mAbs and IL-12 for 36 or 72 hours (n=4 mice per genotype). Data are representative of at least three independent experiments. Error bars, SEM.; P values (Mann–Whitney U test). See also Figure S4
Among all dysregulated genes, Eomes was the second (T12h) and first (T24h) most suppressed gene in Foxo3−/− CD4+ T cells. Analyses by RT-qPCR and flow cytometry confirmed that Foxo3-deficient CD4+ T cells exhibited a decreased expression of Eomes after activation (Figure 3C–D). Although Eomes expression is lower in CD4+ T cells than in CD8+ T cells, its expression increases after activation (Figure S4E). Indeed, TCR-dependent signal intensity controlled Eomes expression in CD4+ T cells, and this expression was largely Foxo3-dependent (Figure 3E). Eomes expression by CD4+ T cells was detected after 18 hours of stimulation and rose substantially between 36 and 72 hours, correlating with the expression of Foxo3 (Figure 3F). We next assessed Eomes expression in other Th subsets. In Th1 cell polarizing conditions, Foxo3 also controlled Eomes expression (Figure 3G). Nevertheless, Eomes expression is IL-12 independent (Figure 3H) and its expression rose between 36 and 72 hours as observed for Th0 cells (Figure 3I). Finally, Eomes expression was low in Th17 and Treg cells as compared to Th0 cells (Figure S4F). These results collectively show that Foxo3 expression is required for TCR-induced Eomes expression in CD4+ T cells.
Foxo3 indirectly controls Ifng and Csf2 genes in CD4+ T cells through the regulation of Eomes expression
Since Foxo3 expression was highly increased in CD4+ T cells expressing Eomes (Figure 4A), we hypothesized that Foxo3 might directly control Eomes transcription. To assess this possibility, we first performed in silico analysis to identify conserved Foxo-binding sites (FBS) in mouse and human EOMES loci. We found three putative FBS: one (FBS1) located in the promoter of Eomes gene (Chr9: 118,478,419) and the other two (FBS2 and FBS3) positioned downstream of the 3′UTR of Eomes (Chr9: 118,487,803), in a region enriched in transcription factor binding sites that might therefore represent a putative 3′UTR enhancer region (p3′UTR-E) (Figure 4B). To determine whether Foxo3 can directly bind within the Eomes locus, we conducted chromatin immuno-precipitation experiments using primer sets designed to amplify regions located at each identified FBS. We found that Foxo3 could bind to the FBS1, although binding was more pronounced for FBS2 and FBS3 (Figure 4C).
Figure 4. Eomes is a direct target gene of Foxo3.
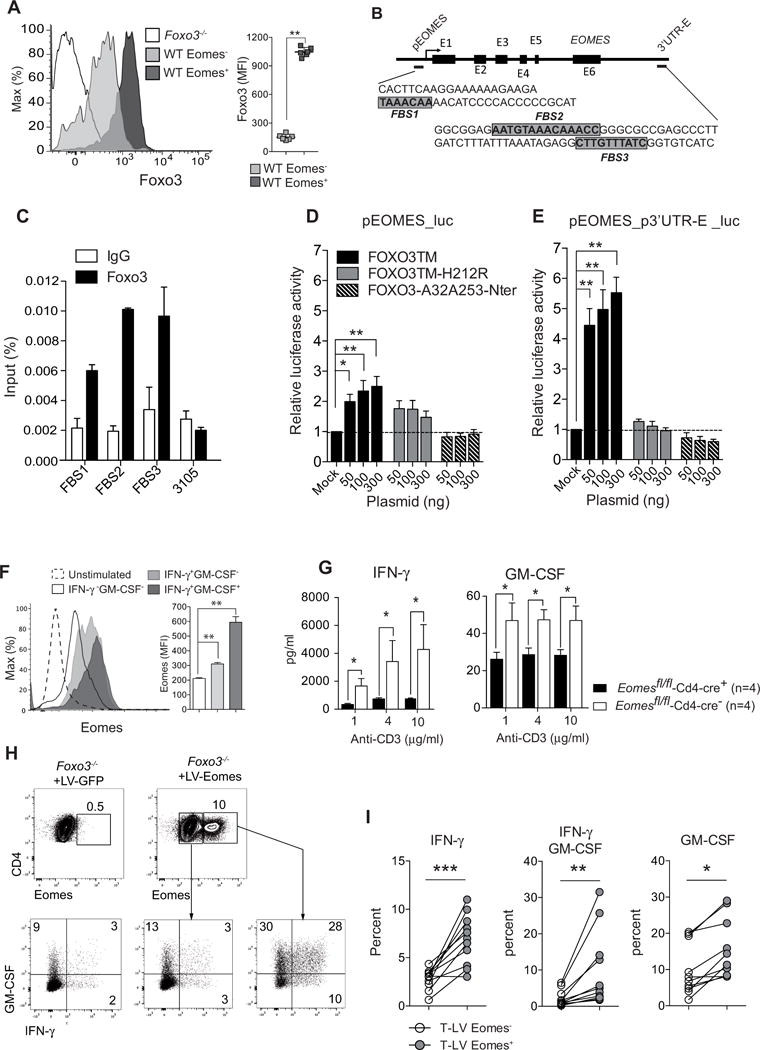
(A) Foxo3 expression gated on Eomes+ (dark gray) and Eomes− (light gray) WT CD4+ T cells stimulated with 2μg/mL of anti-CD3 mAbs (n=6–7 mice per genotype) (B) Schematic structure of the EOMES gene, the arrow represents transcriptional start site of a gene, the black boxes represent exon position (E1 to E6), the positions and sequences of the putative Forkhead-binding sites (FBS) are represented in highlighted in grey. (C) Chromatin immunoprecipitation analysis of Foxo3 binding to the Eomes locus in purified CD4+ T cells stimulated for 24h with 2μg/mL anti-CD3 mAbs. Results are expressed as percentage of input. (D) HEK293 T cells were co-transfected with reporter plasmids containing the human promoter region of EOMES cloned into the pGL3-Basic vector (pEOMES_luc) or (E) plasmids containing the human promoter region of EOMES with the 3′UTR region containing the 2 putative FBS (pEOMES-p3′UTR-E_luc) together with plasmids coding for different forms of FOXO3: the constitutively active FOXO3a mutant (FOXO3TM, black bars), the constitutively active FOXO3TM mutated for the DNA binding domain (FOXO3TM-H212R, grey bars), the constitutive active FOXO3TM deleted for the transactivation domain (FOXO3-A32A253-Nter, dashed bars) or empty vector (Mock). All luciferase activities were normalized to the expression of the co-transfected Renilla luciferase. (F) Eomes expression gated on IFN-γ−GM-CSF− or IFNγ+GM-CSF− and IFN-γ +GM-CSF+ producing CD4+ T cells stimulated with 2 μg/mL anti-CD3 mAbs under non-polarizing condition. (n=5 mice per genotype). (G) Naive CD4+ T cells purified from Eomesfl/fl-Cd4-cre+ (black bars) or Eomesfl/fl-Cd4-cre− (open bars) were stimulated with anti-CD3 mAbs and the secretion of IFN-γ and GM-CSF was analyzed by ELISA in the supernatant after 3 days of culture (n=4 mice per genotype). (H) IFN-γ and GM-CSF expression in naive Foxo3−/− CD4+ T cells transduced with lentiviral particles expressing the GFP alone (LV-GFP) or Eomes and the GFP (LV-Eomes) and gated on either GFP-transduced (LV-GFP left panel), non-transduced (LV-Eomes, middle panel) or Eomes-transduced CD4+ T cells (LV-Eomes, right panel). (I) Frequency of IFN-γ +, IFN-γ +-GM-CSF+ or GM-CSF+ cells among Foxo3−/− CD4+ T cells either non-transduced (LV-Eomes-,open dots) or transduced (LV-Eomes+, grey dots) (n=11 mice, from 3 independent experiments). Data are representative of at least three independent experiments or two independent experiments (C). Error bars, SEM.; P values (Mann–Whitney U test). See also Figure S5
To address whether these FBS regions are involved in the regulation of Eomes expression, we conducted luciferase reporter assays. HEK293T cells were transfected with a reporter plasmid in which a 1 Kb fragment located upstream of the human promoter region of EOMES was cloned into the pGL3-Basic vector (prEomes_luc) (Li et al., 2013b). Cells were co-transfected with plasmids coding for different forms of V5-tagged-FOXO3: the constitutively active form of FOXO3 (FOXO3TM (Brunet et al., 1999)); the Nt fragment from FOXO3TM used as dominant negative (FOXO3-A32A253-Nt (Charvet et al., 2003)); or the active FOXO3TM mutated in the DNA binding domain (FOXO3TM-H212R). Transfection of FOXO3TM induced a 2-fold increase in luciferase activity, whereas the transfection of FOXO3-A32A253-Nt had no impact (Figure 4D). To assess whether the 3′UTR region is involved for EOMES expression, an 81 bp fragment of the p3′UTR-E region containing the 2 putative FBS was sub-cloned into the pEomes-luc vector (pEomes_p3′UTR-E_luc). Using this construct, we found a 6-fold increased luciferase activity in the presence of FOXO3TM, whereas the mutant FOXO3TM-H212R failed to affect luciferase activity indicating that FOXO3 bound directly to the FBS in the p3′UTR-E_region of EOMES (Figure 4E). Altogether, these results show that FOXO3 binds to FBS present in the 3′UTR region of EOMES and that EOMES is a direct transcriptional target gene of FOXO3.
