

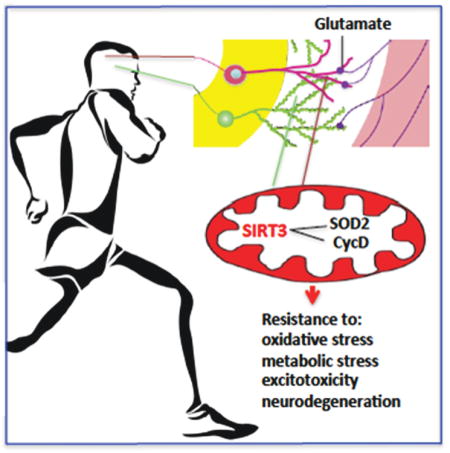
Abstract
The impact of mitochondrial protein acetylation status on neuronal function and vulnerability to neurological disorders is unknown. Here we show that the mitochondrial protein deacetylase SIRT3 mediates adaptive responses of neurons to bioenergetic, oxidative and excitatory stress. Cortical neurons lacking SIRT3 exhibit heightened sensitivity to glutamate-induced calcium overload and excitotoxicity, and oxidative and mitochondrial stress; AAV-mediated Sirt3 gene delivery restores neuronal stress resistance. In models relevant to Huntington’s disease and epilepsy, Sirt3−/− mice exhibit increased vulnerability of striatal and hippocampal neurons, respectively. SIRT3 deficiency results in hyperacetylation of several mitochondrial proteins including superoxide dismutase 2 and cyclophilin D. Running wheel exercise increases the expression of Sirt3 in hippocampal neurons, which is mediated by excitatory glutamatergic neurotransmission and is essential for mitochondrial protein acetylation homeostasis and the neuroprotective effects of running. Our findings suggest that SIRT3 plays pivotal roles in adaptive responses of neurons to physiological challenges and resistance to degeneration.
Keywords: CypD, SOD2, ROS, mPTP, mitochondria, excitotoxicity, neurodegeneration, voluntary exercise
Graphical Abstract
INTRODUCTION
Glutamate, the major excitatory neurotransmitter in the vertebrate brain triggers membrane depolarization, influx of Na+ and Ca2+, and increased mitochondrial oxidative phosphorylation and superoxide production in neurons (Mattson et al., 2008). Neurons normally recover rapidly from excitation by restoring the transmembrane ion gradients, replenishing energy substrates, and removing reactive oxygen species (ROS). This recovery depends critically on properly functioning mitochondria which generate ATP and buffer Ca2+ transients. However, in pathological conditions where neurons are excited excessively (epilepsy), deprived of oxygen and glucose (stroke and cardiac arrest) or suffer more insidious metabolic and oxidative stress (Alzheimer’s, Parkinson’s and Huntington’s diseases), mitochondria fail to counteract the stress and neurons therefore degenerate and die (Mattson, 2003).
When transiently exposed to mild to moderate levels of metabolic, oxidative and excitatory stress, neurons respond adaptively by engaging signaling pathways that bolster their bioenergetics, antioxidant defenses, and abilities to prevent and repair molecular damage (Mattson, 2008). For example, exposure of neurons to a low level of glutamate can protect them from being killed by a higher level of glutamate, and intermittent exercise and fasting can counteract neurodegenerative disease processes in animal models of Alzheimer’s disease (AD), Parkinson’s disease (PD) and Huntington’s disease (HD), epilepsy and stroke (Mattson, 2012; Zigmond and Smeyne, 2014). Evidence supports the involvement of several underlying mechanisms including up-regulation of neurotrophic factors, DNA repair enzymes, protein chaperones and autophagy (Mattson, 2012). Mitochondria might play several roles in adaptive responses of neurons to excitatory, metabolic and oxidative stress (Mattson et al., 2008). For example, manganese superoxide dismutase (SOD2) is an essential antioxidant enzyme that serves as the first line of defense against the superoxide free radicals generated by the electron transport chain. SOD2 can protect neurons against degeneration in experimental models of neurodegenerative disorders (Keller et al., 1998; Andreassen et al., 2001). While mitochondria are crucial for neuronal stress resistance, they are also the organelle that mediates apoptosis, a form of programmed cell death implicated in many acute and chronic neurodegenerative conditions (Mattson, 2000). Excessive DNA damage, Ca2+ overload and oxidative stress causes the association of cyclophilin D with mitochondrial membrane permeability transition pores (PTP) which triggers opening of the PTP, through which cytochrome C passes into the cytosol where it activates caspase enzymes that execute the cell death process (Galluzzi et al., 2009).
Sirtuins are a family of enzymes that remove acetyl groups from lysine residues on specific protein substrates. SIRT1, the most intensively studied sirtuin, is located in the nucleus where it regulates adaptive changes in gene expression in response to bioenergetic challenges (Chalkiadaki and Guarente, 2012). In contrast, SIRT3 is located in mitochondria, where its functions are beginning to be revealed. Recent findings suggest that SIRT3 deacetylates and activates mitochondrial enzymes involved in fatty acid-β-oxidation, amino acid metabolism, the electron transport chain, and antioxidant defenses (He et al., 2012; Kincaid and Bossy-Wetzel, 2013). Studies of non-neuronal cells suggest that SIRT3 mediates adaptations of mitochondria to increased energy demand (Palacios et al., 2009; Kincaid and Bossy-Wetzel, 2013). SIRT3-mediated deacetylation of cyclophilin D suppresses age-related cardiac hypertrophy (Hafner et al., 2010). Moreover, it was recently reported that SIRT3 overexpression protects cultured motor neurons against the cell death-promoting effect of an SOD1 mutation that causes familial ALS (Song et al., 2013) and protects mice against noise-induced hearing loss (Someya et al., 2010), suggesting neuroprotective roles for SIRT3. However, the mechanisms that regulate the expression of SIRT3 in neurons, and its potential roles in neuronal responses to stress are unknown.
RESULTS
SIRT3 deficiency renders neurons vulnerable to excitatory, oxidative and metabolic stress
In preliminary analyses we established that Sirt3−/− mice lack SIRT3 protein in the cerebral cortex, and that SIRT3 is present in high amounts in the mitochondrial fraction and absent from the cytosolic fraction of cortical cells (Supplemental Figure 1A – C). To investigate how SIRT3 can influence neuronal survival, we established cultures of primary neocortical neurons that were isolated and cultured from embryonic day 15 Sirt3+/+ and Sirt3−/− mice. Immunostaining of cells on culture day 7 using antibodies against the neuronal marker beta III tubulin (Tuj1), and the astrocyte marker glial fibrillary acidic protein (GFAP) indicated that the cultures are predominantly Tuj1+ neurons (approximately 95%) for both Sirt3+/+ and Sirt3−/− mice and that cultured cortical neurons from Sirt3+/+ mice exhibit a subcellular distribution of SIRT3 immunoreactivity consistent with a mitochondrial localization (Supplemental Figure 1D and E). There were no apparent differences in neuronal morphology (neurites illustrated by Tuj1 immunostaining) under basal culture conditions. Cultured cortical neurons from Sirt3+/+ mice and Sirt3−/− mice were incubated in the presence of H2O2, glutamate, N-methyl-D-aspartate (NMDA), kainic acid (KA) or 3-nitropropionic acid (3-NPA; succinate dehydrogenase inhibitor) at various concentrations as indicated. After 24 h treatment, live cultures were stained with Hoechst and neuronal cell death was quantified by determining the percentage of neurons that exhibited condensed nuclear DNA with intense Hoechst fluorescence (apoptotic cells) (Figure 1A). The Results were further confirmed by double staining with Tuj1 antibody and Hoechst dye in fixed cells (Figure 1B). Neurons in vehicle-treated control cultures exhibited very weak diffuse nuclear Hoechst staining and intact neurites under phase-contrast microscopy with no differences between neurons from Sirt3+/+ mice and Sirt3−/− mice (Figure 1A, Supplemental Figure 1D). Neurons damaged and killed by glutamate, KA, NMDA, H2O2 and 3NPA exhibited highly condensed nuclear DNA-associated fluorescence and neurite fragmentation or loss depending on the severity of damage (Figure 1A, B), whereas the surviving cells exhibited relatively intact healthy Tuj1+ neurites (Figure 1B). Neurons lacking SIRT3 exhibited a significant increase in vulnerability to excitotoxic death induced by glutamate, KA and NMDA across a broad range of concentrations of each excitatory amino acid (Figure 1A, C, D, E). SIRT3 deficiency also significantly increased the percentage of neurons killed by H2O2 concentrations from 1–50 μM (Figure 1F) and 3-NPA concentrations of 5 and 10 mM (Figure 1G).
