

Abstract
Background
Tribbles proteins are conserved pseudokinases that function to control kinase signalling and transcription in diverse biological processes. Abnormal function in human Tribbles has been implicated in a number of diseases including leukaemia, metabolic syndromes and cardiovascular diseases. Caenorhabditis elegans Tribbles NIPI-3 was previously shown to activate host defense upon infection by promoting the conserved PMK-1/p38 mitogen-activated protein kinase (MAPK) signalling pathway. Despite the prominent role of Tribbles proteins in many species, our knowledge of their mechanism of action is fragmented, and the in vivo functional relevance of their interactions with other proteins remains largely unknown.
Results
Here, by characterizing nipi-3 null mutants, we show that nipi-3 is essential for larval development and viability. Through analyses of genetic suppressors of nipi-3 null mutant lethality, we show that NIPI-3 negatively controls PMK-1/p38 signalling via transcriptional repression of the C/EBP transcription factor CEBP-1. We identified CEBP-1’s transcriptional targets by ChIP-seq analyses and found them to be enriched in genes involved in development and stress responses. Unlike its cell-autonomous role in innate immunity, NIPI-3 is required in multiple tissues to control organismal development.
Conclusions
Together, our data uncover an unprecedented crosstalk involving multiple tissues, in which NIPI-3 acts as a master regulator to inhibit CEBP-1 and the PMK-1/p38 MAPK pathway. In doing so, it keeps innate immunity in check and ensures proper organismal development.
Electronic supplementary material
The online version of this article (doi:10.1186/s12915-016-0320-z) contains supplementary material, which is available to authorized users.
Keywords: Tribbles, C/EBP transcription factor, p38 MAP kinase, Genetic suppression, Transcriptional regulation, Signal transduction, Development, Stress responses
Background
The Tribbles genes encode a family of highly conserved pseudokinases which lack key catalytic amino acids in the kinase domain [1–3]. Functional studies in multiple organisms have shown that these pseudokinases play diverse roles in innate immunity, cell signalling, energy homeostasis and cell division [2, 4]. The Drosophila tribbles gene is required for cell proliferation and migration in embryogenesis and oogenesis [5–8]. The mammalian Tribbles family includes three genes, Trib1, Trib2 and Trib3, each of which plays unique roles in signalling networks regulating adipose tissue, metabolic homeostasis and the immune system [2, 4]. Abnormal function in human Tribbles has been implicated in a number of diseases including leukaemia, metabolic syndromes and cardiovascular disease [9].
Mammalian Tribbles can interact with components of the mitogen-activated protein (MAP) kinase pathway and act as adaptor proteins to modulate the strength and output of kinase signalling cascades [10–12]. Like fly Tribbles [7, 13], mammalian Tribbles bind to the basic leucine zipper (bZIP) transcription factors. Trib1 and Trib2 can induce degradation of several CCAAT/enhancer-binding protein (C/EBP) members in a context-dependent manner [1, 14, 15]. Trib3 binds to and inhibits the transcriptional activity of both activating transcription factor 4 (ATF4) and C/EBP homologous protein (CHOP) in cultured cell lines [16–19], although the in vivo functional relevance of such protein interactions remains unknown.
Given its extensive connections to different cellular processes, many questions remain to be answered regarding the role of Tribbles at the organismal level. No induction of peptide after Drechmeria infection 3 (NIPI-3) is the single Tribbles protein in Caenorhabditis elegans [20]. A role for nipi-3 in the innate immune response was previously uncovered through the isolation of a partial loss-of-function mutation, which contains a missense mutation in the pseudokinase domain [20]. NIPI-3 is required for the upregulation of antimicrobial peptide (AMP) gene expression following infection by the fungus Drechmeria coniospora [20]. It acts upstream of a p38 MAP kinase (MAPK) pathway consisting of NSY-1/MAPKKK, SEK-1/MAPKK and PMK-1/MAPK [20, 21]. Both NIPI-3 and all components of the MAPK cascade are required cell autonomously in the epidermis during the immune response [20].
In this study, we generated null mutations of nipi-3 and uncovered a novel role in animal development and viability. The lethality of nipi-3 null animals is completely suppressed by loss of function in CEBP-1, a C. elegans member of the C/EBP family, previously known to be required for adult sensory axon regeneration and neuronal stress responses [22, 23]. Unexpectedly, loss of function in components of the PMK-1/p38 MAPK cascade also suppresses the lethality of nipi-3 null animals. In nipi-3 mutants, the levels of activated PMK-1 are increased, in a cebp-1-dependent manner. Through chromatin immunoprecipitation and deep sequencing (ChIP-seq) analyses and identification of target genes, we found that CEBP-1 binds to a conserved DNA motif. Our analyses of candidate target genes of CEBP-1 suggest a functional enrichment in development and stress responses. Importantly, in contrast to its role in innate immunity, we show that NIPI-3 acts in multiple tissues to negatively regulate transcriptional expression of cebp-1. This inhibition of CEBP-1 by NIPI-3 is also required across multiple tissues to enable larval development and maintain fecundity. The coordinated inhibition of CEBP-1 by NIPI-3 in multiple tissues reveals novel requirements for systemic regulation of signalling pathways in organism development.
Results
C. elegans Tribbles nipi-3 is required for larval development and viability
To better understand the biological roles of nipi-3, we used clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 genome editing [24–27] to generate two deletion alleles of nipi-3 (fr148 and ju1293) and a green fluorescent protein (GFP) knock-in (KI) (fr152) (Fig. 1a; see Methods and below). The fr148 and ju1293 deletion alleles remove 1.6 kb and 0.6 kb of the 5′ region of the gene, respectively (Fig. 1a), resulting in molecular nulls for nipi-3. The phenotypes of homozygous mutants of either nipi-3 deletion allele, designated as null (0), were indistinguishable (Fig. 1c, d). Mutants arrested development at the second to third larval stages (L2–L3) (see below) and eventually died between 5–10 days after hatching. When compared to wild-type larvae at the same stage (2 days post-hatching), nipi-3(0) arrested larvae displayed a small and dumpy body morphology. At 3 days post-hatching, wild-type animals reached the adult stage, as evidenced by fusion of seam cells (lateral epidermal cells), formation of adult alae and the vulva (Fig. 1b, f). By contrast, all age-matched nipi-3(0) animals were arrested at L2–L3, as the seam cells did not fuse, adult alae were not observed and the vulval invagination did not occur (Fig. 1c, d, f). In these mutant animals, the germline also appeared to be arrested, generally at L3 based on the size of the gonad and the number of germ cells (Fig. 1c, d, g). Occasionally, in nipi-3(0) animals with longer bodies, we observed some sperm or a few unfertilized oocytes. The nipi-3(0) animals also exhibited an abnormal pharyngeal morphology (Additional file 1: Figure S1). We rescued the larval lethality and sterility of nipi-3(0) by expressing the wild-type nipi-3 genomic DNA as high-copy-number extrachromosomal arrays (Fig. 1a, e, h; Methods). As expression from such transgenes is silenced in the germline [28], this result indicates that the larval lethality and germline development defects of nipi-3(0) are both primarily due to its function in somatic tissues. Thus, the analyses of nipi-3 null animals indicate an essential somatic role of nipi-3 in organism development.