We next assessed whether Eomes expression was also linked to GM-CSF and IFN-γ secretion in CD4+ T cells. Intracellular staining showed that the expression of Eomes was higher in GM-CSF+-IFN-γ+ cells as compared to GM-CSF-IFN-γ+ or GM-CSF−-IFN-γ− (Figure 4F). Moreover, when naïve CD4+ T cells purified from mice with a T-cell specific deletion of Eomes (Eomesfl/flCd4-cre) were stimulated in vitro with increased concentration of anti-CD3 mAbs, both GM-CSF and IFN-γ secretion were reduced in Eomesfl/flCd4-cre+ CD4+ T cells as compared to Eomesfl/flCd4-cre− cells (Figure 4G) whereas the proliferation and survival were similar (data not shown). Therefore, the decreased Eomes expression associated with Foxo3 deficiency might explain the defect GM-CSF and IFN-γ secretion in Foxo3−/− CD4+ T cells. To address this issue directly, we tested whether lentiviral-based overexpression of Eomes could overcome the defect in IFN-γ and GM-CSF production. We showed that Eomes transduction of Foxo3-deficient T cells restored the expression of both IFN-γ and GM-CSF. This finding supports the notion that Foxo3 indirectly regulates Ifng and Csf2 genes in CD4+ T cells through the regulation of Eomes expression (Figure 4H–I).
In addition, we address whether Eomes directly controls ifng and Csf2 expression. We performed an in silico analysis and found 6 highly conserved noncoding sequences enriched in DNaseI hypersensitivity sites and putative transcription factor binding sites positioned downstream of the 3′UTR of CSF2. Next, luciferase reporter assays were performed by coupling these elements to the proximal CSF2 promoter. Using this technique, we were unable to demonstrate a direct regulation of CSF2 by EOMES (Figure S5A). In contrast, the same technique revealed that EOMES, but not FOXO3, directly transactivates the promoter of IFNG (Figure S5B). Moreover, we showed that Foxo3 was unable to transactivate the Ifng locus (Figure S5C). Altogether, these data support the concept that the Eomes-Foxo3 axis is required for the polarization of effector CD4+ T cells into IFN-γ and GM-CSF producing cells.
Eomes acts independently of T-bet for GM-CSF regulation in CD4+ T cells
Since Foxo3 deficiency affects both T-bet and Eomes expression by CD4+ T cells, we next wondered whether Eomes and T-bet could be co-regulated and to what extent diminished GM-CSF and IFN-γ secretion resulted from decreased T-bet expression in Foxo3-deficient CD4+ T cells. A time course analysis showed that Eomes expression precedes that of T-bet and the defect in Eomes and IFN-γ preceded the reduction of T-bet expression in Foxo3−/− CD4+ T cells suggesting that initial production of IFN-γ by CD4+ T cell might be Eomes-dependent but T-bet independent. (Figure 5A). In this regard, previous studies demonstrate that the first wave of IFN-γ is T-bet independent and causes the autocrine induction of T-bet (Schulz et al., 2009). Therefore, the decreased T-bet expression in Foxo3−/− cells might be due to the decreased Eomes-dependent IFN-γ secretion.
Figure 5. Eomes acts independently of T-bet for GM-CSF regulation in CD4+ T cells.
