

Abstract
Objective:
To report on a novel neuronal target antigen in 3 patients with autoimmune cerebellar degeneration.
Methods:
Three patients with subacute to chronic cerebellar ataxia and controls underwent detailed clinical and neuropsychological assessment together with quantitative high-resolution structural MRI. Sera and CSF were subjected to comprehensive autoantibody screening by indirect immunofluorescence assay (IFA) and immunoblot. Immunoprecipitation with lysates of hippocampus and cerebellum combined with mass spectrometric analysis was used to identify the autoantigen, which was verified by recombinant expression in HEK293 cells and use in several immunoassays. Multiparameter flow cytometry was performed on peripheral blood and CSF, and peripheral blood was subjected to T-cell receptor spectratyping.
Results:
Patients presented with a subacute to chronic cerebellar and brainstem syndrome. MRI was consistent with cortical and cerebellar gray matter atrophy associated with subsequent neuroaxonal degeneration. IFA screening revealed strong immunoglobulin G1 reactivity in sera and CSF with hippocampal and cerebellar molecular and granular layers, but not with a panel of 30 recombinantly expressed established neural autoantigens. Neurochondrin was subsequently identified as the target antigen, verified by IFA and immunoblot with HEK293 cells expressing human neurochondrin as well as the ability of recombinant neurochondrin to neutralize the autoantibodies' tissue reaction. Immune phenotyping revealed intrathecal accumulation and activation of B and T cells during the acute but not chronic phase of the disease. T-cell receptor spectratyping suggested an antigen-specific T-cell response accompanying the formation of antineurochondrin autoantibodies. No such neurochondrin reactivity was found in control cohorts of various neural autoantibody-associated neurologic syndromes, relapsing-remitting multiple sclerosis, cerebellar type of multiple system atrophy, hereditary cerebellar ataxias, other neurologic disorders, or healthy donors.
Conclusion:
Neurochondrin is a neuronal target antigen in autoimmune cerebellar degeneration.
Autoantibodies against neuronal constituents are associated with several severe immune-mediated CNS disorders. These disorders may predominantly affect gray matter structures of different brain regions such as the archicortex of the limbic system, neocortex, and basal ganglia, as well as cerebellar cortex and brainstem.1
In recent years, a significant number of autoantibodies against neuronal surface membrane antigens such as neurotransmitter receptor and ion channel proteins as well as adhesion molecules with direct pathogenic potential and often without an association to cancer have been reported. They include antibodies against aquaporin 4,2 NMDA receptor,3 AMPA receptors 1 and 2,4 GABAA and GABAB receptors,5,6 LGI1,7,8 CASPR2,8,9 glycine receptor,10 DPPX,11 metabotropic glutamate receptors 1 and 5,12,13 and IgLON5.14
If directed against intracellular neuronal antigens like Hu, Yo, Ri, Ma/Ta, and CV2/CRMP5, autoantibodies are generally considered to be epiphenomena of a T-cell-driven paraneoplastic autoimmune reaction.1 However, autoantibodies against intracellular autoantigens without a tight connection to cancers have also been described.15 Because of their limited access to their target antigens, they probably bear no pathogenic potential in vivo. Experimental transfer of such autoantibodies to model animals does not conclusively produce clinical symptoms and antibody-depleting treatments in patients in most cases do not lead to lasting improvement.1
We report on a novel intracellular neuronal target antigen in 3 patients with autoimmune cerebellar degeneration.
METHODS
Standard protocol approvals, registrations, and patient consents.
All patients were recruited at the Department of Neurology, University of Münster, Germany. All patients gave written informed consent to the study, which was approved by the local ethics committee (AZ 2013 350-f-S) and includes scientific evaluation and publication of all clinical, paraclinical, and scientific data obtained.
Patients.
All patients were assessed clinically by experienced neurologists (K.S.G., C.S., T.W., N.M.). Cerebellar dysfunction was rated using the Scale for the Assessment and Rating of Ataxia (SARA)16 and documented by videography following written informed consent of the patients.
Control collectives included 37 healthy donors, 33 patients with neurologic symptoms and defined antineural autoantibodies (5× anti-NMDAR, 5× anti-Hu, 2× anti-Hu/anti-Ri, 3× anti-Yo, 2× anti-Yo/anti-Ri, 3× anti-Ri, 5× anti-AQP4, 5× anti-LGI1, 3× anti-CASPR2), 36 treatment-naive patients with relapsing-remitting multiple sclerosis (RRMS), 20 patients with the cerebellar type of multiple system atrophy (MSA-c), 35 patients with hereditary cerebellar ataxias, and 150 consecutive patients with various neurologic disorders collected from all participating neurologic departments.
Details on neuropsychological assessment and quantitative high-resolution structural MRI can be found in the e-Methods at Neurology.org/nn.
Identification of the antigen.
Immunoprecipitation, identification of the antigen, cloning of the expression vector, and heterologous expression of neurochondrin in HEK293 cells was performed as described in the e-Methods.
Multicolor flow cytometry of peripheral blood (PB) and CSF.