Fig. 1.
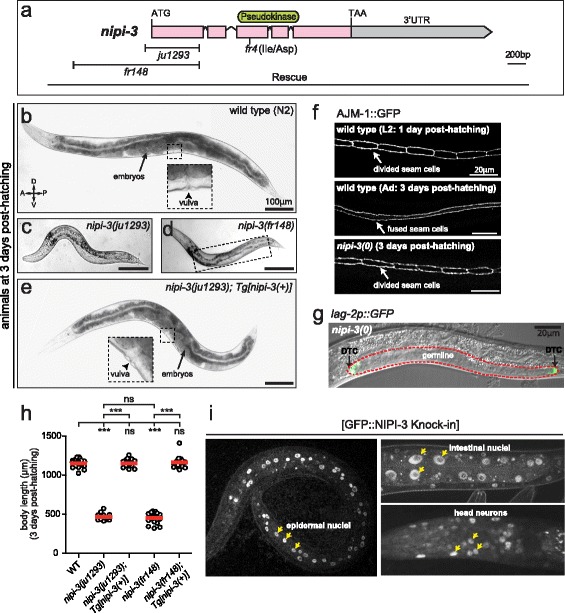
C. elegans Tribbles nipi-3 is required for larval development and viability. a The nipi-3 locus. Top, nipi-3 encodes a pseudokinase of the Tribbles family. Middle, nipi-3 deletions generated using CRISPR-Cas9 genome editing. Bottom, the extent of the nipi-3 genomic region used to rescue the deletion mutants. b–e Bright-field images of worms at 3 days post-hatching; wild type (b), nipi-3 null mutants (c,d) and a transgenic animal (Tg) expressing the wild-type nipi-3 genomic DNA in a nipi-3(0) background (e). f Fluorescence images of worms expressing AJM-1::GFP reporter in the epithelial cells to allow visualization of seam cells. g Overlaid differential interference contrast (DIC) and fluorescence image of a nipi-3(fr148) mutant at 3 days post-hatching expressing lag-2p::GFP reporter. This image corresponds to the boxed region in d. Two distal tip cells (DTC) are shown in green (arrow), and the germline of arrested nipi-3 null larva is denoted by a dotted red line. h Body length (μm) of worms at 3 days post-hatching. Each dot represents a single animal measured as shown; each red line represents the mean value. ***P < 0.001; ns, not significant (one-way ANOVA with Tukey’s post hoc tests). i Fluorescence images of endogenous NIPI-3 expression visualized in the GFP KI strain (fr152). Expression is observed in the nuclei (yellow arrows) of the epidermis (left panel), the intestine (upper right panel) and the head neurons (lower right panel)
Knocking-in gfp to the nipi-3 locus, which produced a protein tagged at its N-terminus (GFP::NIPI-3), had no adverse effect; KI animals (fr152) were fully viable and indistinguishable from wild type in growth and movement. We observed GFP expression in the epidermis, intestine and in neurons (Fig. 1i), consistent with the previously reported expression pattern obtained using transgenic transcriptional reporters [20]. Interestingly, although NIPI-3 does not have a clearly identifiable nuclear-localization signal, GFP::NIPI-3 expression was observed predominantly in the nuclei (Fig. 1i), with an overall intensity peaking at the L2–L3 stages. This nuclear localization suggests a role for NIPI-3 in regulation of gene expression.
Loss of cebp-1 suppresses the lethality, but not the innate immune response defect, of nipi-3 null animals
To dissect the molecular mechanisms involving NIPI-3, we undertook a yeast two-hybrid screen using the full-length NIPI-3 protein as bait (Cypowyj S. et al., manuscript in preparation). One prominent candidate interacting partner was CEBP-1, a member of the C/EBP family of transcription factors [22]. The CEBP-1 protein consists of 319 amino acids, with a bZIP domain at its C-terminus. In further analyses using the yeast two-hybrid assay, we found that the N-terminal region (amino acids 1–115) of CEBP-1 was sufficient for binding NIPI-3 (Additional file 2: Figure S2). This interaction is reminiscent of those observed for fly and vertebrate Tribbles proteins, which bind and degrade C/EBP family proteins [1, 7, 13–15, 29], with human Trib1 binding the N-terminus of C/EBPα [1]. Thus, the ability of Tribbles and C/EBP proteins to interact directly is likely conserved from C. elegans to humans.
To understand the functional significance of the observed protein interaction, we next performed genetic analysis using null mutations of nipi-3 and cebp-1. Null mutants of cebp-1 show normal development and body appearance. Remarkably, cebp-1(0) completely suppressed the growth and fertility defects of nipi-3(0) mutants (Fig. 2b, h; Methods). cebp-1(0) also completely suppressed the body size defect and the developmental delay observed at 25 °C of the partial loss-of-function nipi-3(fr4) mutants (Additional file 3: Figure S3). NIPI-3 is also known to be necessary for the epidermal innate immune response upon fungal infection [20]. The expression of AMP genes after fungal infection is highly induced in wild-type animals and abrogated in nipi-3(fr4) mutants [20]. However, cebp-1(0) nipi-3(0) animals did not exhibit an induction of the AMP gene nlp-34 upon infection (Additional file 4: Figure S4a). Further, cebp-1(0) nipi-3(0) animals expressing wild-type cebp-1 in a tissue-specific manner in either the epidermis or neurons also showed no AMP gene induction (Additional file 4: Figure S4a). These results indicate that loss of cebp-1 does not suppress the immune response defect in nipi-3(0). As cebp-1(0) strongly impairs sensory axon regeneration after laser axotomy [22], we also tested whether nipi-3 affected posterior lateral microtubule (PLM) axon regeneration. Neither nipi-3(0) nor nipi-3(fr4) showed significant effects in axon regeneration, and cebp-1(0) nipi-3(0) double mutants showed impaired axon regeneration similar to cebp-1(0) (Additional file 4: Figure S4b). These results show that the genetic interaction between nipi-3 and cebp-1 is highly specific for larval development and organism fecundity.
Fig. 2.