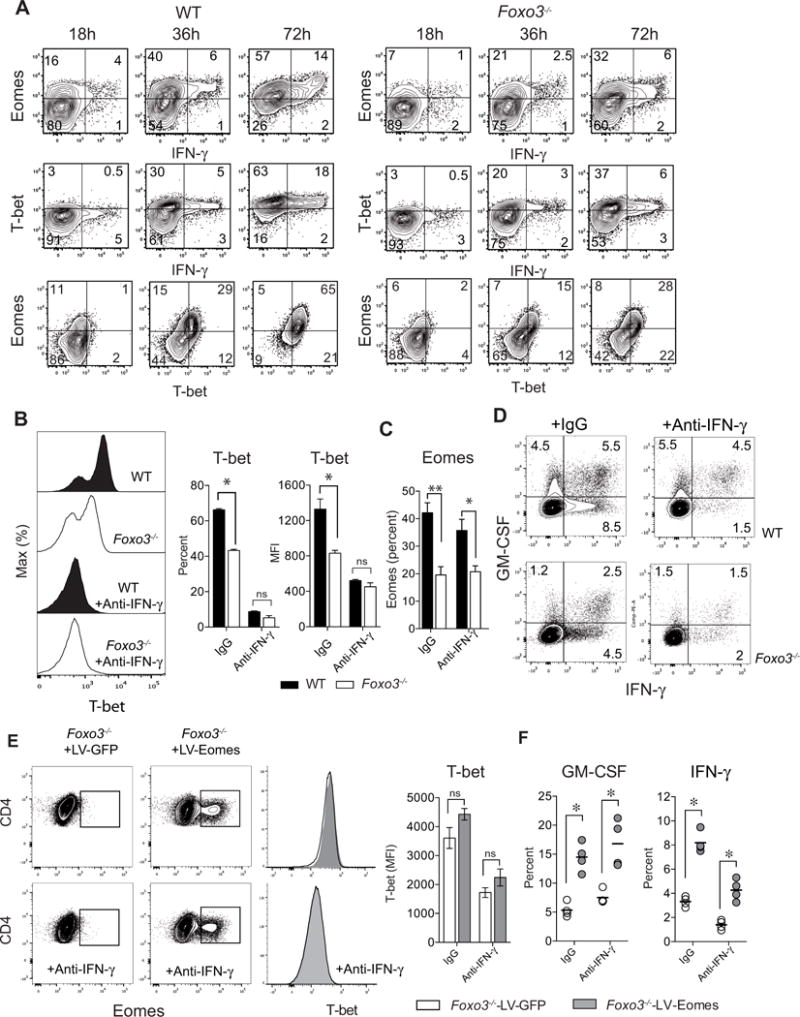
(A) Kinetics of T-bet, Eomes and IFN-γ expression in naive WT or Foxo3−/− CD4+ T cells stimulated with 2μg/ml of anti-CD3 mAbs for 18, 36 or 72 hours (n= 4 mice per genotype). (B) T-bet expression or (C) Eomes expression in naive WT (black bars/histograms) or Foxo3−/− (white bars/histograms) CD4+ T cells stimulated with anti-CD3 mAbs in the absence or presence of anti-IFN-γ blocking mAbs. (n= 4–5 mice per group). (D) Frequency of IFN-γ +, IFN-γ +-GM-CSF+ and GM-CSF+ producing cells in naive WT or Foxo3−/− CD4+ T cells cultured in the absence or presence of anti-IFN-γ neutralizing mAbs. (E) Eomes and T-bet expression in naive WT or Foxo3−/− CD4+ T cells transduced with either control (LV-GFP) or Eomes (LV-EOMES) expressing lentiviral particles in presence or absence of anti-IFN-γ mAbs. (F) Frequency of GM-CSF and IFN-γ producing cells in naive Foxo3−/− CD4+ T cells transduced either with control (LV-GFP, open bars/dots) or Eomes (LV-EOMES, grey bars/dots). Data are representative of at least three independent experiments. Error bars, SEM.; P values (Mann–Whitney U test). See also Figure S5
To test this hypothesis, WT and Foxo3−/− CD4+ T cells were stimulated in the presence of neutralizing anti-IFNγ monoclonal antibody (mAb) to prevent T-bet induction by IFN-γ. Upon IFN-γ neutralization, a clear reduction of T-bet expression was observed, leading to similar expression of T-bet in both WT and Foxo3−/− CD4+ T cells (Figure 5B). These results establish that Foxo3 has no direct impact on T-bet expression and further indicate that decreased T-bet resulted from decreased IFN-γ secretion by Foxo3−/− CD4+ T cells. We also showed that the expression of Eomes was independent of the signaling pathway downstream of IFN-γ since the expression of Eomes was not affected by blocking IFN-γ (Figure 5C).
We next analyzed cytokine secretion in presence of blocking anti-IFNγ, antibody. While suppressing the IFN-γ autocrine effect strongly impacted the production of IFN-γ, it had no effect on GM-CSF production. Yet, GM-CSF secretion was diminished in Foxo3−/− cells whereas T-bet expression remained unchanged (Figure 5D and S5D). These results further support the notion that the Foxo3-Eomes pathway, but not T-bet, is critical for GM-CSF regulation. Similar results were obtained in T cells overexpressing Eomes following lentiviral transduction. Under conditions in which the IFN-γ was blocked and expression of T-bet was low, Eomes overexpression still resulted in increased IFN-γ and GM-CSF expression. (Figure 5E–F). These data further demonstrate that Eomes can act independently of T-bet to control IFN-γ and GM-CSF secretion.
Foxo3 controls the severity to neuroinflammation
We next addressed the in vivo relevance of the Eomes-Foxo3 pathway by assessing whether Foxo3 deficiency modifies the susceptibility to experimental autoimmune encephalomyelitis (EAE), a well-characterized mouse model for multiple sclerosis (MS). Hence, Foxo3−/− female mice and their wild type littermates were immunized with MOG35–55 peptide emulsified in CFA. While the incidence of EAE disease and the mean day of onset were similar, Foxo3−/− mice developed a much less severe disease than their wild-type counterparts (Figure 6A). Comparable results were obtained when male mice were used, showing that there was no gender bias (Figure S6A). To assess whether this decreased EAE severity was the consequence of a bias in the TCR repertoire, Foxo3−/− mice were crossed to 2D2 TCR transgenic mice in which the CD4+ T cell population expresses an I-Ab-restricted TCR specific for the immune-dominant MOG35–55 peptide (Bettelli et al., 2003). Foxo3 deficiency in 2D2 mice also led to a reduction of disease severity (Figure 6B). To exclude the implication of Foxo3 expression by the resident cells of the CNS, passive EAE was induced in Foxo3−/− and WT recipients by transfer of WT MOG-specific CD4+ T cells differentiated in vitro into encephalitogenic Th1 and Th17 cells. The analysis of clinical scores showed that the incidence and severity of EAE induced was similar between the two genotypes (Figure S6B), thereby excluding any implication of Foxo3 expression in the target organ.
Figure 6. Total Foxo3-deficient mice and mice with a T-cell specific deletion of Foxo3 are less susceptible to EAE.
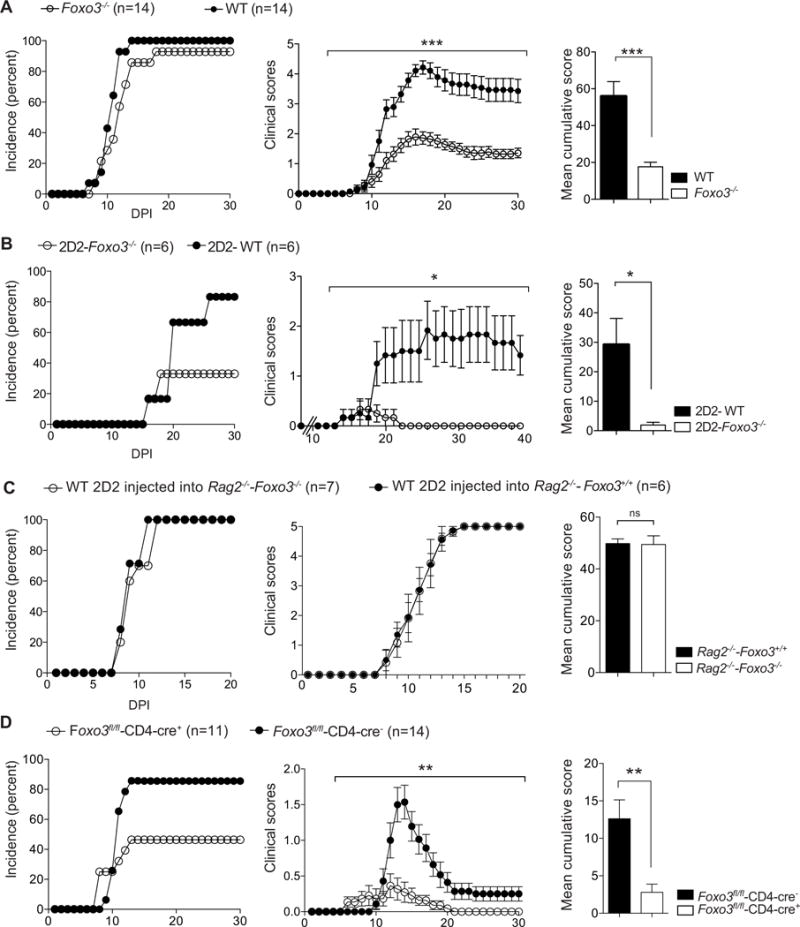