Figure 1. Neurons lacking SIRT3 exhibit increased vulnerability to excitotoxic, oxidative and metabolic stress.

(A) Representative merged phase-contrast and Hoechst dye fluorescence (blue) images of cultured cortical neurons from Sirt3+/+ and Sirt3−/− mice that had been exposed for 24 hours to the indicated agents. Viable neurons (indicated by yellow arrows) exhibit weak diffuse nuclear DNA-associated (Hoechst) fluorescence and intact neurites, whereas dying/dead neurons (indicated by red arrows) exhibit intense punctate Hoechst fluorescence as a result of nuclear chromatin condensation, and fragmented neurites. Treatment concentrations: 200 μM glutamate, 200 μM kainic acid (KA), 200 μM N-methyl-D-aspartate (NMDA). Scale bar = 20 μm. (B) Representative images of Hoechst staining (blue), and merged Tuj1 fluorescent immunostaining (green) and Hoechst staining of cultured neurons from Sirt3+/+ and Sirt3−/− mice that had been exposed to 200 μM glutamate for 24 h. Arrows indicate surviving Tuj1+ neurons and arrowheads indicate neurite fragmentation. (C–G) Results of quantitative analysis of neuronal death. Values are mean ± SEM of cell counts performed on cultures established from 4 or 5 mice. *p<0.05, **P<0.01.
SIRT3 deficiency endangers striatal and hippocampal neurons in mouse models of Huntington’s disease and temporal lobe epilepsy
To determine whether SIRT3 plays a role in protecting striatal neurons against 3-NPA, we first administered 3-NPA to Sirt3−/− and Sirt3+/+ mice on a daily basis (30 mg/kg/day) and evaluated survival as an end point (see Methods). Whereas all Sirt3−/− mice succumbed within 7–11 days of the onset of 3-NPA administration, more than 50% of the Sirt+/+ mice survived through day 14, indicating an acceleration of the clinical progression to end-stage in this HD model (Figure 2A). In a second experiment, the motor performance of Sirt3−/− and Sirt3+/+ mice was evaluated on a rotarod apparatus, and then they were administered 3-NPA (30 mg/kg) once daily for 7 days and their performance on the rotarod test was again evaluated. The latency to the first fall from the rotarod and the number of falls over 5 min was recorded in three consecutive trials and the average of these three trials was calculated. Prior to 3-NPA administration the Sirt3−/− and Sirt3+/+ mice exhibited similar levels of performance on the rotarod as indicated by equivalent time intervals to fall off the rotarod and numbers of falls during the testing session (Figure 2B, C). After one week of 3-NPA administration, the rotarod performances of all Sirt3+/+ and Sirt3−/− were significantly impaired. However, the magnitude of motor impairment was significantly greater in Sirt3−/− mice compared to Sirt3+/+ mice (Figure 2B, C). Immediately after evaluation of motor function, the mice were euthanized and their brains prepared for histochemical analysis of damage to striatal neurons. Neurons stained with NeuN appeared to be homeogenously distributed in the cortex and striatum of control Sirt3+/+ and Sirt3−/−. Wild type mice treated with 3-NPA exhibited loss of some striatal neurons but retention of cortical neurons (Figure 2D, E). In contrast, the same dosage of 3-NPA caused a much larger lesion area in the striatum with complete neuronal loss in the central region of the lesion (Figure 2D, E). The extent of neuronal loss caused by 3-NPA in Sirt3−/− mice was significantly greater than that of Sirt3+/+ mice as quantified by measuring the area of cell loss in NeuN-stained sections (Figure 2D, F) or by counting NeuN immunoreactive neurons in vertical bins across the striatal throughout the dorsal - ventral axis of the striatum (Figure 2E, G). These findings indicate that SIRT3 increases the resistance of striatal neurons to degeneration in this animal model of HD.
Figure 2. Striatal neurons of mice lacking SIRT3 exhibit hypersensitivity to degeneration in the 3-nitropropionic acid (3-NPA) model of Huntington’s disease.

(A) Modified Kaplan-Meier survival curves for Sirt3+/+ and Sirt3−/− mice. Mice (10 of each genotype) were given 3-NPA (30 mg/kg) once daily and mortality was quantified as described in Materials and Methods. (B and C) Results of rotarod testing. Before the initial 3-NPA injection, and after the 7th daily 3-NPA injection (30 mg/kg/day) motor performance was evaluated by measuring the latency to the first fall from the rotarod (B) and the total number of falls during a 300 second period (C). Values are mean ± SEM. (8–10 mice per group). (D and E) Confocal images showing a cerebral hemisphere in a coronal brain section immunostained with NeuN (green) to label neuronal nuclei (after 7 days of 3-NPA injection). The lower images show the border (white dotted line) between the area of neuronal damage and the relatively undamaged area. In Sirt3+/+ mice there were many NeuN+ cells in the area of cell loss compared to Sirt3−/− mice which exhibited nearly complete neuronal loss. The area demarcated by the red dotted line indicates the damaged area in the same images of Sirt3+/+ and Sirt3−/− mice, respectively as shown in the corresponding upper panel. Bars = 200 μm. Panel F shows higher magnification confocal images of brain sections stained with NeuN with a vertical column of 200 x 200 μm bins overlayed on the striatum. (F) Graph showing values for area of striatal cell loss determined from analysis of brain sections from Sirt3+/+ and Sirt3−/− mice (after 7 days of 3-NPA injection). Values are mean ± SEM (n = 5–6 mice per group). **P<0.01. (G) The number of neurons (NeuN+ cells) were counted in bins of a vertical strip of each tissue section as showed in (F) that spanned the dorsal-ventral axis of the striatum. Control Sirt3+/+ and Sirt3−/− mice were injected with PBS. Values are mean ± SEM. (n = 5–6 mice per group). *P<0.05, **P<0.01 compared to Sirt3+/+ mice treated with 3-NPA.
To determine whether SIRT3 influences neuronal vulnerability to excitotoxic stress we used a model relevant to temporal lobe epilepsy in which infusion of KA into the dorsal hippocampus causes severe seizures and degeneration of pyramidal neurons in regions CA1 and CA3. In control sham mice, there were no significant differences in the density of CA1 or CA3 neurons between Sirt3+/+ and Sirt3−/− mice. The KA-induced loss of CA1 and CA3 neurons was quantified as described in the Methods Section. The extent of KA-induced loss of CA1 and CA3 neuron was significantly greater in Sirt3−/− mice compared to Sirt3+/+ mice in both CA1 and CA3 regions (Figure 3), indicating that SIRT3 deficiency increases the vulnerability of hippocampal neurons to exictiatory stress in vivo.
Figure 3. Hippocampal neurons of SIRT3-deficient mice exhibit hypersensitivity to seizure-induced degeneration.

(A) Low magnification images showing NeuN immunostaining in coronal sections of hippocampus of Sirt3+/+ and Sirt3−/− mice that had been administered kainic acid (KA) 7 days prior to euthanasia. Boxes 1 and 2 enclose pyramidal neurons in regions CA1 and CA3, respectively. (B and C) Higher magnification confocal images showing NeuN immunostaining (green) and propidium iodide staining (PI, red) in the CA1 (B) and CA3 (C) regions of the hippocampus of mice that had been administered KA 7 days previously. Arrows in the right panels point alive cells with evenly distributed and dimmer PI staining in contrast to the surrounding dead cells with condensed and bright PI staining of nuclei (arrow head). (D and E) Results of quantitative analysis of the number of undamaged neurons in bin areas (200 μm x 50 μm) of CA1 and CA3. Values are mean ± SEM (n= 5 mice per group; analysis was performed on 2 sections anterior to and 2 sections posterior to the needle track. *p < 0.05 and **p <0.01.