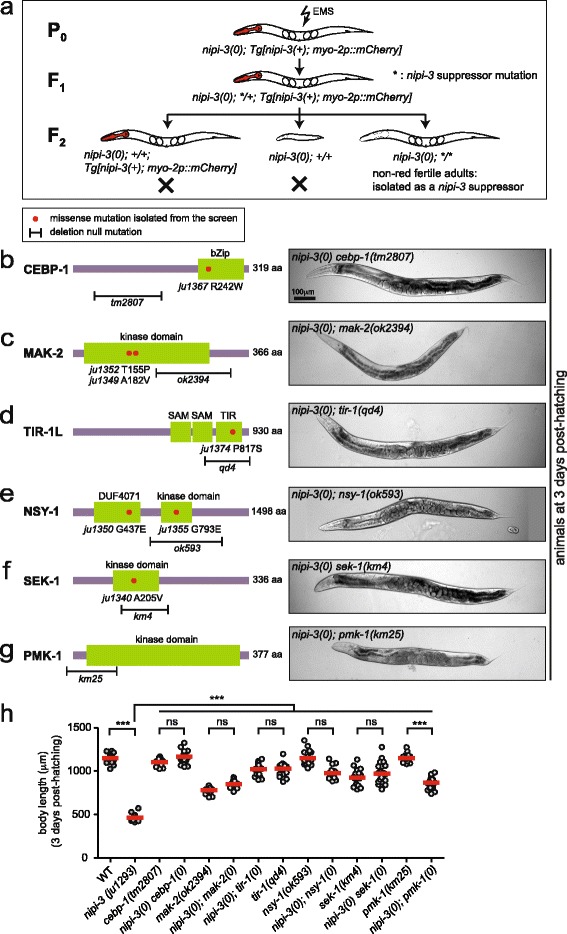
nipi-3(0) lethality is suppressed by loss of cebp-1 or components of a PMK-1/p38 MAPK cascade. a Schematic overview of the forward genetic screen designed to identify the suppressors of nipi-3(0) larval arrest and lethality. b–g The mutations isolated from the nipi-3(0) suppressor screen and the deletion null mutations tested for nipi-3(0) suppression assay are shown in the left column and bright-field images of worms at 3 days post-hatching are shown in the right column. h Body length (μm) of worms at 3 days post-hatching. Each dot represents a single animal measured as shown; each red line represents the mean value; some data are replicated from Fig. 1 as shown with darker grey dots. ***P < 0.001; ns, not significant (one-way ANOVA with Tukey’s post hoc tests)
Loss of the PMK-1/p38 MAPK pathway also suppresses nipi-3(0) lethality
To gain further insight into the mechanism underlying nipi-3’s role in animal development, we performed a forward genetic screen for suppressors of nipi-3(0) lethality. We mutagenized nipi-3(ju1293); Tg[nipi-3(+); myo-2p::mCherry] animals (Methods). Among their F2 progeny, we isolated fertile animals that had lost the rescuing transgene (Fig. 2a) and established multiple suppressor lines (genotypes designated as nipi-3(0); suppressor). We screened ~16,000 haploid genomes and identified 7 independent suppressor alleles. All the identified suppressors in this study were associated with a full reversion of the nipi-3(0) lethality; fertile adults could be propagated without the nipi-3(+) rescuing transgene.
We characterized several suppressor mutations using candidate gene analyses in combination with whole genome sequencing. One suppressor caused a missense mutation in the bZIP domain of cebp-1, and behaved like cebp-1(0) (Fig. 2b, h). Among the other suppressors of nipi-3(0), we found missense alterations in TIR-1/the sterile alpha (SAM) and Toll-interleukin receptor (TIR) motif-containing protein SARM), NSY-1/MAPKKK, SEK-1/MAPKK and MAK-2/MAPK-activated protein kinase (MAPKAPK) (Fig. 2c–f). These mutations are located in known functional domains, including the TIR domain for TIR-1, the kinase domains for NSY-1, SEK-1 and MAK-2, and the DUF4071 domain, commonly found at the N-terminus of many serine-threonine kinase-like proteins, for NSY-1 (Fig. 2c–f). We then tested known null mutants in each of these genes and found them to suppress nipi-3(0) to the same degree as our suppressor mutations, as shown by quantification of the body length (Fig. 2b–f, h; Additional file 5: Figure S5a). Thus, loss of function in tir-1 and these three kinase genes causes strong suppression of nipi-3(0).
NIPI-3 has a specific role in the regulation of epidermal defense genes. It acts together with TIR-1, NSY-1 and SEK-1, as well as several other genes including tpa-1, pmk-1 and sta-2 [20, 21, 30]. We therefore tested mutants for these three latter genes for their genetic interaction with nipi-3(0). We found that loss of pmk-1, but not tpa-1 or sta-2, suppressed the lethality of nipi-3(0) (Fig. 2 g, h for pmk-1; Additional file 5: Figure S5b for tpa-1 and sta-2). Loss of pmk-1 resulted in suppression of nipi-3(0) phenotypes similar to the other suppressor mutants such that nipi-3(0); pmk-1(0) double mutants developed into fertile adults with adult alae and vulva. The rescue of body size, however, was not complete (Fig. 2 g, h). As cebp-1 is involved in two other MAPK cascades known for their roles in adult axon regeneration [22, 31, 32], we tested mutations in several other candidate genes and found that loss of function in dlk-1, pmk-3, mlk-1 or kgb-1 did not suppress nipi-3(0) defects (Additional file 5: Figure S5b). Together, our analyses from both the forward genetic screening and test of candidate mutants reveal a previously unknown role of CEBP-1 in larval development mediated by NIPI-3, and a novel genetic interaction between NIPI-3 and the PMK-1/p38 MAPK cascade.
NIPI-3 inhibits PMK-1 phosphorylation via CEBP-1
To dissect the mechanism underlying the interaction between PMK-1/p38 MAPK and NIPI-3, we first asked how the levels of active PMK-1 might be altered in nipi-3(0) suppressor animals. We performed a Western blot analysis using an anti-phospho-p38 MAPK antibody that specifically recognizes phosphorylated, active PMK-1 [30]. We made protein lysates from animals at 1 day post-hatching (L2) because nipi-3(0) animals at this stage were as healthy as the wild type. We observed that levels of active PMK-1 were significantly increased in nipi-3(0), but remained similar to wild type in cebp-1(0) and cebp-1(0) nipi-3(0) animals (Fig. 3a, b). Phosphorylated PMK-1 was undetectable in nipi-3(0) sek-1(0) animals, consistent with PMK-1 being activated by SEK-1 (Fig. 3a, b). The total PMK-1 levels were likely unchanged in nipi-3(0), as the mRNA levels of pmk-1 were comparable between nipi-3(0) and wild type when assessed by quantitative RT-PCR (Fig. 3c). We note that mak-2(0) did not affect phosphorylated PMK-1 (Additional file 6: Figure S6), suggesting that MAK-2 likely acts downstream of, or in parallel to, PMK-1. Together, these results suggest that the abnormally high levels of phosphorylated PMK-1 in nipi-3(0) are dependent on cebp-1.
Fig. 3.