Multicolor flow cytometry of peripheral blood (PB) and CSF of all patients and controls were performed as described.17,18
Details on indirect immunofluorescent assay (IFA) and T-cell-receptor (TCR) Vb spectratyping can be found in the e-Methods.
Statistics.
If not stated otherwise, all statistical analyses were performed using Sigma Plot 11 (Systat, Erkrath, Germany) or GraphPad Prism (Graphpad Software, Inc., La Jolla, CA). Data were tested for normality using the D'Agostino Pearson Omnibus test. All normally distributed data are given as mean with SD. All not normally distributed data are given as median with interquartile range. If not stated otherwise, the pre-chosen significance level for all confirmatory tests was set to p < 0.05. Levels of significance are indicated as p > 0.05 (not significant), p < 0.05, and p < 0.01.
RESULTS
Characterization of the patients.
Three patients presented with a pronounced cerebellar and brainstem syndrome of unknown origin (SARA score: patient 1 27/40, patient 2 25/40, patient 32/40; video e-1–e-3; a detailed case description can be found in appendix e-1). Patients 1 and 2 had short disease durations of 21 and 54 months, respectively, whereas patient 3 had clinical symptoms for about 120 months. Symptoms were still progressive in patients 1 and 2 but essentially stable for a couple of years in patient 3. Patient 2 also reported one episode of blurred vision, reduced color vision, and pain upon movement of the right eye for several weeks.
Quantitative high-resolution structural MRI at 3T showed pronounced cerebellar atrophy in all patients (figure e-1, A–C) with pronounced supratentorial symmetric atrophy of cortical more than subcortical gray matter and largely preserved white matter volumes in all patients compared to a cohort of 38 age-matched healthy controls (figures e-1, D–F, and e-2 “pre”). Moreover, infratentorial cerebellar gray more than white matter atrophy was evident in all patients as compared to a cohort of 38 age-matched healthy controls. Diffusion tensor imaging (DTI; figures e-3 and e-4 “pre”) revealed widespread reduction of fractional anisotropy (FA; figures e-3, A–E, and e-4 “pre”) together with increased axial diffusivity (AD; figures e-3F and e-4 “pre”) and radial diffusivity (RD; figures e-2G and e-4 “pre”) in white matter areas to different degrees in all patients as compared to a cohort of 38 age-matched healthy controls.
Despite these widespread structural changes visible on MRI, neuropsychological assessment revealed normal performance in verbal working memory, verbal short-term and long-term memory, as well as executive function (tables e-1 and e-2). Executive function could not be assessed in patient 3 due to severe cerebellar motor symptoms.
All patients had chronic inflammatory changes (i.e., pleocytosis, ≥3 CSF-specific oligoclonal immunoglobulin G [IgG] bands19) upon routine CSF analysis and were thus assumed to have autoimmune or paraneoplastic cerebellar degeneration. Routine testing of serum and CSF for known antineuronal antibodies was unremarkable. However, binding to an unknown intracellular submembrane neuronal antigen both in serum and CSF of all 3 patients was detected. Repeated tumor searches using whole-body FDG-PET/CT together with gynecologic or urologic examination did not reveal an underlying tumor in any of the patients for more than 2–10 years.
Patients had shown no sustained response to preceding antibody-depleting (IV immunoglobulin [IVIg], immunoadsorption, plasma exchange) or short-term immunosuppressive treatments (cyclophosphamide, rituximab, mitoxantrone; appendix e-1). Due to persistent clinical progression, consequent long-term immunosuppressive treatment using cyclophosphamide pulse therapy (750–1,000 mg/m2 body surface area every 4–6 weeks) for 6–12 months was introduced in patients 1 and 2. In patient 3, immunotherapy was continued using azathioprine (150 mg/day) and recurrent IVIg treatments (0.4 g/kg body weight for 5 consecutive days every 3 months), under which clinical stability was present for a couple of years.
Clinical follow-up after 6–12 months showed stable or improved disease in all patients under the respective treatment regimen (SARA score: patient 1 25/40, patient 2 23/40, patient 3 32/40) with definitively halted progression in patients 1 and 2 and continued stability in patient 3. Follow-up quantitative high-resolution structural MRI (figure e-2 “post”) revealed partial regression of cortical and cerebellar gray matter atrophy in all patients. However, DTI (figure e-4 “post”) showed largely unaltered widespread reduction of FA (figure e-4, first row) together with increased AD (figure e-4, second row) and RD (figure e-4, third row) in white matter in all 3 patients consistent with a partial response to long-term immunosuppressive treatment but persistent microstructural damage.
Characterization of the patients' autoantibodies.