SIRT3 suppresses oxidative stress, sustains bioenergetics, stabilizes mitochondrial membranes and improves neuronal Ca2+ handling
We first determined whether neurons lacking SIRT3 exhibited increased oxidative stress. Relative levels of global cellular reactive oxygen species (ROS) and mitochondrial ROS (principally superoxide) were evaluated by imaging of 2,7-dichlorofluorescein diacetate (DCF) and MitoSOX Red fluorescence, respectively, in Sirt3−/− and Sirt3+/+ cortical neurons. Levels of DCF and MitoSOX Red fluorescence were significantly greater in neurons lacking SIRT3 compared to wild type neurons (Figure 4A – D). Oxidative stress can compromise the ability of cells to generate ATP. We found that cellular ATP levels were significantly lower in cultured Sirt3−/− neurons compared to cultured Sirt3+/+ neurons (Figure 4E). Moreover, ATP levels were significantly lower in homogenates of cortical and hippocampal tissues of adult Sirt3−/− mice compared to Sirt3+/+ mice (Figure 4F).
Figure 4. Evidence that SIRT3 suppresses mitochondrial oxidative stress, sustains ATP levels, inhibits mitochondrial membrane permeability transition and improves cellular Ca2+ handling in neurons.

(A and B). Representative images of DCF fluorescence (A) and MitoSox Red fluorescence (B) in primary Sirt3+/+ and Sirt3−/− cortical neurons (8 days in culture). The left panels are merged phase-contrast and fluorescence images to enable visualization of neurons that exhibit very low levels of fluorescence. (C and D) Results of measurements of DCF fluorescence (C) and MitoSox Red fluorescence (D) intensities (arbitrary units; AU), expressed as a percentage of the values for Sirt3+/+ neurons. As a positive control for DCF measurement, Sirt3+/+ neurons were exposed to 100 μM H2O2. Values are the mean ± SEM (n = 5 mice). **p<0.01 (Student’s t-test). (E) ATP levels (normalized to cellular protein levels) in primary cultured Sirt3+/+ and Sirt3−/− cortical neurons (8 days in culture). Values are mean ± SEM (n = 4–5 mice). (F) ATP levels (normalized to cellular protein levels) in cerebral cortex and hippocampus tissues from 10 month old Sirt3+/+ and Sirt3−/− mice. Values are mean ± SEM (n = 9–12 mice). **p<0.01 (Student’s t-test). (G and H) Mitochondrial swelling assays. Mitochondria were isolated from the cerebral cortex of adult (12 month old) Sirt3+/+ and Sirt3−/− mice (G) or primary cultured Sirt3+/+ and Sirt3−/− neurons (H), and were then treated with vehicle or cyclosporin A (CsA; 2 μM) and then exposed to 800 μM CaCl2 and the optical density (OD 450 nm) of the mitochondria was monitored. Values are mean ± SEM (separate mitochondrial preparations from 4 or 5 mice). *p<0.05 and **p<0.01. (I and J) Cytosolic and mitochondrial Ca2+ concentrations were measured by imaging of the probes Fluo-8 and Rhod-2, respectively, prior to and during exposure of Sirt3+/+ and Sirt3−/− cortical neurons to 5 μM glutamate (C and D) (n = 15–30 neurons for each trace) without or with 20 min pretreatment with 1 μM CsA. Arrowheads mark the time of glutamate stimulation. Values are mean ± SEM. (K and L) Fluo-8 and Rhod-2 fluorescence (F/F0 value) at 300 s after exposure to glutamate. Values are mean ± SEM. *P<0.05 and **P<0.01.
To further evaluate the roles of SIRT3 in mitochondrial function and stress resistance, we isolated mitochondria from cortical tissues of adult Sirt3−/− and Sirt3+/+ mice, and from primary cultured embryonic cortical neurons from Sirt3−/− and Sirt3+/+ mice. Because excessive glutamate receptor activation and impaired mitochondrial bioenergetics can render cells vulnerable to mitochondrial PTP opening and cell death, we exposed the isolated mitochondria to 800 μM Ca2+ and monitored light scattering using a well-established assay for mitochondrial swelling and PTP opening. Mitochondria isolated from cortical tissue or cultured cortical neurons of Sirt3−/− mice and were significantly more prone to Ca2+-induced mitochondrial swelling compared to mitochondria from Sirt3+/+ mice (Figure 4G, H). The mitochondrial swelling was completely prevented by the PTP inhibitor cyclosporin A. Because PTP opening is critically sensitive to mitochondrial Ca2+ overload, we determined whether SIRT3 deficiency alters subcellular Ca2+ regulation following glutamate-induced Ca2+ influx. Cortical neurons from Sirt3−/− and Sirt3+/+ mice were loaded with fluorescent probes that enable imaging of cytoplasmic free Ca2+ ([Ca2+]i; Fluo-8) and intramitochondrial Ca2+ ([Ca2+]m; Rhod 2). The neurons were then imaged prior to and during exposure to glutamate. When exposed to a low concentration of glutamate (5 μM), neurons from Sirt3+/+ mice exhibited a rapid rise of [Ca2+]i and [Ca2+]m which both peaked within several seconds of glutamate application and then recovered towards baseline levels over the next 2 minutes (Figure 4I, J). In Sirt3−/− neurons the initial peak [Ca2+]i and [Ca2+]m responses to 5 μM glutamate were similar to the responses of Sirt3+/+ neurons; however, in contrast to Sirt3+/+ neurons, neurons lacking SIRT3 exhibited a subsequent sustained elevation of both [Ca2+]i and [Ca2+]m (Figure 4I, J). Pretreatment of neurons with1 μM cyclosporin A (20 min) resulted in recovery of [Ca2+]I in Sirt3−/− neurons back to baseline levels within 5 minutes of exposure to 5 μM glutamate (Figure 4I, K). In Sirt3−/− neurons pretreatment with 1 μM cyclosporin A (20 min) significantly attenuated, but did not completely prevent the delayed elevation of [Ca2+]m measured at 5 min after exposure to 5 μM glutamate (Figure 4J, L). Collectively, the Ca2+ imaging data suggest that SIRT3 deficiency impairs subcellular Ca2+ regulation following glutamate-induced Ca2+ influx, and that inhibition of the mPTP counteracts this adverse effect of SIRT3 deficiency on neuronal Ca2+ homeostasis.
SIRT3 expression is increased in neurons in response to mild excitation and running wheel exercise
Our data above (Figures 1 – 4) suggested that SIRT3 is important for the maintenance of mitochondrial functions by suppressing oxidative stress, sustaining bioenergetics, stabilizing mitochondrial membranes and improving neuronal Ca2+ handling. Previous studies have shown that neurons can be protected against excitotoxic and metabolic stress by challenging them with low levels of excitatory and energetic stress (Mattson, 2008). To determine whether SIRT3 might play a role in such adaptive responses of neurons to excitatory and metabolic challenges, we first exposed cultured wild type neurons for 24 hours to increasing concentrations of glutamate, or the glutamate receptor subtype-specific agonists NMDA and KA, and then measured relative levels of SIRT3 by immunoblot analysis. In the case of each glutamate receptor agonist, neurons treated with a low concentration(s) exhibited elevated SIRT3 levels, whereas neurons treated with higher (excitotoxic) concentrations exhibited reduced SIRT3 levels (Figure 5A–C). We next measured SIRT3 levels in cerebral cortical and hippocampal tissues from Sirt3+/+ mice that had been maintained for 30 days in cages without (sedentary) or with running wheels. Levels of SIRT3 protein and Sirt3 mRNA were elevated by approximately 50% and 350%, respectively, in the cerebral cortex of runner mice compared to sedentary mice (Figure 5D, E). Levels of SIRT3 protein and Sirt3 mRNA were each elevated by approximately 2-fold in the hippocampus of runner mice compared to sedentary mice (Figure 5F, G). Exercise did not significantly affect levels of the mitochondrial marker proteins voltage-dependent anion channel (VDAC: a porin ion channel protein located on the outer mitochondrial membrane) and cytochrome C oxidase subunit I (COXI) (Supplemental Figure 2), suggesting that SIRT3 induction in runner mice was not the result of increased mitochondrial mass. Examination of SIRT3 immunoreactivity in brain sections from sedentary and running Sirt3+/+ mice revealed granular SIRT3 immunoreactivity in NeuN+ cells throughout the hippocampus, cerebral cortex and other regions (Figure 5H). Lack of any SIRT3 immunostaining from Sirt3−/− mice confirmed the specificity of SIRT3 immunostaining in Sirt3+/+ mice. Quantification of SIRT3 immunofluorescence intensity in hippocampal and cortical neurons demonstrated that SIRT3 levels were significantly increased in runner mice compared to sedentary mice.