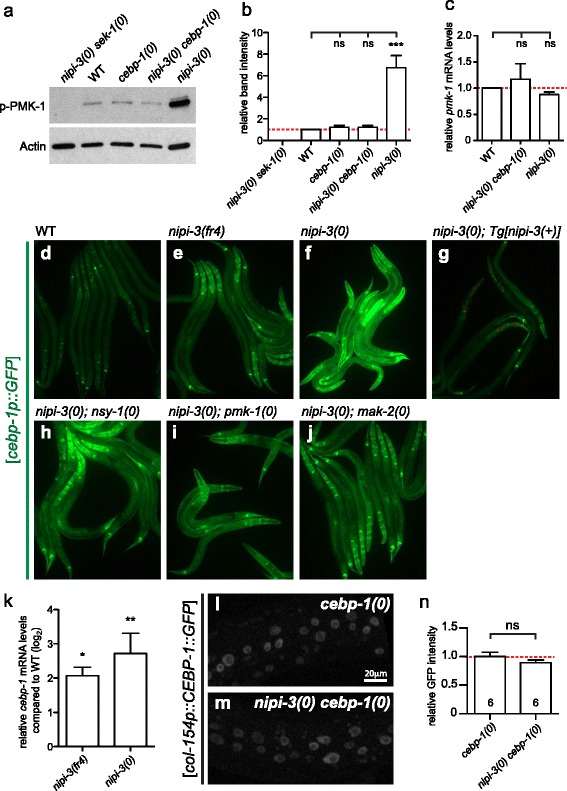
NIPI-3 represses PMK-1 phosphorylation via cebp-1 and represses cebp-1 transcription. a Western blot analysis on total protein lysate from various animal strains using the indicated antibodies, α-phospho-p38 MAPK antibody to detect a phosphorylated form of PMK-1 proteins (p-PMK-1) or α-actin antibody as a loading control. b Densitometric quantifications of immunoblot signals normalized to actin. n = 3; error bars represent standard error of the mean (SEM); *** P < 0.001 (one-way ANOVA with Tukey’s post hoc tests). c Quantitative RT-PCR (qRT-PCR) analysis of pmk-1. Relative abundance of pmk-1 mRNA normalized to actin mRNA. n = 3; error bars represent SEM; ns, not significant (one-way ANOVA with Tukey’s post hoc tests). d–j Fluorescence images of cebp-1p::GFP reporter animals at L2. k qRT-PCR analysis of cebp-1 in WT, nipi-3(fr4), nipi-3(0) animals. Relative abundance of cebp-1 mRNA normalized to actin mRNA. n = 3; error bars represent SEM; *P < 0.05; **P < 0.01; ns, not significant (one-way ANOVA with Tukey’s post hoc tests). l, m Confocal fluorescence images (z-stack) of col-154p(epidermis)::CEBP-1::GFP reporter animals at L2. n Quantification of GFP intensity measured in each epidermal nucleus (10 per animal). n = 6; error bars represent SEM; ns, not significant (Student’s unpaired t test). Primary data for panels (b, c, and k) are provided in Additional file 14
NIPI-3 represses the transcription of cebp-1 in multiple tissues
To dissect how NIPI-3 inhibits CEBP-1, we examined whether cebp-1 levels were altered in nipi-3(0) and in each nipi-3(0) suppressor. Transcriptional reporters of cebp-1 (Tg[cebp-1p::GFP]) were broadly expressed in most post-embryonic tissues, including epidermis, muscles, pharynx, intestine and neurons (Fig. 3d–j). Strikingly, the expression of the cebp-1 transcriptional reporter was highly and significantly increased in both nipi-3(fr4) and nipi-3(0) mutants, compared with wild-type animals (Fig. 3d–f). Quantitative RT-PCR analysis also showed significantly increased expression of cebp-1 mRNAs in nipi-3 mutant animals (Fig. 3 k). Consistent with the observed transcriptional regulation, we found that a translational CEBP-1::GFP reporter driven by a heterologous epidermal promoter showed no detectable differences in GFP expression in a nipi-3(0) background (Fig. 3 l–n). The increased expression of cebp-1p::GFP in nipi-3(0) was reduced to normal levels when a nipi-3(+) transgene was introduced (Fig. 3 g), indicating that NIPI-3 represses the transcription of cebp-1. Additionally, the transcriptional repression of cebp-1 by NIPI-3 was largely independent of the PMK-1 pathway since cebp-1p::GFP expression in nipi-3(0) remained high in animals that also carried a null mutation of nsy-1, pmk-1 or mak-2 (Fig. 3 h–j). Together, the results show that NIPI-3 negatively regulates expression of cebp-1 at the transcriptional level and that CEBP-1 acts upstream of the PMK-1 pathway.
The tight regulation of cebp-1’s expression level is critical for animal viability. We found that suppression of nipi-3(0) by cebp-1(0) was semi-dominant, as cebp-1(0)/+ caused partial but significant suppression of the short body length of nipi-3(0) mutants (Additional file 7: Figure S7). Moreover, in a wild-type background, the transgene eft-3p::CEBP-1::GFP, which drives strong and ubiquitous expression of CEBP-1, caused dose-dependent lethality (see Methods). In addition, expression of a full-length functional translational reporter of cebp-1 (cebp-1p::CEBP-1::GFP) in nipi-3(0) mutant animals exacerbated developmental defects and accelerated larval lethality, while the same transgene showed no such effects in a wild-type background. Together, these results support the conclusion that the lethality observed in nipi-3(0) mutants is a direct consequence of cebp-1 overexpression.
Overexpression of truncated forms of CEBP-1 suppresses nipi-3(0) lethality
To dissect the molecular basis of CEBP-1’s role in animal development, we expressed truncated forms of CEBP-1 lacking the bZIP domain (amino acids 1–230 or 1–115), or lacking the N-terminus (amino acids 237–319) in nipi-3(0) mutants. Surprisingly, in stark contrast to the strong lethality caused by overexpressing full-length CEBP-1 in nipi-3(0) mutants, we found that expression of either the N-terminal fragment or the bZIP domain of CEBP-1 alone resulted in significant suppression of nipi-3(0) defects (Fig. 4a–d). Overexpression of either the N- or C-terminal truncated protein caused no defects either in the wild-type or cebp-1(0) backgrounds. Among the three CEBP-1 fragments, the expression of C-terminal CEBP-1 (amino acids 237–319) showed the most effective rescue, judged by the fecundity of the transgenic lines and quantitative comparisons of body length (Fig. 4c, d). The expression levels of CEBP-1(amino acids 1–230)::GFP were markedly increased in nipi-3(0), presumably reflecting the transcriptional regulation of cebp-1 described above. We noticed the fluorescence intensity was most strongly increased in the epidermis and neurons, throughout the head region and in the ventral nerve cords (Fig. 4e–g). These observations suggest that the truncated forms of CEBP-1 act in a dominant negative manner to inhibit the activity of the endogenous CEBP-1.
Fig. 4.
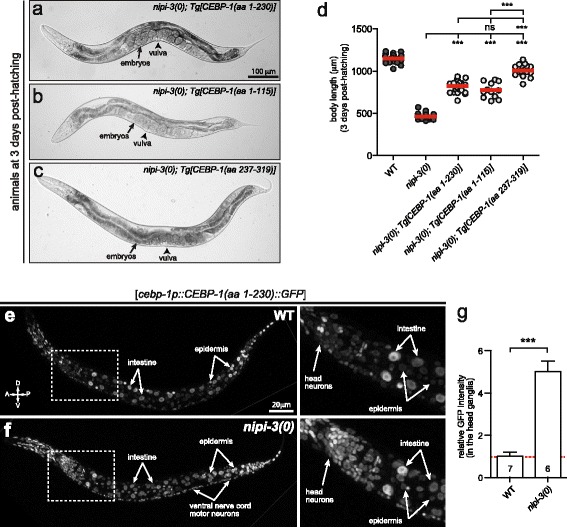
Overexpression of truncated forms of CEBP-1 protein suppresses nipi-3(0) lethality. a–c Bright-field images of worms at 3 days post-hatching expressing truncated forms of CEBP-1 proteins. d Body length (μm) of worms at 3 days post-hatching. Each dot represents a single animal measured as shown; each red line represents the mean value; some data are replicated from Fig. 1 as shown with darker grey dots. ***P < 0.001; ns, not significant (one-way ANOVA with Tukey’s post hoc tests). e, f Confocal fluorescence images (z-stack) of cebp-1p::CEBP-1(aa 1-230)::GFP reporter animals at L2. g Quantification of GFP intensity measured in the region of head neurons. Error bars represent SEM; ***P < 0.001 (Student’s unpaired t test)
CEBP-1 binds conserved DNA motifs in genes regulating development and stress response
To gain further insight into CEBP-1’s function in animal development, we next sought candidate target genes of CEBP-1 by performing ChIP-seq analysis on transgenic animals expressing a functional FLAG-tagged CEBP-1 protein in a cebp-1(0) background (Methods). We found 209 CEBP-1 ChIP-seq peaks in the genome that were associated with 212 coding genes (Additional file 8: Table S1). CEBP-1 peaks were preferentially located within the promoter regions of the target genes (169 genes, 79 %), less frequently within introns (43 genes, 21 %) and never within exons.