Indirect IFA of patients' sera and CSF using permeabilized cryosections of brain tissue showed a fine-granular IgG1 staining of the granular and molecular layer that was stronger on rat hippocampus compared to rat and primate cerebellum (figure 1). Sagittal sections of murine whole brain showed autoantibody reactivity distributed over all parts of the brain (figure e-5). In addition, primate hypothalamus, cerebrum, medulla spinalis, and hippocampus were stained, similarly. The patients had serum and CSF titers of 1:320/1:32 (patient 1), 1:320/1:1 (patient 2), and 1:1,000/1:32 (patient 3) as determined with rat hippocampus corresponding to specific antibody indices (AI) >4 in patients 1 and 3 indicating intrathecal antibody synthesis. Titers were largely unaltered during immunosuppressive treatment. Further monospecific analyses were conducted with recombinant HEK293 cells expressing 30 neural autoantigens: Hu, Yo, Ri, CV2, SOX1, PNMA1, PNMA2, ITPR1, Homer 3, CARP VIII, ARHGAP26, ZIC4, DNER/Tr, GAD65, GAD67, amphiphysin, recoverin, GABAB receptor, glycine receptor, DPPX, glutamate receptors (types NMDA, AMPA, mGluR1, mGluR5), LGI1, CASPR2, AQP4 (M1 and M23), MOG, ATP1A3. None revealed any specific reactivity.
Figure 1. Immunofluorescence staining of central nervous tissues.

Cryosections were incubated with patient or control sera (1:100) or CSF (undiluted) in the first step, and with Alexa488-labelled goat anti-human immunoglobulin G in the second step. Nuclei were counterstained by incubation with TO-PRO-3 iodide (blue). A fine granular staining of both granular and molecular layers was obtained with the strongest reaction on the granular cell layer of rat hippocampus. ×200 magnification.
Identification of neurochondrin as the target neuronal autoantigen.
Immunoprecipitates from homogenized rat hippocampus and cerebellum obtained with the patients' sera presented a protein of approximately 75 kDa in sodium dodecyl sulfate polyacrylamide gel electrophoresis, which was absent if the homogenates were incubated with normal control sera (figure 2A). Using matrix-assisted laser desorption/ionization–time of flight, the 75 kDa protein fraction was identified as neurochondrin from Rattus norvegicus (UNIPROT acc. #O35095).
Figure 2. Immunoprecipitation and antigen identification.
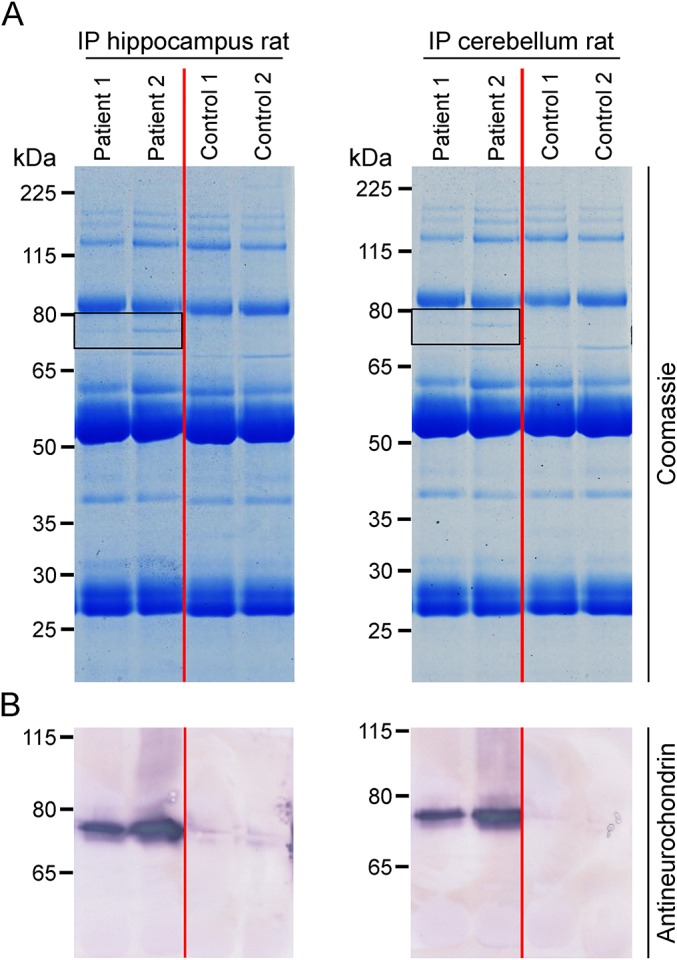
Lysates of rat hippocampus and cerebellum were incubated with patient or control sera (1:33). Immunocomplexes were isolated with protein-G-coated magnetic beads, eluted by sodium dodecyl sulfate, and subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis analysis followed by (A) staining with colloidal Coomassie or (B) Western blot using polyclonal rabbit antineurochondrin. Frames indicate the position of the immunoprecipitated antigen at about 75 kDa.
Western blot analysis of the immunoprecipitates received with the patients' sera but not controls revealed a strong reaction at 75 kDa using a polyclonal rabbit antineurochondrin antibody (figure 2B). When used in IFA, rabbit antineurochondrin produced a fluorescence pattern on rat hippocampus matching that generated by the patients' sera (figure 3A). In IFA with permeabilized hippocampal neurons, both the patient sera and the rabbit antineurochondrin antibody showed a predominant reactivity against the cytoplasm of the neuronal cell body but also stained the neurites to a weaker extent (figure 3B).
Figure 3. Double-staining of hippocampal tissue and cells with patient serum and rabbit antineurochondrin antibody.