Figure 5. Low levels of glutamate receptor stimulation, and voluntary wheel running, induce SIRT3 expression in brain cells.<.

br>(A – C) Immunoblot analysis of SIRT3 protein levels in primary cortical neurons that had been exposed for 24 hours to vehicle (control) or the indicated concentrations of glutamate (Glut), kainate acid (KA) or NMDA. Each lane is a sample from an individual culture, and quantitative data include results of three separate experiments. Values were normalized to the actin level in the same sample, and values are expressed as percentage difference from the value for vehicle control cultures. *p<0.05 and **P<0.01 (ANOVA with Student Newman–Keuls post-hoc tests). (D – G) Immunoblot analysis of SIRT3 protein levels (D and F) and quantitative RT-PCR analysis of Sirt3 mRNA levels (E and G) in cerebral cortical (D, E) and hippocampal (F, G) tissues of wild type mice that were maintained for one month without (sedentary) or with running wheels in their cages. Values for SIRT3 protein and Sirt3 mRNA were normalized to actin protein or mRNA, respectively. Values are expressed as a percentage of the average value for sedentary mice and are mean ± SEM (n= 6–8 mice). *p < 0.05 and **p <0.01 (Student’s t-test). (H) Representative confocal images of double-label fluorescence staining of brain sections with a SIRT3 antibody (green) and NeuN (red) showing cerebral cortex of Sirt3−/− and Sirt3+/+ mice, and the hippocampal CA3 region in both sedentary and runner mice. Bar = 50 μm. The results of SIRT3 immunofluorescence intensity quantification are shown in the graph. The mean value in areal bins in the cortex (100 x 100 μm) and hippocampal CA3 region (50 x 100 μm) are expressed as a percentage of the mean value for sedentary mice (mean ± SEM; n= 4 mice). **p <0.01 (Student’s t-test). (I) Immunoblot analysis of SIRT3 in samples of hippocampal tissues of Sirt3+/+ and Sirt3−/− sedentary or running mice that received intraperitoneal injections of saline (control condition) or 0.5 mg/kg of the NMDA receptor antagonist MK80l once daily for 1 week. Sirt3 levels (densitometry analysis) (right panel) were normalized to actin protein levels and expressed as a percentage of the value for Sirt3+/+ sedentary mice. Values are mean ± SEM (n= 6 mice). **p <0.01.
Because we found that SIRT3 expression is increased in cultured neurons in response to low to moderate concentrations of glutamate and NMDA, and because exercise is known to increase glutamatergic neuronal network activity in many brain regions including the hippocampus (Nishijima et al., 2013), we asked whether excitatory glutamatergic synaptic activity was responsible for exercise-induced SIRT3 expression. To test this possibility both sedentary and runner mice received intraperitoneal injections of saline (control condition) or 0.5 mg/kg of the NMDA receptor antagonist MK80l once daily for 1 week. The SIRT3 level increased robustly in response to running in mice in the saline control group, whereas the effect of running on SIRT3 expression was largely abolished in MK801-treated mice (Figure 5I). These findings indicate that glutamatergic signaling mediates the up-regulation of SIRT3 in response to wheel running.
SIRT3 is essential for mitochondrial protein acetylation homeostasis
Our immunoblot analysis of cortical tissue from adult animals using an acetyl-lysine antibody revealed numerous proteins that exhibited increased acetylation in Sirt3−/− mice compared to Sirt3+/+ mice (Figure 6A – C). Previous studies reported that SIRT3 (~28 kDa) is located in mitochondria of non-neuronal cells (Schwer et al. 2002). We found that, in samples from Sirt3+/+ mice, SIRT3 (~28 kDa) was localized exclusively to the mitochondria and, as expected, no SIRT3 protein was detected in samples from the Sirt3−/− mice, confirming the specificity of the antibody for endogenous SIRT3 protein (Supplemental Figure 1). Indeed, whereas multiple proteins were hyperacetylated in the mitochondrial fraction of cortical tissue from Sirt3−/− mice, protein acetylation was not different in cytosolic fractions of cortical tissues from Sirt3−/− and Sirt3+/+ mice (Figure 6B). Densitometric analysis of all acetylated proteins detected by immunoblot analysis using an antibody against acetylysine, revealed that the total acetylation level in Sirt3−/− hippocampal tissue was approximately 2.5 fold of that of Sirt3+/+ hippocampal tissue (Figure 6C). Interestingly, the protein acetylation level in Sirt3+/+ runners appeared unchanged compared to that of Sirt3+/+ sedentary mice, whereas the acetylation level in Sirt3−/− runners appeared greater than in sedentary Sirt3−/− mice (Figure 6C). We next performed a total acetylation assay in the mitochondrial fractions isolated from the cerebral cortex of sedentary or runner (7 days of running) Sirt3+/+ and Sirt3−/−. The acetylation level in the mitochondria of Sirt3+/+ runners was similar to that of sedentary Sirt3+/+ mice. In contrast, running resulted in a significant increase in the acetylation level in the mitochondrial fraction of Sirt3−/− runners compared to sedentary Sirt3−/− mice; the NMDA receptor antagonist MK801 administration did not block this effect of running (Supplemental Figure 3A, B).
Figure 6. Evidence that SIRT3 regulates neural cell mitochondrial protein acetylation homeostasis.

(A) Immunoblot analysis of acetylated proteins, detected using an acetyl-lysine antibody, in samples of cortical tissue of adult (10 months old) Sirt3+/+ and Sirt3−/− mice. (B) Immunoblot analysis of protein acetylation in mitochondrial and cytosolic fractions of cerebral cortex tissue from 10 month-old Sirt3+/+ and Sirt3−/− mice. (C) Immunoblot analysis of acetylated proteins, detected using an acetyl-lysine antibody, in samples of hippocampal tissues of Sirt3+/+ and Sirt3−/− sedentary and runner mice. Actin was used as a loading control. The middle panel shows the densitometry curves for the 8 lanes of the blot in the left panel for the entire span of immunoreactive bands. Total protein acetylation levels (densitometry analysis) (right panel) were normalized to actin protein levels and expressed as a percentage of the value for Sirt3+/+ sedentary mice. Values are mean ± SEM (n= 6 mice). *p<0.05 and **p <0.01. (D and E) Proteins in lysates of hippocampal tissues from sedentary and runner Sirt3+/+ and Sirt3−/− mice were immunoprecipitated with SOD2 or cyclophilin D (CypD) antibodies or normal rabbit IgG (IgG) as negative control, respectively, and subjected to immunoblotting with antibodies to acetyl-lysine (Ac-SOD2 and Ac-CypD). The blots were reprobed with SOD2 and CypD antibodies to control for protein loading. Input means direct immunoblotting of tissue lysate using antibody against SOD2 (D) or CyPD (E) as positive controls. Arrows show the specific acetylated SOD2 (ac-SOD2) and CypD (ac-CypD) bands. Acetylated SOD2 (D) or CypD (E) levels (densitometry analysis) (right panel) are expressed as percentage of the value for sedentary Sirt3+/+ mice. Values are mean ± SEM (n = 6 mice). *p < 0.05, **p <0.01.
We evaluated the acetylation status of SOD2 and cyclophilin D in hippocampal tissue from Sirt3−/− and Sirt3+/+ mice. Whereas levels of total SOD2 and cyclophilin D proteins in brain tissues were unaffected by SIRT3 deficiency (Supplemental Figure 4), both proteins were hyperacetylated in Sirt3−/− mice compared to Sirt3+/+ mice (Figure 6D, E). Running did not affect the acetylation level of SOD2 in Sirt3+/+ mice, but did result in a small significant increase in SOD2 acetylation in Sirt3−/− mice. Collectively, these data suggested that SIRT3 is critical for mitochondrial protein acetylation homeostasis and mitochondrial stress resistance.