We then performed motif analysis of the genomic regions bound by CEBP-1 using the motif discovery tools Multiple Em for Motif Elicitation (MEME) [33] and Regulatory Sequence Analysis Tools (RSAT) [34]. The most over-represented motif, NTTDYGAAAH, was found in 139 out of 209 CEBP-1 ChIP-seq peak regions (Fig. 5a). We then compared this motif with published motifs using the motif comparison tool Tomtom [35] and found the most statistically significant similarities to vertebrate C/EBP binding motifs [36]. The conservation of CEBP-1 binding motif further reinforces the functional parallels between C. elegans CEBP-1 and vertebrate C/EBPs.
Fig. 5.
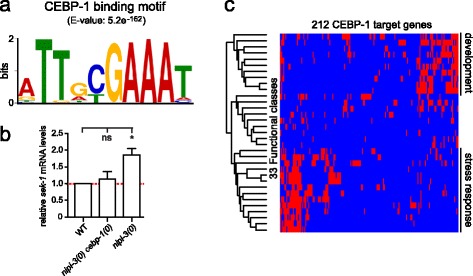
CEBP-1 binds conserved DNA motifs in genes regulating development and stress response. a Motif logo of the most over-represented motif among CEBP-1 ChIP-seq peaks. b qRT-PCR analysis of sek-1. Relative abundance of pmk-1 mRNA normalized to actin mRNA. n = 3; error bars represent SEM; *P < 0.05; ns, not significant (one-way ANOVA with Tukey’s post hoc tests). Primary data are provided in Additional file 14. c Hierarchical clustering of genes and functional classes (see Additional file 10: Figure S9 and Additional file 8: Table S1 for class labels and full data); the presence of a gene in a class is represented by a red rectangle, its absence by blue
As CEBP-1 likely acts upstream of the PMK-1 pathway, we searched among the targets of CEBP-1 for components of the PMK-1 pathway and found CEBP-1 ChIP-seq peaks present in the promoter of sek-1 (Additional file 9: Figure S8). When we examined the mRNA levels of sek-1 by quantitative RT-PCR in nipi-3(0) animals where cebp-1 is overexpressed, we observed increased sek-1 mRNA levels (Fig. 5b). In contrast, in nipi-3(0) cebp-1(0) animals the levels of sek-1 mRNAs were similar to those of the wild type. These results suggest that the abnormally high levels of cebp-1 in nipi-3(0) can promote the expression of sek-1, which in turn promotes the phosphorylation of PMK-1 [30], leading to abnormal larval development and lethality.
We then asked whether the list of potential CEBP-1 targets was enriched for genes with specific functions. To this end, we searched for enriched categories through an Expression Analysis Systematic Explorer (EASE) analysis [37, 38], using our in-house database of functional annotations as described [39]. This includes 4600 datasets automatically updated from multiple sources including WormBase, FlyBase, the Kyoto Encyclopedia of Genes and Genomes (KEGG) and relevant RNAi databases. Most (80 %) of CEBP-1 target genes were associated with at least one of 33 enriched functional classes (P < 10-5; Additional file 8: Table S1). Hierarchical clustering of the genes in each of the enriched classes identified two main groups (Fig. 5c; Additional file 10: Figure S9; Additional file 8: Table S1). One group is related to development with phenotypic classes such as 'larval lethal’ or 'slow growth’ and includes genes involved in basic cellular processes, transcription, translation or endocytosis. The second group is related to the response to biotic or abiotic stress, including response to cadmium, hygromycin or bacterial toxins. Interestingly, McEwan et al. have found that most of the genes upregulated in nipi-3(fr4) mutants are also induced by the translational inhibitory toxin ToxA. Out of the 14 stress-related classes associated with the cebp-1 targets, 10 are shared with those found for genes upregulated in nipi-3(fr4) (D.L. McEwan, personal communication; P < 10-10; Additional file 8: Table S1). On the other hand, consistent with the fact that the nipi-3(fr4) allele does not provoke larval lethality, only 1 out of the 10 classes in the development cluster is shared between the cebp-1 targets and the genes upregulated in nipi-3(fr4). These analyses suggest that CEBP-1 regulates genes functioning in development and in stress responses, and might have a particularly important impact on organismal physiology when overexpressed in a nipi-3 mutant context.
NIPI-3 is required in multiple tissues to ensure proper larval development
We next asked in which tissue the expression of nipi-3(+) is required for animal viability. We expressed nipi-3(+) in a tissue-specific manner, using intestinal, epidermal and pan-neuronal promoters, in the nipi-3(0) background (Fig. 6a–f). In contrast to the complete rescue of body size and lethality in nipi-3(0) mutants expressing nipi-3(+) under its own promoter (Fig. 1e, h), expressing nipi-3(+) in individual tissues failed to rescue the developmental arrest (Fig. 6a–c, f). Pan-neuronal expression of nipi-3(+) resulted in slightly increased body length of nipi-3(0) 3 days post-hatching (Fig. 6c, f). Since nipi-3 activity strongly inhibits cebp-1 transcription in the epidermis and neurons (Fig. 4e–g), we expressed nipi-3(+) in both of these tissues together, and found that these transgenic animals showed an increased body size (Fig. 6e, f), compared to those expressing nipi-3(+) in each tissue alone, or in the intestine and epidermis simultaneously (Fig. 6a–d, f). Some of the transgenic animals with a body length closer to that of wild-type animals showed improved somatic and germline development, with formation of vulva and adult alae, and produced a few viable but infertile progeny. When we expressed nipi-3(+) in all three tissues together, the transgenic animals showed no further improvement of body size compared to those expressing nipi-3(+) in both the epidermis and neurons (Fig. 6f), and did not recapitulate the rescue of lethality associated with the expression of nipi-3 under its own promoter. Thus, we conclude that NIPI-3 is required in multiple tissues, particularly epidermis and neurons, for animal growth and development.
Fig. 6.