(A) Foxo3−/− (open circles, bars) and WT littermate mice (black circles, bars) were immunized with 50μg of peptide MOG35–55 emulsified in CFA at day 0 and 200 ng of pertussis toxin was injected iv. on day 0 and day 2 (n=14 mice per genotype) (B) 2D2-Foxo3−/− (open circles, bars) or 2D2-WT (black circles, bars) were injected iv. with 150 ng of Pertussis Toxin at day 0. (n=6 mice per genotype) (C) Rag2−/−-Foxo3−/− (open circles, bars) or Rag2−/−-Foxo3+/+ (black circles, bars) mice were injected iv. with 2.104 2D2-WT naive CD4+ T cells mixed with 4.106 WT CD4+ T cells. Mice were then immunized with 50μg of peptide MOG35–55 emulsified in CFA and injected iv. with 100 ng of pertussis toxin. (n=6–7 mice per genotype). (D) Foxo3fl/fl-Cd4-cre+ (open circles, bars) or Foxo3fl/fl-Cd4-cre− (black circles, bars) littermate controls were immunized as in A. Incidence and mean cumulative clinical scores are shown (n=11–14 per genotype). Incidence, clinical scores and mean with SEM of cumulative clinical scores were calculated. Error bars, s.e.m.; P values (Mann–Whitney U test); P values for clinical Scores (2way ANOVA). Data are representative of at least three independent experiments. See also Figure S6
Additional experiments were conducted to decipher the relative contribution of Foxo3 in T cells vs APCs during EAE. The impact of a Foxo3 deficiency in non-T cells was assessed by transferring WT MOG35–55 specific 2D2 CD4+ T cells into Foxo3-deficient or -sufficient Rag2−/− mice. Mice were next immunized and disease severity was evaluated. Both groups of mice developed EAE with similar incidence, kinetics and severity (Figure 6C). These data point to a minimal role of Foxo3 in non-T cells during EAE development. Furthermore, EAE experiments were next conducted on genetically engineered mice harboring a T-cell specific deletion of Foxo3 (Foxo3fl/fl -Cd4-cre). Foxo3fl/fl -Cd4-cre+ mice developed disease with a reduced incidence and severity as compared to Foxo3fl/fl -Cd4-cre− control mice demonstrating that Foxo3 controls the susceptibility to EAE in a T cell-intrinsic manner (Figure 6D). Altogether, these results reveal that Foxo3 expression in CD4+ T cells plays a critical role in the susceptibility to CNS inflammation.
Foxo3 drives the differentiation of pathogenic IFN-γ+and GM-CSF+ CD4+ T cells during EAE
We next assessed whether the outcome of EAE in Foxo3-deficient mice was accompanied by differences in polarization of both peripheral and CNS-infiltrating CD4+ T cells. In agreement with our results obtained in vitro, MOG-specific Foxo3-deficient CD4+ T cells produced lower amounts of the effector cytokines IFN-γ and GM-CSF whereas the production of IL-17, TNF and other cytokines was not affected (Figure 7A and S7A). Intracellular staining was performed to identify which Th cell subset was impacted by Foxo3 deficiency. We observed a large decrease in the proportion of both IFN-γ+ GM-CSF− and IFN- γ + GM-CSF+ CD4+ T cells. The frequency of IL17+ GM-CSF− cells was not impacted, whereas Foxo3 deficient CD4+ T cell exhibited a slight decreased frequency of IL17+ GM-CSF+ cells (Figure 7B). The frequency of Foxp3 Treg cells was unaffected in immunized Foxo3-deficient mice (Figure 7C).
Figure 7. Foxo3-deficiency in T cells is associated with reduced differentiation of IFN-γ and GM-CSF pathogenic CD4+ T cells during EAE.
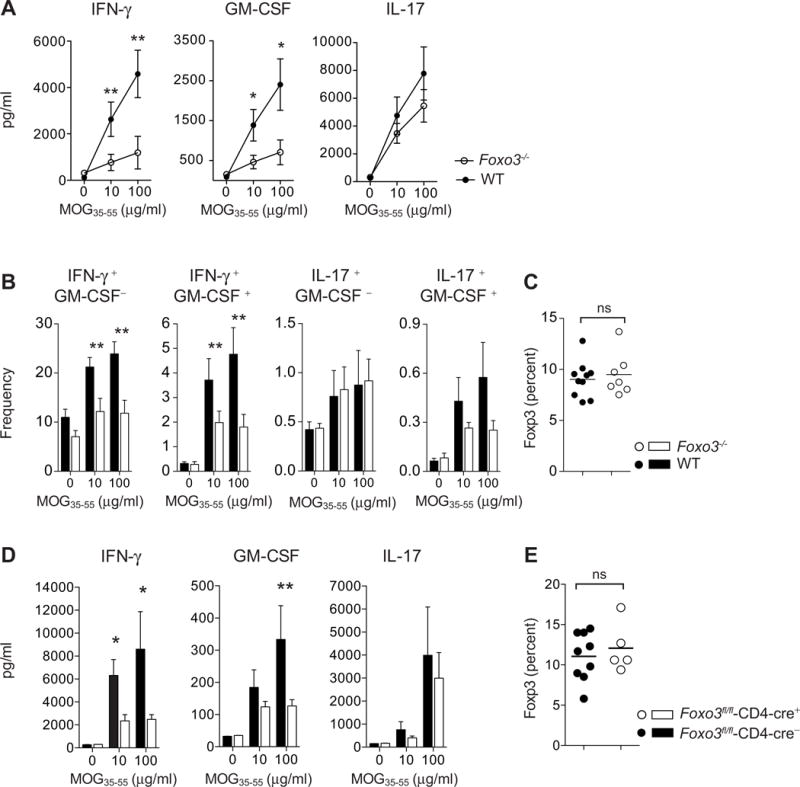
(A) Foxo3−/− (open circles, n=8) and WT littermate mice (black circles, n=8) were immunized with 50μg of peptide MOG35–55 emulsified in CFA. At day 9 post-immunization, CD4+ T cells were purified from spleens and restimulated in vitro with WT APC and MOG35–55 peptide: The secretion of IFN-γ, GM-CSF and IL-17 was analyzed by ELISA in the supernatant after 3 days of culture (n=4 mice per genotype) (B) Frequency of IFN-γ, GM-CSF and IL-17 producing CD4+ T cells was determined by intracellular staining after overnight restimulation with MOG35–55 peptide (n=8 mice per genotype). (C) The expression of Foxp3 by splenic CD4+ T cells from WT and Foxo3−/− mice was assessed by intracellular staining (n=8 mice per genotype) (D) Foxo3fl/fl-Cd4-cre+ or Foxo3fl/fl-Cd4-cre− littermate controls were immunized with 100μg of peptide MOG35–55 emulsified in CFA. At day 9 post-immunization, splenocytes were restimulated in vitro with MOG35–55 peptide and IFN-γ, GM-CSF and IL-17 secretion was analyzed by ELISA (n=9 mice per genotype). (E) The expression of Foxp3 by splenic CD4+ T cells from Foxo3fl/fl-Cd4-cre+ or Foxo3fl/fl-Cd4-cre− mice was assessed. (n=8 mice per genotype). Data are representative of at least two independent experiments. Error bars, s.e.m.; P values (Mann–Whitney U test). See also Figure S7
To address whether this defective CD4+ T cell differentiation was also observed in CNS infiltrating leukocytes, mononuclear infiltrating cells from the spinal cord and brain of Foxo3−/− and WT littermate mice were isolated and characterized by flow cytometry. Analysis of T cell distribution in brain vs spinal cord showed that Foxo3-deficient T cells migrated preferentially to the brain at the expense of the spinal cord (Figure S7B–D). As for their peripheral counterparts, Foxo3-deficient CD4+ T cells from the brain and spinal cord exhibited a decreased capacity to secrete IFN-γ and GM-CSF (Figure S7C–E). The proportion of CNS-infiltrating Foxp3+ CD4+ T cells was not altered by Foxo3 deficiency (Figure S7F).