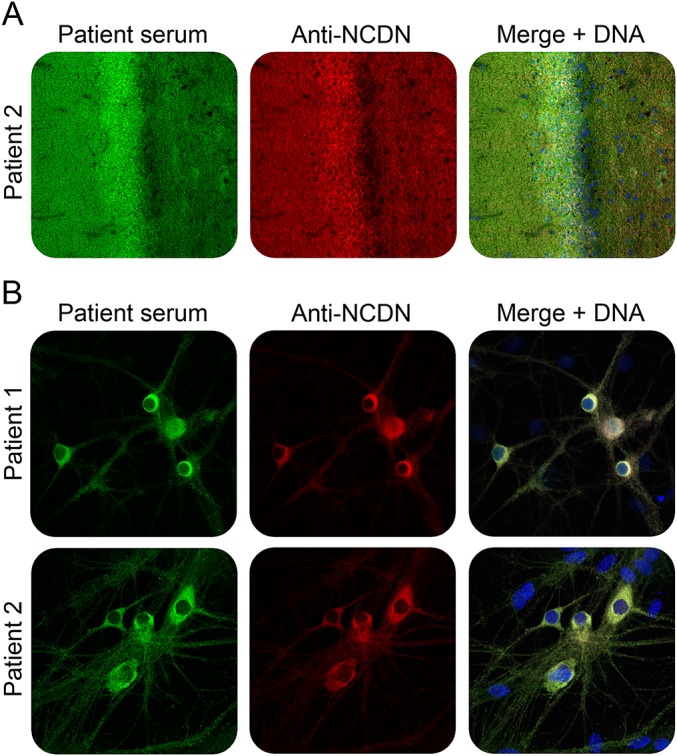
Immunofluorescence staining of rat hippocampus tissue section (A) or formalin-fixed and TritonX-100 permeabilized rat hippocampal neurons (B) with patient sera (green) and antineurochondrin antibody (red). Nuclei were counterstained by incubation with TO-PRO-3 iodide (blue). (A, B) ×200 magnification.
As a proof for correct antigen identification, the patients' samples were then tested by IFA using transfected HEK293 cells that expressed neurochondrin (figure 4A). All patients' sera and CSFs reacted with the neurochondrin-expressing cells with 10-fold higher endpoint titers than on tissue and, consistently, the antibody subclass was IgG1. All samples also reacted with recombinant neurochondrin in immunoblot using HEK293-neurochondrin lysate. In contrast, mock-transfected cells did not demonstrate any specific antibody binding. Specific antibody indices as determined with HEK293-neurochondrin were also AI >4 in patients 1 and 3.
Figure 4. Verification of neurochondrin as the novel autoantigen by indirect immunofluorescence.

(A) Indirect immunofluorescence using acetone-fixed neurochondrin or mock-transfected HEK293 cells incubated with 1:1,000 diluted serum or 1:10 diluted CSF of patient 1, patient 2, or a healthy control (green). (B) Neutralization of immunofluorescence reaction on neuronal tissues. Patient serum (green) was preincubated with extracts of HEK293 cells transfected with neurochondrin or with empty vector as control. The extract containing neurochondrin abolished the immune reaction. Nuclei were counterstained by incubation with TO-PRO-3 iodide (blue). (A) Hippocampus tissue section: ×200 magnification. (B) Rat hippocampal neurons: ×400 magnification.
The reaction of the patients' autoantibodies on tissue could be abolished by preincubation with HEK293 lysate containing neurochondrin (figure 4B). Antibody binding was unaffected when a comparable fraction from mock-transfected HEK293 cells was used.
Anti-neurochondrin autoantibodies are not detected in several control cohorts.
Sera from 33 patients with various neural autoantibody-associated neurologic syndromes in part also involving cerebellum and brainstem (anti-NMDA receptor, anti-Hu, anti-Yo, anti-Ri, anti-AQP4, anti-LGI1, anti-CASPR2), 36 treatment-naive patients with RRMS, most of them with infratentorial lesions, 20 patients with MSA-c, 35 patients with hereditary cerebellar ataxias, 150 consecutive patients with various other neurologic disorders, and 37 healthy controls were analyzed by IFA and with HEK293-neurochondrin in parallel to the samples of the patients. None of the control sera produced a similar immunofluorescence pattern as the patients' sera on rat brain tissue, and all were negative when tested on HEK293 cells expressing neurochondrin. Hence, we consider IgG antibodies against neurochondrin specific for patients with autoimmune cerebellar degeneration. Moreover, these autoantibodies do not seem to merely represent an unspecific secondary epiphenomenon of cerebellar and brainstem inflammation or degeneration, although this cannot be fully excluded.
Characterizing the autoimmune response in neurochondrin antibody-associated cerebellitis.