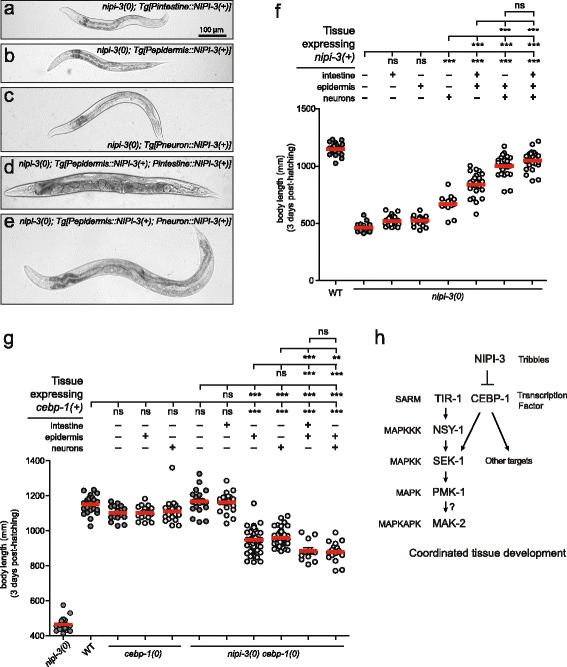
Tight regulation of both NIPI-3 and CEBP-1 is required in multiple tissues for proper organism development. a–e Bright-field images and (f) the body length of worms expressing tissue-specific nipi-3(+) driven by the intestinal (mtl-2), epidermal (col-12) or pan-neuronal (rgef-1) promoters in a nipi-3(0) background. g The body length of worms expressing tissue-specific cebp-1(+) driven by the intestinal (ges-1), epidermal (col-154) or pan-neuronal (rgef-1) promoters in a nipi-3(0) cebp-1(0) background. f, g Each dot represents a single animal measured as shown; each red line represents the mean value; some data are replicated from Figs. 1 and 2 as shown with darker grey dots. **P < 0.01; ***P < 0.001; ns, not significant (one-way ANOVA with Tukey’s post hoc tests). h Working model for NIPI-3 function in C. elegans development. In wild type, presence of NIPI-3 keeps cebp-1 expression level optimal for coordinated tissue development. In nipi-3(0), however, cebp-1 and sek-1 are overexpressed and in turn PMK-1 is hyperactivated
Suppression of nipi-3(0) by cebp-1(0) also requires a block of CEBP-1 activity in multiple tissues
Conversely, we asked whether expression of cebp-1(+) in a single tissue might cause the viable nipi-3(0) cebp-1(0) animals to die. We expressed cebp-1(+) using the intestinal, epidermal and pan-neuronal promoters in cebp-1(0) nipi-3(0) double mutants (Fig. 6 g). Expression of cebp-1(+) in individual tissues was insufficient to produce larval lethal phenotypes in cebp-1(0) nipi-3(0) animals. Interestingly, epidermal or neuronal expression of cebp-1(+) in cebp-1(0) nipi-3(0) double mutants caused short body length, but not in cebp-1(0) mutants (Fig. 6 g). Co-expression of cebp-1(+) in the epidermis and neurons resulted in further reductions in body length, although these were not as severe as those in nipi-3(0) animals (Fig. 6 g). In addition, we noticed that the same transgenes expressing cebp-1(+) in both epidermis and neurons caused an abnormal pharyngeal morphology in cebp-1(0) nipi-3(0) animals, similar to that seen in nipi-3(0) mutants (Additional file 1: Figure S1). Together, our data suggest that the tight regulation of both NIPI-3 and CEBP-1 in multiple tissues is required in a systemic manner for normal animal growth and development.
Discussion
C. elegans Tribbles NIPI-3 was identified on the basis of its roles in host defense [20, 21]. Here, through generation and analyses of null alleles, we find nipi-3 to be essential for animal development and viability. Remarkably, the larval arrest and lethality caused by complete loss of nipi-3 is fully suppressed by loss of cebp-1, a C/EBP bZIP transcription factor, or by loss of function in the PMK-1/p38 MAPK cascade including tir-1/SARM, nsy-1/MAPKKK, sek-1/MAPKK and pmk-1/MAPK. Our data show that complete elimination of the function of nipi-3 causes abnormally high expression of CEBP-1, and activation of PMK-1 MAPK. This then disrupts development and leads to death. The level of sek-1 mRNA is increased in nipi-3(0) mutants but not in cebp-1(0) or in nipi-3 cebp-1 animals. The level of phosphorylated (active) PMK-1 follows the same trend. Coupled with our ChIP-seq analyses and genetic epistasis data, this suggests that CEBP-1 acts as a direct positive regulator of sek-1. The PMK-1 pathway is therefore activated when CEBP-1 expression is high in nipi-3(0). On the other hand, cebp-1 expression levels remain high in nipi-3(0); pmk-1(0) animals, confirming that CEBP-1 does not act downstream of the PMK-1 pathway. Together, these results suggest that NIPI-3 negatively regulates the PMK-1 MAPK cascade, via CEBP-1, to promote animal viability and development (Fig. 6 h).
In innate immunity, however, nipi-3 cell-autonomously promotes or enhances the same p38 kinase cascade to activate host defense in the epidermis [20]. It has been shown that overexpression of sek-1 in the epidermis rescues the block of AMP induction in nipi-3 mutants upon fungal infection [20], and an overexpression of nipi-3 provokes an increase in the constitutive expression of AMP which is dependent on the p38 cascade [21]. It is intriguing that NIPI-3 appears to be capable of activating or inhibiting PMK-1/p38 in the epidermis at different times or under different conditions (infection versus development). How might NIPI-3 achieve this dual role under different stresses and in altered cellular contexts? As Tribbles proteins are well known to act as adaptors, NIPI-3 might be regulated via binding with other co-factors only present under specific circumstances. Indeed, we find that other upstream and downstream components of the epidermal immune response cascade are not involved in the developmental regulation described here. Thus, the core PMK-1/p38 MAPK cassette has evolved context-specific functions depending on different upstream regulators or co-factors [40, 41]. Members of the Tribbles family in other species have been mostly studied in the context of cell proliferation, adipocyte tissue differentiation, energy metabolism and immunity, where they function in a cell-autonomous manner. Our discovery of the opposing roles of NIPI-3 in development and in the immune response illustrates how cellular context can alter the function of highly conserved signalling molecules.
Negative regulation of C/EBP by Tribbles has been observed throughout the animal kingdom. Drosophila and mammalian Tribbles bind and degrade C/EBP proteins [1, 7, 13–15]. We find that C. elegans NIPI-3 represses the transcription of cebp-1, which has important functional consequences in vivo. This form of regulation has not been reported in other organisms. Given its nuclear localization, NIPI-3 may inhibit the transcription of cebp-1 by interfering with other transcription factor(s). The promoter of cebp-1 contains putative CEBP-1 binding consensus motifs, raising the possibility that NIPI-3, by binding to CEBP-1, may also alter the transcriptional activity of CEBP-1.
NIPI-3 is required to control CEBP-1 levels in multiple tissues for animal development and viability. Consistent with the inhibition of CEBP-1 expression by NIPI-3 in the epidermis and neurons, simultaneous expression of nipi-3(+) in both tissues makes a noticeable contribution to animal development in nipi-3(0) mutants, compared with nipi-3(+) expression in single tissues. Conversely, simultaneous expression of cebp-1(+) in both epidermis and neurons causes noticeable defects in animal development in nipi-3(0) cebp-1(0) mutants, compared with cebp-1(+) expression in single tissues. Thus, a tightly regulated coordination of these two genes’ interactions in multiple tissues is required to ensure proper development.