As described in total Foxo3−/− mice, MOG-specific CD4+ T cells from Foxo3fl/fl -Cd4-cre+ exhibited decreased secretion of IFN-γ and GM-CSF whereas IL-17 secretion was unchanged (Figure 7D). Again, the frequency of Foxp3 Treg cells was unaltered in both periphery and CNS (Figure 7E and S7G). Altogether, these results reveal the T cell intrinsic control of Foxo3 on encephalitogenic CD4+ T cell differentiation and susceptibility to CNS autoimmunity.
DISCUSSION
Up to now, the role of Foxo3 in CD4+ T cell has been unappreciated, mainly because of its low expression in lymphoid cells and also because of the dominant role of Foxo1. The present study showed that TCR engagement results in increased expression of Foxo3 in CD4+ T cells and that this increase correlates with TCR signaling strength. Moreover, this increased Foxo3 expression has a functional impact on CD4 T cells. Foxo3 deletion in primary CD4+ T cells specifically impaired their ability to secrete IFN-γ and GM-CSF. Importantly, microarray analyses showed that decreased expression of genes involved in the IFN-γ pathway was not associated with global defect of CD4+ T cell activation or changes in expression of genes from Th2, Th17 or Treg cell programs further demonstrating that Foxo3 plays a specific role in the polarization of pathogenic CD4+ T cells. These results are consistent with our in vitro and in vivo results showing that, after anti-CD3 stimulation or immunization with MOG35–55 peptide, CD4+ T cells from Foxo3−/− mice showed a decreased production of IFN-γ and GM-CSF whereas the ability of these cells to secrete IL-17, type 2 cytokines or IL-10 was not affected. We therefore conclude that Foxo3 deficiency is not associated with a general defect in CD4+ T cell activation but rather impacts Th polarization by specifically disturbing the production of both IFN-γ and GM-CSF.
Several studies show that Foxo factors are crucial for Foxp3 Treg cell development and function (Kerdiles et al., 2010; Ouyang et al., 2010; Ouyang et al., 2012). We demonstrated here that the Treg cell program is not altered in Foxo3-deficient cells and that Foxo3-deficient Treg cells are as suppressive as WT Treg cells. Moreover, Foxo3 deficiency did not impact the proportion of peripheral or CNS-infiltrating Foxp3 Treg cells during EAE. Therefore, Foxo3 is not necessary for development, differentiation, migration or function of Foxp3 Treg cells.
Analysis of the molecular mechanism underlying these phenotypes revealed that Foxo3 induces expression of the TF Eomes. We showed that, Eomes expression is controlled by TCR signaling strength and correlates with the dynamics of Foxo3 expression in CD4+ T cells, supporting the notion that Foxo3 might regulate Eomes in CD4+ T cells. Eomes was indeed a direct target gene of Foxo3 in CD4+ T cells. Transactivation of Eomes by Foxo3 was dependent upon a 3′UTR distal region containing two FBS and may correspond to an enhancer region. Accordingly, the analysis of Foxo3 genome-wide binding profile showed that this TF acts as a transcriptional activator, regulating target gene expression through transcription initiation by binding preferentially to enhancer regions with increased conservation (Eijkelenboom et al., 2013a; Eijkelenboom et al., 2013b).
In CD4+ T cells, most of the described roles for Eomes are redundant with T-bet (Steiner et al., 2011; Suto et al., 2006; Yang et al., 2008). Here, we have provided information on the critical role of Eomes, independent of T-bet, in CD4+ T cell polarization. Overexpression of Eomes overcame the defect in IFN-γ and GM-CSF production by Foxo3-deficient CD4+ T cells supporting the notion that Eomes is involved in Ifng and Csf-2 regulation in CD4+ T cells. Moreover, under conditions in which T-bet upregulation was blocked, Eomes overexpression still resulted in increased IFN-γ and GM-CSF expression. These results are in agreement with data showing that Eomes is responsible for the T bet-independent production of IFN-γ in T-bet deficient or GATA3-deficient CD4+ T cells (Yagi et al., 2010; Yang et al., 2008). Therefore, the Foxo3-Eomes axis is part of the signaling events responsible for the first wave of IFN-γ. As a consequence, decreased Eomes expression by Foxo3-deficient cells led to reduction of IFN-γ and disrupted the positive feedback loop by which IFN-γ supports T-bet expression. Indeed, our results demonstrated that neither Eomes nor Foxo3 were able to directly regulate T-bet expression. Moreover, inhibition of the IFN-γ autocrine loop had no effect on GM-CSF secretion, further demonstrating that the Foxo3-Eomes pathway, but not T-bet, is critical for GM-CSF regulation (O’Connor et al., 2013).
Uncontrolled CD4+ T cell polarization may have pathological consequences and lead to autoimmune diseases. We showed that Foxo3 deficiency diminished disease severity, that this phenotype is T-cell intrinsic and correlated with the reduced ability of Foxo3-deficient CD4+ T cells to differentiate into IFN-γ and GM-CSF producing CD4+ T cells. IFN- γ, IL-17 and GM-CSF are the main effector cytokines in the pathophysiology of both EAE and MS (Codarri et al., 2010; Goverman, 2009; Korn et al., 2009). In immunized Foxo3-deficient animals, the frequency of MOG-specific Th17 cells was unaffected, excluding the involvement of Th17 cells in the observed phenotype. Decreased IFN-γ production by Foxo3-deficient CD4+ T cells may impact T cell distribution within the CNS. Indeed, Foxo3-deficient T cells migrated preferentially to the brain rather than spinal cord. These results are consistent with studies showing that the Th17-Th1 cell ratio of infiltrating T cells determines the topography of CNS inflammation (Goverman, 2009; Stromnes et al., 2008). However, we can not exclude that Foxo3 might have a direct role in T cell migration and homing since microarray analysis showed that Foxo3-deficient CD4+ T cells exhibited increased expression of Klf2, S1pr1, Sell and decreased expression of Ccr8.