SIRT3 plays roles in exercise-induced resistance of neurons to excitotoxicity
Sirt3+/+ and Sirt3−/− mice were housed with or without running wheels for 30 days; the average distance run per day by the mice was 16.8 ± 5.4 km with no significant difference in running distance or time between Sirt3+/+ and Sirt3−/− mice. On day 31, sedentary and runner Sirt3+/+ and Sirt3−/− mice received an intra-hippocampal injection of KA, and additional cohorts of sedentary Sirt3+/+ and Sirt3−/− mice were subjected to a sham operation. Seven days after KA administration, the mice were euthanized and their brains processed for analysis of neuronal survival in regions CA1 and CA3 of the hippocampus in brain sections stained with propidium iodide to label all cells and with a NeuN antibody to label neurons (Figure 7A). The results of cell counts revealed that significantly more neurons survived KA exposure in Sirt3+/+ runner mice compared to Sirt3+/+ sedentary mice (Figure 7B, C), indicating the running can protect CA1 and CA3 neurons against KA-induced cell death. Fewer neurons survived exposure to KA in Sirt3−/− mice compared to Sirt3+/+ mice, consistent with data shown in Figure 3. However, running was ineffective in protecting hippocampal neurons in Sirt3−/− mice against KA-induced death (Figure 7B, C).
Figure 7. SIRT3 deficiency abolishes the excitoprotective effect of running, and neuronal vulnerability to excitotoxicity can be ameliorated by Sirt3 gene therapy, a mitochondria-targeted superoxide scavenger and a PTP inhibitor.

(A) Representative confocal images showing NeuN immunostaining (green) and propidium iodide staining (PI; red) in the CA1 region of the hippocampus of Sirt3+/+ and Sirt3−/− mice that had been running for 30 days prior to, and for 7 days after, administration of KA. (B and C) Results of counts of undamaged neurons remaining in regions CA1 (B) and CA3 (C) of Sirt3+/+ and Sirt3−/− mice that had been maintained for 30 days without (sedentary) or with running wheels in their cages prior to intra-hippocampal infusion of KA, or sham surgery. Values are mean ± SEM (n= 5 mice per group). *p < 0.05, **p <0.01. (D) Immunoblot of SIRT3 and acetylysine in the hippocampal tissues from Sirt3+/+ mice after 24 h saline or KA stereotaxic injections. Actin was used as a loading control. (E) Immunoblot analysis of SIRT3 protein levels and protein acetylation levels in wild type cortical neurons 72 h after infection with AAV-GFP or AAV-Sirt3-IRES-GFP. The SIRT3 protein level and total acetylated protein levels (densitometric analysis) (graphs in panels D and E) are normalized to actin protein levels and expressed as percentage of the value for Sirt3+/+ mice. Values are mean ± SEM (n = 4 mice). *p < 0.05, **p <0.01. (F) Results of quantitative analysis of cell death in cultures of Sirt3+/+ and Sirt3−/− cortical neurons that had been infected with AAV-GFP or AAV-Sirt3-IRES-GFP) for 3 days and then exposed for 24 hours to glutamate, KA or NMDA (200 μM each). Cell death was quantified as described in the Figure 1 legend. (G – I) Sirt3+/+ and Sirt3−/− cortical neurons were pretreated with the indicated agents and then exposed to 200 μM glutamate (G), KA (H) or NMDA (I) for 24 hours. Cell death was quantified as described in the Figure 1 legend. NAC, N-acetyl-L-cysteine (1 mM); mitoTEMPO, a mitochondrial superoxide scavenger (1 μM); CsA, the mitochondrial PTP inhibitor cyclosporin A (1 μM). Values are mean ± SEM (separate cultures from 4 or 5 mice; 6 images per culture were analyzed). *p<0.05 and **P<0.01.
As demonstrated above (Figure 5A – C), neurons treated with high (excitotoxic) concentrations of glutamate receptor agonists exhibited reduced SIRT3 levels. In the KA-induced seizure model of temporal lobe epilepsy we found that SIRT3 levels were significantly decreased within 24 h of KA administration in Sirt3+/+ mice (Figure 7D). The reduction of SIRT3 levels was associated with a significant increase in mitochondrial protein acetylation levels in KA injected mice compared with control mice (Figure 7D). Moreover, in the 3-NPA mouse model of Huntington’s disease SIRT3 levels were significantly reduced and total mitochondrial protein acetylation level was significantly increased in 3-NPA treated mice compared to control mice (Supplemental Figure 3C). These data are consistent with the hypothesis that deacetylation of SIRT3 target proteins is neuroprotective. To test this hypothesis primary cultured Sirt3+/+ and Sirt3−/− neurons were infected with adeno-associated virus (AAV) to increase the levels of SIRT3 protein (Figure 7E) and then total protein acetylation levels in cell lysates were evaluated. When neurons were exposed to an optimal concentration of AAV, over 90% of the cells were infected as indicated by GFP fluorescence (Supplemental Figure 5). Immunoblot analysis showed that 3 days after exposure to virus neurons infected with AAV-GFP-Sirt3 expressed approximately 1.5–2.5 fold higher amounts of SIRT3 protein compared to control neurons infected with AAV-GFP (Figure 7E and Supplemental Figure 5). Examination of neurons in cultures exposed to AAV-GFP-Sirt3 revealed that infected neurons exhibited robust SIRT3 immunoreactivity with a subcellular localization consistent with the expected localization in mitochondria, whereas uninfected neurons exhibited much weaker SIRT3 immunoreactivity (Supplemental Figure 5). We next quantified neuronal cell death in cultured Sirt3+/+ and Sirt3−/− neurons that had been infected with AAV-GFP or AAV-GFP-Sirt3 and then 3 days later were exposed to neurotoxic concentrations of glutamate, KA or NMDA or 3-NPA for 24 hours. Sirt3−/− neurons were significantly more vulnerable than Sirt3+/+ neurons to glutamate, KA, NMDA and 3-NPA. Overexpression of SIRT3 resulted in highly significant reductions in neuronal death induced by each glutamate receptor agonist and 3-NPA in both Sirt3+/+ and Sirt3−/− neurons (Figure 7F and Supplemental Figure 6B).
Superoxide removal and cyclophilin D inhibition protect neurons against excitotoxicity
We next determined whether pharmacological agents known to reduce superoxide levels or inhibit cyclophilin D could protect Sirt3+/+ and/or Sirt3−/− neurons against excitotoxicity. Similar to Sirt3−/− mouse brain tissues, cultured embryonic cortical neurons from Sirt3−/− mice exhibited hyperacetylation of multiple proteins including SOD2 and cyclophilin D compared to cultured cortical neurons from Sirt3+/+ mice (Figure 7E and Supplemental Figure 6A). Cortical neurons from Sirt3+/+ and Sirt3−/− mice were preincubated with N-acetylcysteine (NAC; a glutathione precursor that principally reduces cytosolic ROS), the mitochondria-targeted superoxide scavenger mitoTEMPO or the PTP inhibitor cyclosporin A which acts by inhibiting cyclophilin D. The neurons were then exposed to a neurotoxic concentrations of glutamate, KA, NMDA or 3-NPA for 24 hours and neuronal death was quantified. MitoTEMPO and cyclosporin A afforded significant protection of neurons against death induced by glutamate, KA, NMDA and 3-NPA in both Sirt3+/+ and Sirt3−/− neurons, indicating pivotal roles for mitochondrial superoxide and PTP opening in excitotoxicity and 3-NPA induced cell death (Figure 7G – I, and Supplemental Figure 6B). NAC was moderately effective in protecting neurons against 3-NPA induced cell death, but was ineffective in protecting neurons against excitotoxicity. Interestingly, we did not observe additive effects of MitoTEMPO and cyclosporin A in protecting neurons against different stresses, indicating that oxidative stress and mPTP opening may be coupled events. In contrast to MitoTEMPO and cyclosporin A, NAC was not effective in protecting neurons against excitotoxicity or 3-NPA, although there was a non-significant trend towards protection against 3-NPA (Figure 7G – I, and Supplemental Figure 6B).