Immunophenotyping of PB and CSF (figure e-6) revealed that inflammatory changes were most pronounced in patient 1, patient 2 was intermediate, and patient 3 was largely within normal limits in line with the clinical disease activity. Before long-term immunosuppression with cyclophosphamide, patient 1 exhibited elevated numbers (figure e-6B) and fractions (figure e-6A) of CD4+ T cells and activated CD4+ HLADR+ T cells in PB and CSF. In contrast, numbers (figure e-6B) and fractions (figure e-6A) of CD8+ T cells and activated CD8+ HLADR+ T cells as well as CD19+ B cells and CD138+ CD19+ plasma cells were elevated in CSF but not in PB. Following long-term immunosuppression, these changes were largely regressive (figure e-6). The peripheral and intrathecal accumulation and activation of T cells in addition to B cells might reflect a T-cell response towards neurochondrin-expressing neurons in autoimmune cerebellar degeneration with neurochondrin antibodies.
Indeed, TCR spectratyping in PB (figure e-7) revealed increased fractions of Vβ families with skewed or oligoclonal spectratypes and decreased fractions of Vβ families with normal or shifted spectratypes in CD4+ more than CD8+ T cells in patients 1 and 2 but not patient 3 compared to a cohort of 11 healthy controls. In patients 1 and 2, the degree of TCR repertoire perturbation was comparable or even higher compared to that reported for patients with multiple sclerosis or chronic inflammatory demyelinating polyneuropathy.20,21 Notably, the CD8+ TCR repertoire showed an unusual high degree of perturbation while the CD4+ TCR repertoire was much more normally distributed in healthy controls (figure e-7) compared to published values.20,21 These changes largely persisted throughout the disease course and suggest an antigen-driven activation of CD8 T cells and CD4 T cells in addition to the presence of antineurochondrin antibodies. Whether the target of the T-cell immune response is neurochondrin or not is currently unknown.
DISCUSSION
We present 3 cases of autoimmune cerebellar degeneration associated specifically with the presence of high titers of IgG1 antibodies against neurochondrin in serum and CSF. All patients had chronic inflammatory CSF changes together with an intrathecal synthesis of antineurochondrin antibodies. Consistently, during the active but not the residual state of the disease, B cells and plasma cells as well as activated CD4+ and CD8+ T cells accumulated in the CSF compartment. Moreover, in addition to the B-cell-derived antineurochondrin antibodies, TCR repertoire analysis in the peripheral immune compartment suggested an antigen-driven activation of CD8+ T cells and CD4+ T cells.
Structural MRI data were consistent with cortical and cerebellar inflammatory gray matter atrophy with subsequent neuroaxonal degeneration. Nevertheless, clinical neuropsychological assessment, especially memory performance, was essentially normal in all patients. Patients did not respond to antibody-depleting and short-term immunosuppressive treatments, but long-term immunosuppressive treatment with cyclophosphamide led to clinical stabilization or improvement together with regressive gray matter atrophy on MRI. However, antineurochondrin antibody titers in serum and CSF stayed largely unaltered. Hence, B-cell-derived antibodies do not seem to overtly contribute to disease pathogenesis in contrast to T cells, consistent with the intracellular cytoplasmic localization of the antigen.
Neurochondrin is a 75-kDa protein with an aminoacid identity of about 98% among rat, mouse, and human orthologous proteins.22 In the adult rodent brain, neurochondrin is highly expressed in cerebellum, amygdala, and hippocampus, and more moderately in striatum and cortex.23,24
Neurochondrin is specifically expressed in neurons in a somato-dendritic distribution.24 Subcellular fractionation experiments indicate that the vast majority of neurochondrin indeed displays a cytosolic localization.24 Neurochondrin expression is regulated during long-term synaptic plasticity induction following diverse environmental stimuli. In this context, neurochondrin was found to interact with G-protein-coupled receptors such as metabotropic glutamate receptor (mGluR)1 and mGluR5, both of which are essentially involved in cerebellar and hippocampal long-term synaptic plasticity, respectively.25,26
Autoantibodies against mGluR1 have been described in paraneoplastic cerebellar degeneration associated with Hodgkin lymphoma,13,27,28 where they cause cerebellar motor coordination deficits by a combination of acute effects on synaptic transmission and plasticity and chronic degenerative effects in cerebellar Purkinje cells.29 Disturbance of learning and memory have not been described in these patients. Vice versa, autoantibodies against mGluR5 have been described in paraneoplastic limbic encephalitis associated with Hodgkin lymphoma (Ophelia syndrome).12,30,31 But cerebellar deficits were not reported to be present in these patients. In case of pathogenic effects of antineurochondrin antibodies, impaired hippocampus-based learning and memory would be expected to occur together with impaired cerebellar function due to the target antigen expression pattern, which was not the case in our patients. In contrast, T cells require neuronal antigen processing and presentation to recognize their target antigen, the capability of which may differ between various brain regions.32 Moreover, properties of the blood–brain barrier may be regionally different, potentially contributing to a preferential migration towards cerebellar but not hippocampal gray matter in antineurochondrin antibody–associated cerebellar degeneration.33–36
Our data thus identify neurochondrin as an intracellular neuronal antigen targeted by specific autoantibodies in autoimmune cerebellar degeneration, and suggest an accompanying antigen-specific T-cell response as underlying effector mechanism. They thus add another important disease entity to the spectrum of immune-mediated cerebellar ataxias.37,38
Supplementary Material
ACKNOWLEDGMENT
The authors thank Laura Grossmann, Susann Satow, Beatrice Schneider, Sabrina Voigt, and Beatrice Witt, Euroimmun, Lübeck, Germany, and Verena Schütte, Schumina Säuberlich, and Kerstin Gottschalk, Department of Neurology, University of Münster, Germany, for technical assistance.