Perhaps most importantly, we showed that Foxo3 deficiency also impacted the ability of CD4+ T cell to produce GM-CSF, a key factor in the effector phase of EAE (McQualter et al., 2001; Ponomarev et al., 2007). Both Th1 and Th17 cells can secrete GM-CSF during EAE (Codarri et al., 2011). However, a recent study showed that GM-CSF+ Th cells might represent a unique Th lineage distinct from that of Th1 and Th17 cells (Herndler-Brandstetter and Flavell, 2014; Sheng et al., 2014). The factors regulating Csf2 expression remain to be defined (Croxford et al., 2015). Here, we have shown that GM-CSF-producing CD4+ T cells exhibited high and sustained expression of Eomes and low Eomes expression impaired the differentiation of GM-CSF producing cells. These data suggest the implication of this TF in the gene program of GM-CSF secreting CD4+ T cells. In agreement, recent transcriptomic studies showed that Eomes is among the genes that are specifically expressed by the GM-CSF+ Th lineage (Sheng et al., 2014). The role of this T-Box transcription factor in CNS neuroinflammation has recently been demonstrated. Indeed, mice harboring a T cell-specific deletion of Eomes developed EAE with reduced severity, a similar phenotype as Foxo3-deficient mice (Raveney et al., 2015). Moreover, EOMES has been identified as a susceptibility gene in MS (Parnell et al., 2014; Patsopoulos et al., 2011). In addition, an increased proportion of Eomes+ CD4+ T cells has reported in patients with secondary progressive MS as compare to relapsing remitting MS or healthy controls and these cells accumulates in the CSF from MS patients further supporting the role of this transcription factor in CNS inflammation in human (Raveney et al., 2015).
EXPERIMENTAL PROCEDURES
Mice
Foxo3−/− (Dejean et al., 2009), 2D2 (Bettelli et al., 2003), Eomesfl/flCd4-cre (Zhu et al., 2010), Foxo3fl/flCd4-cre (Paik et al., 2007) and C57BL/6 mice were maintained in the breeding facility of PreCREFRE (Toulouse UMS06) under SPF conditions. All animal procedures were conducted in accordance with institutional guidelines on Animal Experimentation and were under a French Ministry of Agriculture license.
Encephalomyelitis autoimmune experimental (EAE)
To induce active EAE, mice were immunized with 50μg of MOG35–55 peptide (Polypeptide) emulsified with Complete Freund Adjuvant (CFA) containing 2mg/mL of Mycobacterium tuberculosis (Difco). 200 ng/mL of pertussis toxin (COGER) was given at day 0 and day 2 after immunization. For Foxo3fl/fl-Cd4-cre, 100μg of MOG35–55 peptide was used. Clinical score were evaluated on a five-stage scale from 0 to 5.
CD4+ T cell purification, stimulation and Flow cytometry
Naive CD62L+ CD4+ T cells were obtained by negative selection of total CD4+ T cells (Dynal) and positive selection by CD62L+ beads (Myltenyi). Naive CD4+ T cells were stimulated with anti-CD3 antibody (Biolegend) with or without anti-CD28 (BD Biosciences) in non-polarizing condition or with IL-12 and IL-2 (R&D) for Th1 cells polarizing condition. Cytokines and transcription factor expression were measured by intracellular staining using the “Foxp3 staining buffer” (Ebioscience). Antibodies were all purchased from Ebioscience, BD Pharmingen or Cell signaling for anti-Foxo3 mAbs (clone 75D8). All samples were acquired and analyzed with the LSR II flow cytometer (Becton Dickinson) and FlowJo software (TreeStar).
Microarray gene expression study
Gene expression analysis were performed on purified naive CD4+ T cells from Foxo3−/− (n=3–4) or WT (n=4) littermate controls either unstimulated (T0) or stimulated with 2 μg/ml of anti-CD3 mAbs for 12 (T12) or 24 (T24) hours at the GeT facility (GénoToul, Génopole Toulouse Midi-Pyrénées) using Agilent Sureprint G3 Mouse microarrays (8×60K, design 028005) following the manufacturer’s instructions. Microarray data and experimental details are available in the Gene Expression Omnibus (GEO) database (accession GSE86287).
Chromatin Immunoprecipitation
CD4+ T cells were stimulated with anti-CD3 (2 μg/ml) and anti-CD28 (1 μg/ml) mAbs for 24 hours. Foxo3 ChIP experiments were performed using iDeal ChIP-Seq Kit for Transcription Factors (Diagenode, C01010055) with some modifications. Briefly, cells were fixed with 1% PFA during 15 minutes then glycine (0.250 mM) was added. Cells were then lysed with manufacturer’s buffers and sonicated with 15 cycles of 30sec ON/60 sec OFF using a bioruptor pico. Sonicated chromatin was incubated overnight at 4°C either with 5 μg of anti-Foxo3 antibody (Santa-Cruz, sc-11351X) or an IgG control. Chromatin was then washed and eluted using manufacturer’s recommendations. For ChIP analysis, QPCR was performed using SyberGreen Master mix (Roche) on a 480 LightCycler in duplicate with primers listed in Table S1. Percent of Input was calculated using the following formula: 2^(adjusted INPUT-Ct (IP))*100 where adjusted INPUT = Ct INPUT − log2 (1).
Luciferase assay
HEK 293T cells were co-transfected both with Eomes_Luc or pEomes_p3′E_luc plasmids together with plasmids coding for different forms of FOXO3: (FOXO3TM, FOXO3TM-H212R or FOXO3-A32A253-Nter) or with an empty vector using Genejuice (Novagen). Luciferase assays were performed with a dual luciferase assay kit (Promega, Dual-Luciferase Reporter Assay System, E1910) and all luciferase activities were normalized to the expression of the co-transfected Renilla luciferase.
Lentiviral vector transduction of naive CD4+ T cells
The gene encoding eomes was synthetized and fully sequenced by life technologies. The cDNA was then inserted into a pWPXLd-IRES-GFP backbone vector using BamHI and PmeI restriction sites to make the pWPXLD-Eomes-IRES-GPF vector. 5×106 naive Foxo3−/− CD4+ T cells were activated with anti-CD3 (3ug/ml) plus soluble anti-CD28 (2 μg/ml) and IL-2 (10UI/ml) in p24 well plates coated overnight with 40ug/ml of RetroNectin (TAKRA). 18 hours after activation, the medium was replaced by OptiMEM medium containing lentiviral particles (LV-EOMES or LV-GFP). Anti-CD28 and IL-2 were added (10UI/ml and 2ug/ml respectively). Cells were then centrifugated (3000 rpm) for 1 h at 32°C and incubated overnight at 37°C. The next day, supernatant was replaced by complete RPMI medium supplemented with IL-2 (10 UI/ml) and anti-CD28 (2ug/ml). 72H after transduction, infected cells were then activated with PMA plus ionomycin (0.5 μg/ml each) for 4h plus Golgiplug (1/1000). Cells were then stained and analyzed by flowcytometry (FACS LSRII).
Statistical Analysis
p values were determined by Mann-Whitney tests. p values < 0.05 were considered statistically significant (*** = P<0.001, ** = P<0.005, * = P<0.01). All error bars represent the SEM. For EAE clinical scores, p values were determined by 2way ANOVA (*** = P<0.001, ** = P<0.005, * = P<0.01).
Supplementary Material
Acknowledgments
We would like to thank Dr. B. Li (Institut Pasteur of Shanghai, China) for providing us the the pGL3-Eomes-Luc reporter construct; Dr. C. Charvet (Institut Cochin, Paris, France) for FOXO3 plasmids; A. Thouard and Dr. M. Szelechowski for advice in lentiviral production; Dr. V. Adoue and Dr. O. Joffre for their help on ChIP experiments; Dr. S. Kassem for her expertise on EAE experiments; Dr. L.T. Mars for his help with funding; Dr. D. Dunia, Dr. N. Fazilleau and Dr. R. Lesourne for their critical comments on the manuscript. We would also like to thank the flow cytometry and microscopy core facility (CPTP) and the animal house staff members for their technical assistance (UMS 06). This work was supported financially by grants from INSERM (Nouveau-recruté), the ANR (project ANR-09-RPDOC-015-01) and ARSEP Fondation (project R112195BB). C.S was the recipient of a doctoral fellowship from ARSEP Fondation (R14067BB).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
C.S and M.F.M performed experiments, data analysis, and helped to write the manuscript, M.B, N.C and I.B performed experiments, data analysis and helped with in vivo experiments. X-H.N performed experiments related to luciferase constructs. Y.L performed microarray data analysis and design. S.M. H provided mice and helped to write the manuscript. F.D helped with in vitro experiment. R.L and A.S gave advice for experiment procedures and helped to write the manuscript. A.S.D performed and oversaw research, designed experiments, and wrote the manuscript.