DISCUSSION
Our findings provide evidence that SIRT3 plays a critical role in mediating neuroprotective adaptive stress responses. We found that: a) both voluntary running and low levels of glutamate receptor activation induce Sirt3 expression; b) NMDA receptor activation mediates running wheel exercise-induced SIRT3 expression; c) when challenged with metabolic or excitatory stress, neurons lacking SIRT3 exhibit a marked proclivity for dysregulation of mitochondrial function and increased vulnerability to death; d) SIRT3 deficiency abolished the beneficial effects of voluntary running in protecting hippocampal neurons in CA1 and CA3 region against KA-induced cell death; e) overexpression of SIRT3 protects neurons from being killed by metabolic and excitatory stress; f) Mechanistically, SIRT3 is essential for maintaining mitochondrial protein acetylation at low levels under physiological and mild stress conditions. SIRT3 deficiency or SIRT3 reduction under pathological conditions such as in the mouse models of temporal lobe epilepsy, results in hyperacetylation of several mitochondrial proteins including SOD2 and cyclophilin D. Therefore, by deacetylating mitochondrial proteins such as SOD2 and cyclophilin D, SIRT3 protects neurons against metabolic and oxidative stresses by reducing mitochondrial superoxide levels, stabilizing cellular and mitochondrial Ca2+ homeostasis and inhibiting mitochondrial membrane PTP formation to prevent apoptosis.
Our findings suggest that SIRT3 plays important roles in bolstering mitochondrial health, in part by deacetylating SOD2 and cyclophilin D. By deacetylating and thereby activating SOD2, SIRT3 prevents excessive mitochondrial oxidative stress and its potentially damaging sequelae. Second, by deacetylating and inhibiting cyclophilin D, SIRT3 prevents PTP formation, sustains mitochondrial membrane potential and maintains ATP production. Considerable evidence suggests that SOD2 is a direct SIRT3 target (Qiu et al., 2010; Tao et al., 2010, 2014), and that SOD2 is a major guardian of mitochondria in neurons. Indeed, mice with reduced SOD2 levels exhibit features of accelerated aging, neurodegeneration and increased vulnerability to excitotoxicity (Paul et al., 2007; Li et al., 1998) whereas cultured neuronal cells overexpressing SOD2 are resistant to apoptosis induced by amyloid β-peptide and oxidative stress, and cortical neurons in mice overexpressing SOD2 exhibit increased resistance to a focal ischemic stroke (Keller et al., 1998). We found that neurons lacking SIRT3 exhibit hyperacetylation of SOD2 and CypD, and those neurons exhibit increased vulnerability to being killed by excitotoxic, oxidative and metabolic stresses. Conversely, overexpression of SIRT3 protected neurons against excitotoxicity. When subjected to excitatory challenges, neurons lacking SIRT3 exhibited elevated levels of mitochondrial ROS, excessive Ca2+ accumulation in mitochondria and the cytosol, and cell death that was significantly attenuated when PTP opening was inhibited with cyclosporin A or superoxide scavenged by mito-TEMPO, a mitochondrially targeted antioxidant. Whereas mito-TEMPO was effective in protecting against both excitotoxicity and 3-NPA, NAC, which acts by increasing global cellular glutathione levels, was ineffective. Our findings are therefore consistent with suppression of mitochondrial ROS accumulation being a key mechanism by which SIRT3 protects neurons against excitotoxicity and 3-NPA.
Comparisons of the mitochondrial ‘protein acetylomes’ of cultured cell lines in which Sirt3 has been knocked down (Sol et al., 2012) and of liver cells from Sirt3 knockout mice (Hebert et al., 2013) have revealed putative SIRT3 targets including proteins involved in branched chain amino acid catabolism, ketone body synthesis, the citric acid cycle, oxidative phosphorylation, antioxidant responses and mitochondrial biogenesis (Choudhary et al., 2014; Lombard et al., 2007). Our immunoblot analysis of SIRT3 deficient cortical tissue or cultured neurons revealed numerous mitochondrial proteins in a hyperacetylated state. Mitochondrial proteins that were previously shown to be affected by SIRT3-mediated deacetylation include the complex I protein NDUFA9 (Ahn et al., 2008), complex V/ATP synthase (Rahman et al., 2014), the DNA repair enzyme OGG1 (Cheng et al., 2013) and isocitrate dehydrogenase 2 (Yu et al., 2012). Together with SOD2 and cyclophilin D, SIRT3-mediated deacetylation of some of these other proteins may also play roles in adaptive cellular stress responses. In addition, studies of non-neuronal cells suggest that SIRT3 can promote mitochondrial biogenesis (Kong et al., 2010). It is therefore becoming evident that SIRT3 serves as a key regulator of adaptive cellular responses to bioenergetic challenges such as those neurons experience in response to electrical/synaptic activity and vigorous exercise, as demonstrated in the present study. We found that levels of ATP were significantly lower in the hippocampus and cerebral cortex of Sirt3−/− mice compared to wild type mice. Because neurons have high energy requirements to support ongoing synaptic activity, it is possible that there are functional deficits resulting from SIRT3 deficiency. SIRT3 deficiency rendered hippocampal and striatal neurons vulnerable to pathological levels of excitotoxic and metabolic stress that are known to cause neuronal ATP depletion. Whether reduced ATP levels in SIRT3 deficient mice also copromise synaptic function under conditions of physiological challenges remains to be determined.
We found that in the KA-induced seizure model of temporal lobe epilepsy, SIRT3 levels were significantly reduced and acetylation of mitochondrial proteins was increased within 24 hours of seizure induction. Similarly, in the 3-NPA mouse model of Huntington’s disease, SIRT3 levels were decreased and mitochondrial protein acetylation levels were increased in brain cells, and SIRT3 deficiency rendered striatal neurons vulnerable to 3-NPA induced death. We found that in primary cultured neurons and in brain cells in vivo SIRT3 deficiency caused mitochondrial protein hyperacetylations, which could be restored back to physiological levels by overexpression of SIRT3. These findings suggest that SIRT3-mediated deacetylation of SOD2 and cyclophilin D functions as a defense mechanism against excessive oxidative, excitotoxic and metabolic stress. Consistent with a role for SIRT3 in counteracting neurodegeneration in Huntington’s disease, it was recently reported that a phenolic compound increases SIRT3 levels and protects cultured neuroblastoma cells against the toxicity of mutant huntingtin protein (Fu et al., 2012).
Environmental factors and cellular signaling pathways that regulate the expression of SIRT3 in neurons are poorly understood. However, data from studies of skeletal muscle cells suggest roles for SIRT3 in adaptive responses to energetic challenges. As evidence, treadmill running increases SIRT3 expression in soleus an plantaris muscles in rats (Hokari et al., 2010), and transgenic mice that overexpress SIRT3 in skeletal muscle exhibit improved treadmill running performance and a higher percentage of slow oxidative muscle fibers (Lin et al., 2014). Moreover, aerobic exercise training results in an increase in SIRT3 expression in skeletal muscle in overweight human adolescents (Vargas-Ortiz et al., 2015). As with SIRT1, SIRT3 is an NAD+-dependent enzyme, it is likely that energy states that elevate NAD+ levels increase SIRT3 activity. It is therefore likely that, because of their differential subcellular locations, SIRT1 and SIRT3 coordinate inter-organellar responses to energetic challenges. We found that activation of glutamate receptors and running wheel exercise increased the levels of SIRT3 in neurons, consistent with roles for SIRT3 in activity-dependent neuroplasticity and neuroprotection. Previous studies have shown that physiological levels of synaptic glutamate receptor activation can stimulate neurotrophic factor production, up-regulate DNA repair, bolster neuronal bioenergetics, and increase the resistance of neurons to oxidative and excitotoxic stress (Mattson, 2012). Our cell culture and in vivo data suggest that glutamate receptor activation is sufficient to induce SIRT3 expression, and is necessary for up-regulation of SIRT3 expression in hippocampal neurons in vivo in response to running wheel exercise. Our findings further demonstrate pivotal roles for SIRT3 in suppressing mitochondrial oxidative stress, sustaining neuronal bioenergetics and Ca2+ handling. The latter actions of SIRT3 may explain our finding that SIRT3 deficiency negates the neuroprotective effects of running in the KA seizure model. Therefore, up-regulation of the mitochondrial enzyme SIRT3 may be an important mechanism whereby bioenergetics challenges such as exercise and dietary energy restriction bolster brain health.