GLOSSARY
- AD
axial diffusivity
- AI
antibody indices
- DTI
diffusion tensor imaging
- FA
fractional anisotropy
- IFA
immunofluorescent assay
- IgG
immunoglobulin G
- IVIg
IV immunoglobulin
- mGluR
metabotropic glutamate receptor
- MSA-c
cerebellar type of multiple system atrophy
- PB
peripheral blood
- RD
radial diffusivity
- RRMS
relapsing-remitting multiple sclerosis
- SARA
Scale for the Assessment and Rating of Ataxia
- TCR
T-cell receptor
Footnotes
Supplemental data at Neurology.org/nn
AUTHOR CONTRIBUTIONS
R.M. and M. Scharf were involved in immunoprecipitation, antibody testing, microscopy, and writing of the manuscript. K.S.G. and C.S. were involved in recruiting and treating of patients and writing of the manuscript. M.H., U.B., A.S.M., and C.C.G. were involved in functional immunophenotyping, spectratyping of the immune cell repertoire, and writing of the manuscript. K.B. and H.L. were involved in neuropsychological assessment of patients and writing of the manuscript. M.D. performed MRI analysis. L.S. and M. Synofzik identified and recruited one patient, provided control sera and CSFs, and were involved in writing of the manuscript. T.W. performed clinical assessment of patients. Y.D. performed mass spectrometric analysis. S.B. and C.P. performed molecular biology work. I.M.B., S.M., and B.T. were involved in antibody testing and microscopy. K.-P.W. provided control sera and CSFs and was involved in writing of the manuscript. H.W., S.G.M., W.S., L.K., and N.M. were involved in the conception and organization of the research project and in writing of the manuscript.
STUDY FUNDING
This study was supported by the German Research Foundation (DFG)–funded Collaborative Research Centre TR128 “Initiating/Effector vs Regulatory Mechanisms in Multiple Sclerosis: Progress towards Tackling the Disease” (Project Z2 to H.W.).
DISCLOSURE
R. Miske holds a patent for a diagnostic test for the detection of autoantibodies against neurochondrin and has been employed by Euroimmun. C.C. Gross received speaker honoraria and travel expenses from Genzyme, Novartis Pharma GmbH, and Bayer Healthcare; is a review editor for Frontiers in Immunology; and received research support from German Research Foundation, BMBF, and University of Münster. M. Scharf holds a patent for a diagnostic test for the detection of autoantibodies against neurochondrin and has been employed by Euroimmun. K.S. Golombeck spends 5% of her time performing immunoabsorption. M. Hartwig, U. Bhatia, A. Schulte-Mecklenbeck, K. Boente, and C. Strippel report no disclosures. L. Schoels received research support from DFG, BMBF, HSP-Selbsthilfegruppe Deutschland eV, and the Förderverein fuer HSP-Forschung. M. Synofzik received speaker honoraria from Actelion Pharmaceuticals and research support from Actelion Pharmaceuticals, Volkswagen Foundation, the Robert Bosch Stiftung, Katarina Witt Stiftung, Deutsche Hereditäre Ataxie Gesellschaft, and Center for Rare Diseases, Tübingen. H. Lohmann reports no disclosures. I.-M. Dettmann is employed by Euroimmun. M. Deppe received travel funding and speaker honoraria from Sanofi Genzyme Corporation and Novartis Pharma GmbH. S. Mindorf is employed by Euroimmun. T. Warnecke received speaker honoraria from Abbvie, Teva, Bayer, Zamboon, and UCB; and consulted for Abbvie and UCB. Y. Denno is employed by Euroimmun AG. B. Teegen reports no disclosures. C. Probst is employed by and holds stock or stock options from Euroimmun. S. Brakopp is employed by Euroimmun. K.-P. Wandinger reports no disclosures. H. Wiendl served on the scientific advisory board for Bayer Healthcare, Biogen Idec, Sanofi-Genzyme, Merck Serono, Novartis, Roche, and Teva; received travel funding and/or speaker honoraria from Bayer Vital GmbH, Bayer Schering AG, Biogen, CSL Behring, EMD Serono, Fresenius Medical Care, Sanofi-Genzyme, Merck Serono, Omniamed, Novartis, Teva, GlaxoSmithKline, and GW Pharmaceuticals; is an editorial board for PLOS One, Neurotherapeutics, and Recent Patents on Inflammation & Allergy Drug Discovery; consulted for Biogen, Merck Serono, Novartis, Omniamed, Roche, and Sanofi-Genzyme; and received research support from Bayer Healthcare, Bayer Vita, Biogen, Merck Serono, Novartis, Sanofi-Genzyme, Sanofi US, TEVA Pharma, German Ministry for Education and Research, European Union, Interdisciplinary Centre of Clinical Research Muenster, German Research Foundation, Else Kröner Fresenius Foundation, Fresenius Foundation, Hertie Foundation, and RE Children's Foundation. W. Stoecker holds a patent for Verfahren und Vorrichtungen für Untersuchungen an unbeweglich gemachtem biologischem Material, Anti-NMDA-Rezeptor-Autoantikörpern zum Einsatz in Diagnoseverfahren, Diagnosekit sowie ein Verfahren zur Untersuchung einer menschlichen Patientenprobe auf das Vorhandensein von Neuromyelitis-optica-spezifischen Antikörpern; and is a member of the board and a shareholder of Euroimmun AG. S.G. Meuth served on the scientific advisory board for Almirall, Bayer Health Care, Biogen, Genzyme, Merck Serono, Novartis, Novo Nordisk, Roche, Sanofi-Aventis, and Teva; received travel funding and/or speaker honoraria from Almirall, Bayer Health Care, Biogen, Genzyme, Merck Serono, Novartis, Roche, and TEVA; is on the editorial board for PLoS One; consulted for Biogen, Genzyme, Merck Serono, Novartis, Roche, and TEVA; and received research support from Bayer Health Care, Biogen, Genzyme, Merck Serono, Novartis, and TEVA. L. Komorowski is employed by Euroimmun AG. N. Melzer received travel funding and/or speaker honoraria from Biogen Idec, GlaxoSmithKline, TEVA, and Fresenius Medical Care; performs immunoadsorption 5% of the time; and received research support from Fresenius Medical Care and Diamed. Go to Neurology.org/nn for full disclosure forms.