References
- Amin RH, Schlissel MS. Foxo1 directly regulates the transcription of recombination-activating genes during B cell development. Nature immunology. 2008;9:613–622. doi: 10.1038/ni.1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettelli E, Pagany M, Weiner HL, Linington C, Sobel RA, Kuchroo VK. Myelin oligodendrocyte glycoprotein-specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. The Journal of experimental medicine. 2003;197:1073–1081. doi: 10.1084/jem.20021603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- Charvet C, Alberti I, Luciano F, Jacquel A, Bernard A, Auberger P, Deckert M. Proteolytic regulation of Forkhead transcription factor FOXO3a by caspase-3-like proteases. Oncogene. 2003;22:4557–4568. doi: 10.1038/sj.onc.1206778. [DOI] [PubMed] [Google Scholar]
- Codarri L, Fontana A, Becher B. Cytokine networks in multiple sclerosis: lost in translation. Current opinion in neurology. 2010;23:205–211. doi: 10.1097/WCO.0b013e3283391feb. [DOI] [PubMed] [Google Scholar]
- Codarri L, Gyulveszi G, Tosevski V, Hesske L, Fontana A, Magnenat L, Suter T, Becher B. RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nature immunology. 2011;12:560–567. doi: 10.1038/ni.2027. [DOI] [PubMed] [Google Scholar]
- Croxford AL, Spath S, Becher B. GM-CSF in Neuroinflammation: Licensing Myeloid Cells for Tissue Damage. Trends in immunology. 2015;36:651–662. doi: 10.1016/j.it.2015.08.004. [DOI] [PubMed] [Google Scholar]
- Dejean AS, Beisner DR, Ch’en IL, Kerdiles YM, Babour A, Arden KC, Castrillon DH, DePinho RA, Hedrick SM. Transcription factor Foxo3 controls the magnitude of T cell immune responses by modulating the function of dendritic cells. Nature immunology. 2009;10:504–513. doi: 10.1038/ni.1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejean AS, Hedrick SM, Kerdiles YM. Highly Specialized Role of Forkhead Box O Transcription Factors in the Immune System. Antioxid Redox Signal. 2010 doi: 10.1089/ars.2010.3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dengler HS, Baracho GV, Omori SA, Bruckner S, Arden KC, Castrillon DH, DePinho RA, Rickert RC. Distinct functions for the transcription factor Foxo1 at various stages of B cell differentiation. Nature immunology. 2008;9:1388–1398. doi: 10.1038/ni.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eijkelenboom A, Burgering BM. FOXOs: signalling integrators for homeostasis maintenance. Nature reviews. Molecular cell biology. 2013;14:83–97. doi: 10.1038/nrm3507. [DOI] [PubMed] [Google Scholar]
- Eijkelenboom A, Mokry M, de Wit E, Smits LM, Polderman PE, van Triest MH, van Boxtel R, Schulze A, de Laat W, Cuppen E, Burgering BM. Genome-wide analysis of FOXO3 mediated transcription regulation through RNA polymerase II profiling. Molecular systems biology. 2013a;9:638. doi: 10.1038/msb.2012.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eijkelenboom A, Mokry M, Smits LM, Nieuwenhuis EE, Burgering BM. FOXO3 selectively amplifies enhancer activity to establish target gene regulation. Cell reports. 2013b;5:1664–1678. doi: 10.1016/j.celrep.2013.11.031. [DOI] [PubMed] [Google Scholar]
- Goverman J. Autoimmune T cell responses in the central nervous system. Nature reviews Immunology. 2009;9:393–407. doi: 10.1038/nri2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrick SM. The cunning little vixen: Foxo and the cycle of life and death. Nature immunology. 2009;10:1057–1063. doi: 10.1038/ni.1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrick SM, Hess Michelini R, Doedens AL, Goldrath AW, Stone EL. FOXO transcription factors throughout T cell biology. Nature reviews. Immunology. 2012;12:649–661. doi: 10.1038/nri3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herndler-Brandstetter D, Flavell RA. Producing GM-CSF: a unique T helper subset? Cell research. 2014;24:1379–1380. doi: 10.1038/cr.2014.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerdiles YM, Beisner DR, Tinoco R, Dejean AS, Castrillon DH, DePinho RA, Hedrick SM. Foxo1 links homing and survival of naive T cells by regulating L-selectin, CCR7 and interleukin 7 receptor. Nature immunology. 2009;10:176–184. doi: 10.1038/ni.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerdiles YM, Stone EL, Beisner DL, McGargill MA, Ch’en IL, Stockmann C, Katayama CD, Hedrick SM. Foxo transcription factors control regulatory T cell development and function. Immunity. 2010;33:890–904. doi: 10.1016/j.immuni.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MV, Ouyang W, Liao W, Zhang MQ, Li MO. The transcription factor Foxo1 controls central-memory CD8+ T cell responses to infection. Immunity. 2013;39:286–297. doi: 10.1016/j.immuni.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annual review of immunology. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- Laine A, Martin B, Luka M, Mir L, Auffray C, Lucas B, Bismuth G, Charvet C. Foxo1 Is a T Cell-Intrinsic Inhibitor of the RORgammat-Th17 Program. Journal of immunology. 2015;195:1791–1803. doi: 10.4049/jimmunol.1500849. [DOI] [PubMed] [Google Scholar]
- Lee JC, Espeli M, Anderson CA, Linterman MA, Pocock JM, Williams NJ, Roberts R, Viatte S, Fu B, Peshu N, et al. Human SNP links differential outcomes in inflammatory and infectious disease to a FOXO3-regulated pathway. Cell. 2013;155:57–69. doi: 10.1016/j.cell.2013.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Ebert PJ, Li QJ. T cell receptor (TCR) and transforming growth factor beta (TGF-beta) signaling converge on DNA (cytosine-5)-methyltransferase to control forkhead box protein 3 (foxp3) locus methylation and inducible regulatory T cell differentiation. The Journal of biological chemistry. 2013a;288:19127–19139. doi: 10.1074/jbc.M113.453357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Tsun A, Gao Z, Han Z, Gao Y, Li Z, Lin F, Wang Y, Wei G, Yao Z, Li B. 60-kDa Tat-interactive protein (TIP60) positively regulates Th-inducing POK (ThPOK)-mediated repression of eomesodermin in human CD4+ T cells. The Journal of biological chemistry. 2013b;288:15537–15546. doi: 10.1074/jbc.M112.430207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQualter JL, Darwiche R, Ewing C, Onuki M, Kay TW, Hamilton JA, Reid HH, Bernard CC. Granulocyte macrophage colony-stimulating factor: a new putative therapeutic target in multiple sclerosis. The Journal of experimental medicine. 2001;194:873–882. doi: 10.1084/jem.194.7.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkenschlager M, von Boehmer H. PI3 kinase signalling blocks Foxp3 expression by sequestering Foxo factors. The Journal of experimental medicine. 2010;207:1347–1350. doi: 10.1084/jem.20101156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor RA, Cambrook H, Huettner K, Anderton SM. T-bet is essential for Th1-mediated, but not Th17-mediated, CNS autoimmune disease. European journal of immunology. 2013;43:2818–2823. doi: 10.1002/eji.201343689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obsil T, Obsilova V. Structural basis for DNA recognition by FOXO proteins. Biochim Biophys Acta. 2010 doi: 10.1016/j.bbamcr.2010.11.025. [DOI] [PubMed] [Google Scholar]