EXPERIMENTAL PROCEDURES
Mice and primary neuronal cultures
All animal procedures were approved by the Animal Care and Use Committee of the National Institute on Aging Intramural Research Program. Breeding pairs of SIRT3-deficient mice were a generous gift from David Gius (then at the National Cancer Institute and now at Northwestern University). Methods for genotyping can be found in the Supplemental Experimental Procedures and Supplemental Figures 1A and 1B). For some experiments, 6 month-old Sirt3−/− and Sirt3+/+ mice were housed individually with or without access to a running wheel. Primary embryonic cortical neurons were prepared from E15 mice (see Supplemental Experimental Procedures).
Mouse models of Huntington’s disease and epileptic seizures
Methods for the 3-NPA model of HD and the kainic acid-induced seizure model of temporal lobe epilepsy have been described previously (Bruce-Keller et al., 1999) and are detailed in the Supplemental Experimental Procedures). For the HD model, motor performance was evaluated using the ENV-577M Rotarod system (Med Associates, Georgia, VT) as detailed in the Supplemental Experimental Procedures.
Immunohistochemistry and quantification of neuronal damage
Methods for brain fixation and sectioning, and for immunostaining of sections are detailed in the Supplemental Experimental Procedures. For the 3-NPA model, two methods were used to evaluate neuronal loss in coronal sections stained with propidium iodide and NeuN antibody as described in the Supplemental Experimental Procedures. For the KA model, undamaged cells (cells in which the propidium iodide was distributed uniformly in the nucleus, compared to damaged cells with condensed nuclear DNA) in hippocampal regions CA1 and CA3 were quantified as described in the Supplemental Experimental Procedures.
Immunoblot analysis and immunoprecipitation
Immunoblot analysis was performed using 4–10% SDS gradient polyacrylamide gel followed by standard blotting procedure as described previously (Cheng et al., 2012) and detailed in the Supplemental Experimental Procedures. For immunoprecipitation analysis, tissues or cultured neurons were solubilized and protein extracts (500 μg) were used for immunoprecipitation with antibodies against SOD2 or cyclophilin D as detailed in the Supplemental Experimental Procedures.
Quantitative reverse transcriptase PCR amplification
Total RNAs from mouse cortical or hippocampal tissues were extracted and purified using a RNeasy mini kit (Qiagen, Valencia, CA), and analyzed by quantitative RT-PCR as detailed in the Supplemental Experimental Procedures).
Measurement of cellular ROS and Ca2+ levels, and neuronal survival
The fluorescent reactive oxygen species (ROS) indicator 5-(and-6)-carboxy-2′7′-dichlorodihydrofluorescein diacetate (DCF) was used for measuring intracellular accumulation of ROS, and the mitochondrial superoxide indicator MitoSox Red was used for measuring mitochondrial superoxide levels. Fluo-8 AM was used to measure the cytosolic Ca2+ concentration, and Rhod-2 AM was used to measure the mitochondrial Ca2+ concentration. Neuronal survival was evaluated by staining cells with DNA-binding dye Hoechst 33342 and determining the percentage of live and dead cells based on nuclear morphology. Details of methods in this section can be found in the Supplemental Experimental Procedures.
Mitochondrial isolation, and mitochondrial swelling and ATP assays
Cortical tissues or cultured neurons were homogenized in a mitochondria isolation buffer and mitochondria were isolated by multiple centrifugation steps as detailed in the Supplemental Experimental Procedures. Mitochondrial swelling was triggered by the addition of CaCl2 (final concentration of 800 μM) to isolated mitochondria in the presence or absence of 1 μM cyclosporin A. Mitochondrial swelling was monitored by the decrease in light-scattering at 540 nm in a Perkin Elmer spectrophotometer (HTS 7000 plus) for 20 minutes. Brain tissue and primary cultured neurons were lysed and ATP measurements made using a commercially available kit (Promega) as detailed in the Supplemental Experimental Procedures.
Adeno-associated virus (AAV) construction and packaging
Details of the methods for cloning of Sirt3 cDNA into an AAV-GFP vector, and for generation of virus in HEK293 cells can be found in the Supplemental Experimental Procedures.
Statistical analyses
Statistical analyses were performed by one-way ANOVA or T-test using GraphPad Prism (GraphPad, San Diego, CA). Student Newman-Keuls post hoc tests were applied to detect statistical differences between groups. Data are expressed as mean ± SEM.
Supplementary Material
Acknowledgments
We thank Dr. David Gius (NCI) for providing breeding pairs of Sirt3−/− mice. This work was supported by the Intramural Research Program of the National Institute on Aging, and the Glenn Foundation for Biomedical Research.
Footnotes
Author Contributions: A. C., Y. Y. and M. P. M. designed the experiments; A. C., Y. Y., Y. Z., C. M., D. L., W. P., Y. L., R. W., K. M. and M. M., performed the experiments. A. C., Y. Y. and Y. Z. analyzed the data. A. C., Y. Y., V. A. B. and M. P. M. wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahn BH, Kim HS, Song S, Lee IH, Liu J, Vassilopoulos A, Deng CX, Finkel T. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc Natl Acad Sci U S A. 2008;105:14447–14452. doi: 10.1073/pnas.0803790105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreassen OA, Ferrante RJ, Dedeoglu A, Albers DW, Klivenyi P, Carlson EJ, Epstein CJ, Beal MF. Mice with a partial deficiency of manganese superoxide dismutase show increased vulnerability to the mitochondrial toxins malonate, 3-nitropropionic acid, and MPTP. Exp Neurol. 2001;167:189–195. doi: 10.1006/exnr.2000.7525. [DOI] [PubMed] [Google Scholar]
- Bruce-Keller AJ, Umberger G, McFall R, Mattson MP. Food restriction reduces brain damage and improves behavioral outcome following excitotoxic and metabolic insults. Ann Neurol. 1999;45:8–15. [PubMed] [Google Scholar]
- Chalkiadaki A, Guarente L. Sirtuins mediate mammalian metabolic responses to nutrient availability. Nat Rev Endocrinol. 2012;8:287–296. doi: 10.1038/nrendo.2011.225. [DOI] [PubMed] [Google Scholar]
- Cheng A, Wan R, Yang JL, Kamimura N, Son TG, Ouyang X, Luo Y, Okun E, Mattson MP. Involvement of PGC-1α in the formation and maintenance of neuronal dendritic spines. Nat Commun. 2012;3:1250. doi: 10.1038/ncomms2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Ren X, Gowda AS, Shan Y, Zhang L, Yuan YS, Patel R, Wu H, Huber-Keener K, Yang JW, et al. Interaction of Sirt3 with OGG1 contributes to repair of mitochondrial DNA and protects from apoptotic cell death under oxidative stress. Cell Death Dis. 2013;4(7):e731. doi: 10.1038/cddis.2013.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary C, Weinert BT, Nishida Y, Verdin E, Mann M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat Rev Mol Cell Biol. 2014;15:536–550. doi: 10.1038/nrm3841. [DOI] [PubMed] [Google Scholar]
- Fu J, Jin J, Cichewicz RH, Hageman SA, Ellis TK, Xiang L, Peng Q, Jiang M, Arbez N, Hotaling K, Ross CA, Duan W. trans-(−)-ε-Viniferin increases mitochondrial sirtuin 3 (SIRT3), activates AMP-activated protein kinase (AMPK), and protects cells in models of Huntington Disease. J Biol Chem. 2012;287:24460–24472. doi: 10.1074/jbc.M112.382226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Blomgren K, Kroemer G. Mitochondrial membrane permeabilization in neuronal injury. Nat Rev Neurosci. 2009;10:481–494. doi: 10.1038/nrn2665. [DOI] [PubMed] [Google Scholar]