REFERENCES
- 1.Melzer N, Meuth SG, Wiendl H. Neuron-directed autoimmunity in the central nervous system: entities, mechanisms, diagnostic clues, and therapeutic options. Curr Opin Neurol 2012;25:341–348. [DOI] [PubMed] [Google Scholar]
- 2.Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med 2005;202:473–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dalmau J, Gleichman AJ, Hughes EG, et al. . Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol 2008;7:1091–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lai M, Hughes EG, Peng X, et al. . AMPA receptor antibodies in limbic encephalitis alter synaptic receptor location. Ann Neurol 2009;65:424–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lancaster E, Lai M, Peng X, et al. . Antibodies to the GABA(B) receptor in limbic encephalitis with seizures: case series and characterisation of the antigen. Lancet Neurol 2010;9:67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Petit-Pedrol M, Armangue T, Peng X, et al. . Encephalitis with refractory seizures, status epilepticus, and antibodies to the GABAA receptor: a case series, characterisation of the antigen, and analysis of the effects of antibodies. Lancet Neurol 2014;13:276–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lai M, Huijbers MG, Lancaster E, et al. . Investigation of LGI1 as the antigen in limbic encephalitis previously attributed to potassium channels: a case series. Lancet Neurol 2010;9:776–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Irani SR, Alexander S, Waters P, et al. . Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan's syndrome and acquired neuromyotonia. Brain 2010;133:2734–2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lancaster E, Huijbers MG, Bar V, et al. . Investigations of caspr2, an autoantigen of encephalitis and neuromyotonia. Ann Neurol 2011;69:303–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hutchinson M, Waters P, McHugh J, et al. . Progressive encephalomyelitis, rigidity, and myoclonus: a novel glycine receptor antibody. Neurology 2008;71:1291–1292. [DOI] [PubMed] [Google Scholar]
- 11.Boronat A, Gelfand JM, Gresa-Arribas N, et al. . Encephalitis and antibodies to dipeptidyl-peptidase-like protein-6, a subunit of Kv4.2 potassium channels. Ann Neurol 2013;73:120–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lancaster E, Martinez-Hernandez E, Titulaer MJ, et al. . Antibodies to metabotropic glutamate receptor 5 in the Ophelia syndrome. Neurology 2011;77:1698–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sillevis Smitt P, Kinoshita A, De Leeuw B, et al. . Paraneoplastic cerebellar ataxia due to autoantibodies against a glutamate receptor. N Engl J Med 2000;342:21–27. [DOI] [PubMed] [Google Scholar]
- 14.Sabater L, Gaig C, Gelpi E, et al. . A novel non-rapid-eye movement and rapid-eye-movement parasomnia with sleep breathing disorder associated with antibodies to IgLON5: a case series, characterisation of the antigen, and post-mortem study. Lancet Neurol 2014;13:575–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Solimena M, Folli F, Denis-Donini S, et al. . Autoantibodies to glutamic acid decarboxylase in a patient with stiff-man syndrome, epilepsy, and type I diabetes mellitus. N Engl J Med 1988;318:1012–1020. [DOI] [PubMed] [Google Scholar]
- 16.Schmitz-Hubsch T, du Montcel ST, Baliko L, et al. . Scale for the assessment and rating of ataxia: development of a new clinical scale. Neurology 2006;66:1717–1720. [DOI] [PubMed] [Google Scholar]
- 17.Lueg G, Gross CC, Lohmann H, et al. . Clinical relevance of specific T-cell activation in the blood and cerebrospinal fluid of patients with mild Alzheimer's disease. Neurobiol Aging 2015;36:81–89. [DOI] [PubMed] [Google Scholar]
- 18.Golombeck KS, Bonte K, Monig C, et al. . Evidence of a pathogenic role for CD8(+) T cells in anti-GABAB receptor limbic encephalitis. Neurol Neuroimmunol Neuroinflamm 2016;3:e232. doi: 10.1212/NXI.0000000000000307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reiber H, Peter JB. Cerebrospinal fluid analysis: disease-related data patterns and evaluation programs. J Neurol Sci 2001;184:101–122. [DOI] [PubMed] [Google Scholar]