A key conclusion from our study is that the precise control of CEBP-1 and PMK-1/p38 MAPK pathways in multiple tissues is critical for organismal development. NIPI-3 acts as a master regulator to prevent improper activation of CEBP-1 and PMK-1, whose hyperactivation during development has deleterious consequences. Interestingly, hyperactivation of PMK-1/p38 was previously shown to block larval development when the endoplasmic reticulum unfolded protein response was altered [42]. Moreover, innate immune activation with a xenobiotic that provides protection from bacterial infection in the adult has been shown to provoke a growth delay during development [43]. Subsequently, an elegant genetic suppressor screen revealed that mutations in the PMK-1/p38 MAPK pathway suppressed this developmental phenotype [44]. Thus, the NIPI-3/CEBP-1 axis is a key mechanism by which immune effector expression is held in check during nematode development.
During normal development, both CEBP-1 and PMK-1 are maintained at a basal level by NIPI-3. The levels of inducible signalling from these pathways are, however, important for animals to protect themselves or to promote repair. For instance, following fungal infection, NIPI-3 promotes the PMK-1/p38 MAPK signalling pathway in the epidermis [20]. Thus, animals can successfully defend themselves from fungal infection with activated PMK-1 locally in the epidermis, while survival is not affected as PMK-1 remains inactive in other tissues. Similarly, CEBP-1 is known to play a key role in neuronal stress responses [22, 23, 45], and we identified potential CEBP-1 target genes that are involved in different stress responses. Moreover, a concomitant study has identified NIPI-3 as a negative regulator of CEBP-1 in intestinal defense against the bacterial toxin ToxA (McEwan et al., personal communication). An important challenge for the future will be to understand how NIPI-3 regulates its downstream pathways and how NIPI-3 itself is regulated depending on developmental and environmental conditions. Understanding the molecular mechanism of this systemic, coordinated regulation should advance our knowledge of how animal development can be maintained in the face of environmental stresses.
Conclusions
We showed a novel essential role for the C. elegans Tribbles homolog NIPI-3 in animal development and viability, which requires NIPI-3’s function in multiple tissues. NIPI-3 acts as a master regulator to prevent improper activation of a C/EBP transcription factor and a conserved PMK-1/p38 MAPK signalling cascade known to control innate immunity. These findings suggest that innate immune responses are tightly controlled for proper organismal development.
Methods
Strains, transgenes and plasmids
C. elegans strains were maintained under standard conditions at 20 °C unless otherwise mentioned. The wild type was the N2 Bristol strain [46]. New strains were constructed using standard procedures, and all genotypes were confirmed by PCR or sequencing. All strains and their genotypes used in this study are described in Additional file 11: Table S2. Extrachromosomal array transgenic lines were generated as described previously [47]. Expression constructs, transgenes and strain genotypes are also summarized in Additional file 11: Table S2. For all experiments, at least two independent transgenic lines were examined and quantitative data are shown for one. In our studies of cebp-1 dosage effect, we could not generate transgenic animals when injecting 10 ng/μl of eft-3p::CEBP-1::GFP transgenes into wild-type animals, while many transgenic lines were obtained when cebp-1::gfp DNA was injected with lower concentrations (i.e. 1 ng/μl or 0.01 ng/μl). All plasmids used in this study are described in Additional file 12: Table S3a.
CRISPR-Cas9-mediated deletion and GFP KI
We generated the nipi-3(ju1293) deletion allele using the co-CRISPR method [25, 48]. We used four single guide RNAs (sgRNAs) targeting the N-terminus of the nipi-3 gene (Additional file 12: Table S3a). U6p::nipi-3 sgRNAs were generated by Gibson assembly and injected into cebp-1(tm2807) worms, using standard methods, in mixtures composed of 30 ng/μl of each nipi-3 sgRNA, 50 ng/μl of eft-3p::Cas9-SV40NLS::tbb-2 3′UTR, 50 ng/μl of U6p::unc-22 sgRNA and 1.5 ng/μl of myo-2p::mCherry. For the nipi-3(fr148) deletion allele, a single sgRNA targeting the N-terminus of the nipi-3 gene (Additional file 12: Table S3a) was injected into cebp-1(tm2807) worms, in mixtures composed of 50 ng/μl of nipi-3 sgRNA, 30 ng/μl of eft-3p::Cas9-SV40NLS::tbb-2 3′UTR and 30 ng/μl of col-12p::dsRed [49]. Note that we also injected many wild-type worms with various combinations of nipi-3 sgRNAs, but failed to isolate any deletion alleles.
GFP KI in the nipi-3 locus, nipi-3(fr152) was generated with the same mixture as for nipi-3(fr148), providing 30 ng/μl of the nipi-3 repair template pSO1. This template was generated by Gibson assembly in the self-excising (SEC) cassette containing vector pDD282 [50] with 716 bp and 518 bp homology arms upstream and downstream of the nipi-3 start codon, respectively (Additional file 12: Table S3a).
Genetic screen for nipi-3(0) lethality suppressors
nipi-3(ju1293) mutant animals carrying juEx6807[nipi-3 genomic DNA; myo-2p::mCherry] were mutagenized using 45 mM ethyl methane sulphonate (EMS) following standard procedures as described [46]. Animals were distributed onto nematode growth media (NGM) plates seeded with Escherichia coli OP50 and screened in the F2 generation for normal animal growth reaching adulthood without expressing the transgene (no pharyngeal mCherry) under a fluorescence dissecting microscope.
Mapping and cloning of nipi-3 suppressor alleles
We first performed a conventional Sanger sequencing analysis for all suppressor alleles for cebp-1 and determined that the ju1367 allele affected cebp-1. We next sequenced mak-2, which was previously known to act in the same pathway as cebp-1 in neurons [22], and found ju1349 and ju1352 alleles to affect mak-2. All other suppressors were analysed by whole genome sequencing analysis and single-nucleotide polymorphism (SNP) mapping following established methods [51]. Briefly, genomic DNA was prepared using a Puregene Cell and Tissue Kit (Qiagen) according to the manufacturer’s instructions, and 20X coverage of sequences was obtained using a 90 bp paired-end Illumina HiSeq 2000 at Beijing Genomics Institute (BGI Americas). The raw sequences were mapped to the C. elegans reference genome (WS220/ce10) using Burrows-Wheeler Aligner (BWA) [52] in the Galaxy platform (http://usegalaxy.org) [53]. Following subtraction of the nucleotide variants in the original strains, we generated a list of candidate genes containing unique homozygous nucleotide variants that were predicated to alter the function of the gene. We then confirmed the causality of the candidate genes by testing the known null alleles on the suppression of nipi-3(0).
Body length analysis
To examine the body length of animals during development, we obtained synchronized animals. Briefly, 5–15 gravid adults were placed on a seeded NGM plate to allow egg-laying for 3 h. Eggs laid during this period were incubated at 20 °C for 72 h, and the animals were then mounted on 2 % agarose pads containing a drop of 2.5 mM levamisole and photographed with a Zeiss Axioplan compound microscope, using Nomarski-DIC optics and an attached AxioCam digital camera. ImageJ software (National Institutes of Health (NIH), Bethesda, MD, USA) was used to measure body length by drawing a freehand midline from the tip of the nose to the tip of the tail of each animal.