- Hafner AV, Dai J, Gomes AP, Xiao CY, Palmeira CM, Rosenzweig A, Sinclair DA. Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging. 2010;2:914–923. doi: 10.18632/aging.100252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W, Newman JC, Wang MZ, Ho L, Verdin E. Mitochondrial sirtuins: regulators of protein acylation and metabolism. Trends Endocrinol Metab. 2012;23:467–476. doi: 10.1016/j.tem.2012.07.004. [DOI] [PubMed] [Google Scholar]
- Hebert AS, Dittenhafer-Reed KE, Yu W, Bailey DJ, Selen ES, Boersma MD, Carson JJ, Tonelli M, Balloon AJ, Higbee AJ, et al. Calorie restriction and SIRT3 trigger global reprogramming of the mitochondrial protein acetylome. Mol Cell. 2013;49:186–199. doi: 10.1016/j.molcel.2012.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hokari F, Kawasaki E, Sakai A, Koshinaka K, Sakuma K, Kawanaka K. Muscle contractile activity regulates Sirt3 protein expression in rat skeletal muscles. J Appl Physiol (1985) 2010;109:332–340. doi: 10.1152/japplphysiol.00335.2009. [DOI] [PubMed] [Google Scholar]
- Keller JN, Kindy MS, Holtsberg FW, St Clair DK, Yen HC, Germeyer A, Steiner SM, Bruce-Keller AJ, Hutchins JB, Mattson MP. Mitochondrial manganese superoxide dismutase prevents neural apoptosis and reduces ischemic brain injury: suppression of peroxynitrite production, lipid peroxidation, and mitochondrial dysfunction. J Neurosci. 1998;18:687–697. doi: 10.1523/JNEUROSCI.18-02-00687.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kincaid B, Bossy-Wetzel E. Forever young: SIRT3 a shield against mitochondrial meltdown, aging, and neurodegeneration. Front Aging Neurosci. 2013;5:48. doi: 10.3389/fnagi.2013.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong X, Wang R, Xue Y, Liu X, Zhang H, Chen Y, Fang F, Chang Y. Sirtuin 3, a new target of PGC-1alpha, plays an important role in the suppression of ROS and mitochondrial biogenesis. PLoS One. 2010;5:e11707. doi: 10.1371/journal.pone.0011707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Copin JC, Reola LF, Calagui B, Gobbel GT, Chen SF, Sato S, Epstein CJ, Chan PH. Reduced mitochondrial manganese-superoxide dismutase activity exacerbates glutamate toxicity in cultured mouse cortical neurons. Brain Res. 1998;814:164–170. doi: 10.1016/s0006-8993(98)01082-8. [DOI] [PubMed] [Google Scholar]
- Lin L, Chen K, Khalek WA, Ward JL, 3rd, Yang H, Chabi B, Wrutniak-Cabello C, Tong Q. Regulation of skeletal muscle oxidative capacity and muscle mass by SIRT3. PLoS One. 2014;9:e85636. doi: 10.1371/journal.pone.0085636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombard DB, Alt FW, Cheng HL, Bunkenborg J, Streeper RS, Mostoslavsky R, Kim J, Yancopoulos G, Valenzuela D, Murphy A, et al. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol Cell Biol. 2007;27:8807–8814. doi: 10.1128/MCB.01636-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP. Apoptosis in neurodegenerative disorders. Nat Rev Mol Cell Biol. 2000;1:120–129. doi: 10.1038/35040009. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Excitotoxic and excitoprotective mechanisms: abundant targets for the prevention and treatment of neurodegenerative disorders. Neuromolecular Med. 2003;3:65–94. doi: 10.1385/NMM:3:2:65. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Hormesis defined. Ageing Res Rev. 2008;7:1–7. doi: 10.1016/j.arr.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP. Energy intake and exercise as determinants of brain health and vulnerability to injury and disease. Cell Metab. 2012;16:706–722. doi: 10.1016/j.cmet.2012.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Gleichmann M, Cheng A. Mitochondria in neuroplasticity and neurological disorders. Neuron. 2008;60:748–766. doi: 10.1016/j.neuron.2008.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishijima T, Llorens-Martín M, Tejeda GS, Inoue K, Yamamura Y, Soya H, Trejo JL, Torres-Alemán I. Cessation of voluntary wheel running increases anxiety-like behavior and impairs adult hippocampal neurogenesis in mice. Behav Brain Res. 2013;245:34–41. doi: 10.1016/j.bbr.2013.02.009. [DOI] [PubMed] [Google Scholar]
- Palacios OM, Carmona JJ, Michan S, Chen KY, Manabe Y, Ward JL, 3rd, Goodyear LJ, Tong Q. Diet and exercise signals regulate SIRT3 and activate AMPK and PGC-1alpha in skeletal muscle. Aging (Albany NY) 2009;1:771–783. doi: 10.18632/aging.100075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul A, Belton A, Nag S, Martin I, Grotewiel MS, Duttaroy A. Reduced mitochondrial SOD displays mortality characteristics reminiscent of natural aging. Mech Ageing Dev. 2007;128:706–716. doi: 10.1016/j.mad.2007.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X, Brown K, Hirschey MD, Verdin E, Chen D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010;12:662–667. doi: 10.1016/j.cmet.2010.11.015. [DOI] [PubMed] [Google Scholar]
- Rahman M, Nirala NK, Singh A, Zhu LJ, Taguchi K, Bamba T, Fukusaki E, Shaw LM, Lambright DG, Acharya JK, et al. Drosophila Sirt2/mammalian SIRT3 deacetylates ATP synthase β and regulates complex V activity. J Cell Biol. 2014;206:289–305. doi: 10.1083/jcb.201404118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwer B, North BJ, Frye RA, Ott M, Verdin E. The human silent information regulator (Sir)2 homologue hSIRT3 is a mitochondrial nicotinamide adenine dinucleotide-dependent deacetylase. J Cell Biol. 2002;158:647–657. doi: 10.1083/jcb.200205057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sol EM, Wagner SA, Weinert BT, Kumar A, Kim HS, Deng CX, Choudhary C. Proteomic investigations of lysine acetylation identify diverse substrates of mitochondrial deacetylase sirt3. PLoS One. 2012;7:e50545. doi: 10.1371/journal.pone.0050545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Someya S, Yu W, Hallows WC, Xu J, Vann JM, Leeuwenburgh C, Tanokura M, Denu JM, Prolla TA. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell. 2010;143:802–812. doi: 10.1016/j.cell.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W, Song Y, Kincaid B, Bossy B, Bossy-Wetzel E. Mutant SOD1G93A triggers mitochondrial fragmentation in spinal cord motor neurons: neuroprotection by SIRT3 and PGC-1α. Neurobiol Dis. 2013;51:72–81. doi: 10.1016/j.nbd.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao R, Coleman MC, Pennington JD, Ozden O, Park SH, Jiang H, Kim HS, Flynn CR, Hill S, Hayes McDonald W, Olivier AK, Spitz DR, Gius D. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol Cell. 2010;40:893–904. doi: 10.1016/j.molcel.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao R, Vassilopoulos A, Parisiadou L, Yan Y, Gius D. Regulation of MnSOD enzymatic activity by Sirt3 connects the mitochondrial acetylome signaling networks to aging and carcinogenesis. Antioxid Redox Signal. 2014;20:1646–1654. doi: 10.1089/ars.2013.5482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas-Ortiz K, Perez-Vazquez V, Diaz-Cisneros FJ, Figueroa A, Jiménez-Flores LM, Rodriguez-DelaRosa G, Macias MH. Aerobic training increases expression levels of SIRT3 and PGC-1α in skeletal muscle of overweight adolescents without change in caloric intake. Pediatr Exerc Sci. 2015 doi: 10.1123/pes.2014-0112. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Yu W, Dittenhafer-Reed KE, Denu JM. SIRT3 protein deacetylates isocitrate dehydrogenase 2 (IDH2) and regulates mitochondrial redox status. J Biol Chem. 2012;287:14078–14086. doi: 10.1074/jbc.M112.355206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zigmond MJ, Smeyne RJ. Exercise: is it a neuroprotective and if so, how does it work? Parkinsonism Relat Disord. 2014;20(Suppl 1):S123–127. doi: 10.1016/S1353-8020(13)70030-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.