- Oestreich KJ, Mohn SE, Weinmann AS. Molecular mechanisms that control the expression and activity of Bcl-6 in TH1 cells to regulate flexibility with a TFH-like gene profile. Nature immunology. 2012;13:405–411. doi: 10.1038/ni.2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang W, Beckett O, Flavell RA, Li MO. An essential role of the Forkhead-box transcription factor Foxo1 in control of T cell homeostasis and tolerance. Immunity. 2009;30:358–371. doi: 10.1016/j.immuni.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang W, Beckett O, Ma Q, Paik JH, DePinho RA, Li MO. Foxo proteins cooperatively control the differentiation of Foxp3+ regulatory T cells. Nature immunology. 2010;11:618–627. doi: 10.1038/ni.1884. [DOI] [PubMed] [Google Scholar]
- Ouyang W, Li MO. Foxo: in command of T lymphocyte homeostasis and tolerance. Trends in immunology. 2010 doi: 10.1016/j.it.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang W, Liao W, Luo CT, Yin N, Huse M, Kim MV, Peng M, Chan P, Ma Q, Mo Y, et al. Novel Foxo1-dependent transcriptional programs control T(reg) cell function. Nature. 2012;491:554–559. doi: 10.1038/nature11581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik JH, Kollipara R, Chu G, Ji H, Xiao Y, Ding Z, Miao L, Tothova Z, Horner JW, Carrasco DR, et al. FoxOs are lineage-restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell. 2007;128:309–323. doi: 10.1016/j.cell.2006.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parnell GP, Gatt PN, Krupa M, Nickles D, McKay FC, Schibeci SD, Batten M, Baranzini S, Henderson A, Barnett M, et al. The autoimmune disease-associated transcription factors EOMES and TBX21 are dysregulated in multiple sclerosis and define a molecular subtype of disease. Clinical immunology. 2014;151:16–24. doi: 10.1016/j.clim.2014.01.003. [DOI] [PubMed] [Google Scholar]
- Patsopoulos NA, Bayer Pharma, M.S.G.W.G., Steering Committees of Studies Evaluating, I.-b., a, C.C.R.A., Consortium, A.N., GeneMsa, International Multiple Sclerosis Genetics, C. Esposito F, Reischl J, Lehr S, et al. Genome-wide meta-analysis identifies novel multiple sclerosis susceptibility loci. Annals of neurology. 2011;70:897–912. doi: 10.1002/ana.22609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponomarev ED, Shriver LP, Maresz K, Pedras-Vasconcelos J, Verthelyi D, Dittel BN. GM-CSF production by autoreactive T cells is required for the activation of microglial cells and the onset of experimental autoimmune encephalomyelitis. Journal of immunology. 2007;178:39–48. doi: 10.4049/jimmunol.178.1.39. [DOI] [PubMed] [Google Scholar]
- Raveney BJ, Oki S, Hohjoh H, Nakamura M, Sato W, Murata M, Yamamura T. Eomesodermin-expressing T-helper cells are essential for chronic neuroinflammation. Nature communications. 2015;6:8437. doi: 10.1038/ncomms9437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz EG, Mariani L, Radbruch A, Hofer T. Sequential polarization and imprinting of type 1 T helper lymphocytes by interferon-gamma and interleukin-12. Immunity. 2009;30:673–683. doi: 10.1016/j.immuni.2009.03.013. [DOI] [PubMed] [Google Scholar]
- Sheng W, Yang F, Zhou Y, Yang H, Low PY, Kemeny DM, Tan P, Moh A, Kaplan MH, Zhang Y, Fu XY. STAT5 programs a distinct subset of GM-CSF-producing T helper cells that is essential for autoimmune neuroinflammation. Cell research. 2014;24:1387–1402. doi: 10.1038/cr.2014.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner DF, Thomas MF, Hu JK, Yang Z, Babiarz JE, Allen CD, Matloubian M, Blelloch R, Ansel KM. MicroRNA-29 regulates T-box transcription factors and interferon-gamma production in helper T cells. Immunity. 2011;35:169–181. doi: 10.1016/j.immuni.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone EL, Pepper M, Katayama CD, Kerdiles YM, Lai CY, Emslie E, Lin YC, Yang E, Goldrath AW, Li MO, et al. ICOS Coreceptor Signaling Inactivates the Transcription Factor FOXO1 to Promote Tfh Cell Differentiation. Immunity. 2015;42:239–251. doi: 10.1016/j.immuni.2015.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stromnes IM, Cerretti LM, Liggitt D, Harris RA, Goverman JM. Differential regulation of central nervous system autoimmunity by T(H)1 and T(H)17 cells. Nature medicine. 2008;14:337–342. doi: 10.1038/nm1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan JA, Kim EH, Plisch EH, Peng SL, Suresh M. FOXO3 regulates CD8 T cell memory by T cell-intrinsic mechanisms. PLoS pathogens. 2012a;8:e1002533. doi: 10.1371/journal.ppat.1002533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan JA, Kim EH, Plisch EH, Suresh M. FOXO3 regulates the CD8 T cell response to a chronic viral infection. Journal of virology. 2012b;86:9025–9034. doi: 10.1128/JVI.00942-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suto A, Wurster AL, Reiner SL, Grusby MJ. IL–21 inhibits IFN-gamma production in developing Th1 cells through the repression of Eomesodermin expression. Journal of immunology. 2006;177:3721–3727. doi: 10.4049/jimmunol.177.6.3721. [DOI] [PubMed] [Google Scholar]
- Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100:655–669. doi: 10.1016/s0092-8674(00)80702-3. [DOI] [PubMed] [Google Scholar]
- van der Vos KE, Coffer PJ. The Extending Network of FOXO Transcriptional Target Genes. Antioxid Redox Signal. 2010 doi: 10.1089/ars.2010.3419. [DOI] [PubMed] [Google Scholar]
- Yagi R, Junttila IS, Wei G, Urban JF, Jr, Zhao K, Paul WE, Zhu J. The transcription factor GATA3 actively represses RUNX3 protein-regulated production of interferon-gamma. Immunity. 2010;32:507–517. doi: 10.1016/j.immuni.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Xu J, Niu Y, Bromberg JS, Ding Y. T-bet and eomesodermin play critical roles in directing T cell differentiation to Th1 versus Th17. Journal of immunology. 2008;181:8700–8710. doi: 10.4049/jimmunol.181.12.8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Wang Y, Zhu WG. Applications of post-translational modifications of FoxO family proteins in biological functions. J Mol Cell Biol. 2011;3:276–282. doi: 10.1093/jmcb/mjr013. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Ju S, Chen E, Dai S, Li C, Morel P, Liu L, Zhang X, Lu B. T-bet and eomesodermin are required for T cell-mediated antitumor immune responses. Journal of immunology. 2010;185:3174–3183. doi: 10.4049/jimmunol.1000749. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.