- 20.Mausberg AK, Dorok M, Stettner M, et al. . Recovery of the T-cell repertoire in CIDP by IV immunoglobulins. Neurology 2013;80:296–303. [DOI] [PubMed] [Google Scholar]
- 21.Warnke C, Mausberg AK, Stettner M, et al. . Natalizumab affects the T-cell receptor repertoire in patients with multiple sclerosis. Neurology 2013;81:1400–1408. [DOI] [PubMed] [Google Scholar]
- 22.Mochizuki R, Ishizuka Y, Yanai K, Koga Y, Fukamizu A, Murakami K. Molecular cloning and expression of human neurochondrin-1 and -2. Biochim Biophys Acta 1999;1446:397–402. [DOI] [PubMed] [Google Scholar]
- 23.Istvanffy R, Vogt Weisenhorn DM, Floss T, Wurst W. Expression of neurochondrin in the developing and adult mouse brain. Dev Genes Evol 2004;214:206–209. [DOI] [PubMed] [Google Scholar]
- 24.Shinozaki K, Kume H, Kuzume H, Obata K, Maruyama K. Norbin, a neurite-outgrowth-related protein, is a cytosolic protein localized in the somatodendritic region of neurons and distributed prominently in dendritic outgrowth in Purkinje cells. Brain Res Mol Brain Res 1999;71:364–368. [DOI] [PubMed] [Google Scholar]
- 25.Wang H, Westin L, Nong Y, et al. . Norbin is an endogenous regulator of metabotropic glutamate receptor 5 signaling. Science 2009;326:1554–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu YM, Jia Z, Janus C, et al. . Mice lacking metabotropic glutamate receptor 5 show impaired learning and reduced CA1 long-term potentiation (LTP) but normal CA3 LTP. J Neurosci 1997;17:5196–5205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shams'ili S, Grefkens J, de Leeuw B, et al. . Paraneoplastic cerebellar degeneration associated with antineuronal antibodies: analysis of 50 patients. Brain 2003;126:1409–1418. [DOI] [PubMed] [Google Scholar]
- 28.Marignier R, Chenevier F, Rogemond V, et al. . Metabotropic glutamate receptor type 1 autoantibody-associated cerebellitis: a primary autoimmune disease? Arch Neurol 2010;67:627–630. [DOI] [PubMed] [Google Scholar]
- 29.Coesmans M, Smitt PA, Linden DJ, et al. . Mechanisms underlying cerebellar motor deficits due to mGluR1-autoantibodies. Ann Neurol 2003;53:325–336. [DOI] [PubMed] [Google Scholar]
- 30.Mat A, Adler H, Merwick A, et al. . Ophelia syndrome with metabotropic glutamate receptor 5 antibodies in CSF. Neurology 2013;80:1349–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carr I. The Ophelia syndrome: memory loss in Hodgkin's disease. Lancet 1982;1:844–845. [DOI] [PubMed] [Google Scholar]
- 32.Cebrian C, Zucca FA, Mauri P, et al. . MHC-I expression renders catecholaminergic neurons susceptible to T-cell-mediated degeneration. Nat Commun 2014;5:3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Galea I, Bernardes-Silva M, Forse PA, van Rooijen N, Liblau RS, Perry VH. An antigen-specific pathway for CD8 T cells across the blood-brain barrier. J Exp Med 2007;204:2023–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kooij G, Kroon J, Paul D, et al. . P-glycoprotein regulates trafficking of CD8(+) T cells to the brain parenchyma. Acta Neuropathol 2014;127:699–711. [DOI] [PubMed] [Google Scholar]
- 35.Larochelle C, Lecuyer MA, Alvarez JI, et al. . Melanoma cell adhesion molecule-positive CD8 T lymphocytes mediate central nervous system inflammation. Ann Neurol 2015;78:39–53. [DOI] [PubMed] [Google Scholar]
- 36.Ifergan I, Kebir H, Alvarez JI, et al. . Central nervous system recruitment of effector memory CD8+ T lymphocytes during neuroinflammation is dependent on alpha4 integrin. Brain 2011;134:3560–3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saiz A, Arpa J, Sagasta A, et al. . Autoantibodies to glutamic acid decarboxylase in three patients with cerebellar ataxia, late-onset insulin-dependent diabetes mellitus, and polyendocrine autoimmunity. Neurology 1997;49:1026–1030. [DOI] [PubMed] [Google Scholar]
- 38.Becker EB, Zuliani L, Pettingill R, et al. . Contactin-associated protein-2 antibodies in non-paraneoplastic cerebellar ataxia. J Neurol Neurosurg Psychiatry 2012;83:437–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.