Western blot analysis
Worms of each genotype (80–100 individuals of L2 stage worms) were collected and washed with M9 buffer and boiled in sodium dodecyl sulphate (SDS) sample buffer for 10 min and loaded onto SDS-polyacrylamide gel electrophoresis (PAGE) gel (Bio-Rad). A 1:500 dilution of rabbit anti-phospho-p38 MAPK (Cell Signaling, #9211) and a 1:10,000 dilution of mouse anti-actin (MPbio, #08691001) were used as primary antibodies. ImageJ was used to quantify the intensity of the immunoblot bands.
Quantitative RT-PCR
Quantitative real-time PCR was performed as previously described [49]. Sequences of primers are given in Additional file 12: Table S3b. To collect synchronized nipi-3(0) homozygous animals, we maintained nipi-3(0) animals on cebp-1 RNAi plates. Gravid adults were then treated by bleaching solution to collect nipi-3(0) embryos, which were placed directly on regular NGM plates for 2 days in parallel to other strains.
Fluorescence microscopy and axon regeneration by laser axotomy
Animals were mounted on 2 % agarose pads and immobilized with 2.5 mM levamisole. For transcriptional cebp-1p::GFP, GFP expression was imaged with a Zeiss Axioplan compound microscope, using Nomarski optics and an attached AxioCam digital camera. Translational CEBP-1::GFP expression was imaged with a Zeiss LSM710 confocal microscope for quantitative analyses. For confocal images, z stacks were obtained and maximum projection images were created using Zeiss Zen 2012 software. ImageJ was used to measure the GFP intensity at the nerve ring area for cebp-1p::CEBP-1::GFP and at each nucleus (10 per animal) for col-154p::CEBP-1::GFP. GFP::NIPI-3 KI expression was imaged with a spinning disk confocal microscope as described [54] to improve the signal-to-noise ratio.
We cut PLM axons and quantified the length of regrown axons as described [55].
CEBP-1 ChIP-seq analysis
We generated transgenic animals expressing a functional FLAG-tagged CEBP-1 protein in a cebp-1(tm2807) mutant background (cebp-1(0); juIs418 [cebp-1p::FLAG::CEBP-1::cebp-1 3′UTR]) (Additional file 13: Figure S10) and then immunoprecipitated FLAG-CEBP-1-associated DNA fragments using anti-FLAG antibodies (M2 anti-FLAG magnetic beads; Sigma). We collected mixed stage worms grown at 20 °C on NGM plates followed by 2 % formaldehyde and sonicated the samples as described [56]. We next generated ChIP-seq DNA libraries via ligating DNA to specific adaptors and amplification with barcode primers, then sequenced them on the Illumina HiSeq 2000 platform. We performed two independent ChIP-seq experiments with parallel genomic DNA controls prepared from the same strain. We conducted peak calling using a CLC Genomics Workbench 6.0 (CLC bio). To define genes associated with the peaks, we used the annotation of transcription start site (TSS) and transcription end site (TES) from Wormbase WS220 (http://www.wormbase.org) and annotated the peak if it overlapped with the gene or the 3 kb upstream of the TSS. We then manually confirmed the peaks and associated genes using the University of California, Santa Cruz (UCSC) browser and an update to WS252. If the peak was found within the promoter for one isoform and introns for other isoforms, we categorized it as a peak within a promoter. The ChIP-seq data are available at the Gene Expression Omnibus [GEO:GSE83330].
Bioinformatic analyses
MEME [33] and RSAT [34] were used to identify over-represented motifs from 209 CEBP-1 ChIP-seq peak sequences, and then Tomtom [35] was used to compare the most over-represented motif against a database of known motifs in vertebrates. All informatics tools can be found at http://meme-suite.org and http://www.rsat.eu. Enrichment analyses were run on a newly developed database of functional annotations including 4600 datasets [39] updated to Wormbase WS252 (http://www.wormbase.org). Classes considered for enrichment had a maximum size of 2000 genes, and the P value for enrichment was lower than 10-5.
Yeast two-hybrid assay
Full-length or fragments of complementary DNAs (cDNAs) were cloned into the pACT2 (Gal4 activation domain) or pBTM116 (LexA DNA-binding domain) vectors (Clontech) and constructs were co-transformed into yeast strain L40. We grew transformed yeasts on agar plates with synthetic defined (SD) minimal medium lacking leucine and tryptophan; interactions were examined on plates with SD medium lacking leucine, tryptophan and histidine.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 5. Significance was determined using unpaired t tests for two samples, one-way ANOVA followed by Tukey’s multiple comparison tests for multiple samples. For two nominal variables, Fisher’s exact test was used to evaluate the statistical significance. P < 0.05 (*) was considered statistically significant (* P < 0.05; ** P < 0.01; *** P < 0.001).
Acknowledgements
We thank J. Ewbank for support on the project, A.D. Chisholm and members of the Jin and Chisholm laboratories for valuable discussions and critical comments on the manuscript and D.L. McEwan and F.M. Ausubel for discussions and sharing of unpublished data. We thank S. Cypowyj for the initial findings on cebp-1, J. Ruiz for assistance in strain construction, Z. Wu for help in axotomy analyses, M. Bulle and A. Boned for technical assistance, D.J. Dickinson and Z. Wang for CRISPR plasmids, H-R. Li and Y. Zhou in X-D Fu’s lab at UC San Diego for help in Illumina sequencing and advice on mapping ChIP-seq reads, and WormBase as an information resource. YJ gratefully acknowledges the support from IMéRA, Aix-Marseille University. Some strains were provided by the Caenorhabditis Genetics Center (CGC), which is funded by the NIH Office of Research Infrastructure Programs (P40 OD010440), and by the National Bioresource Project of Japan (NBRP).
Funding
This work was supported by institutional grants from AMU, INSERM and CNRS, the ANR (ANR-12-BSV3-0001-01, ANR-11-LABX-0054 (Investissements d’Avenir–Labex INFORM) and ANR-11-IDEX-0001-02 (Investissements d’Avenir–A*MIDEX) and ANR-10-INBS-04-01 (France Bio Imaging)), NIH grant R01 NS035546 to YJ and an American Heart Association postdoctoral fellowship to KWK. YJ is an investigator of the Howard Hughes Medical Institute.
Availability of data and material
All data generated or analysed during this study are included in this published article (and its additional files). The ChIP-seq data have been deposited in the Gene Expression Omnibus database [GEO:GSE83330] (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=avylwwgwvtmrlmz&acc=GSE83330 ). Requests for material should be made to the corresponding authors.
Authors’ contributions
KWK and YJ designed the genetic suppressor screen, and KWK and CAP performed the screen. KWK performed all experiments on nipi-3 suppressors and CEBP-1 ChIP-seq analyses. SO and JP performed experiments, and NT performed the bioinformatic analyses. YJ and NP supervised the experiments. KWK, YJ and NP analysed the data and wrote the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Abbreviations
- AMP
Antimicrobial peptide
- bZIP
Basic leucine zipper
- C/EBP
CCAAT/enhancer-binding protein
- ChIP-seq
Chromatin immunoprecipitation and deep sequencing
- CRISPR
Clustered regularly interspaced short palindromic repeats
- EASE
Expression Analysis Systematic Explorer
- GO
Gene ontology
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- MAPK
Mitogen-activated protein kinase
- MEME
Multiple Em for Motif Elicitation
- NIPI
No induction of peptide after Drechmeria infection
- RSAT
Regulatory Sequence Analysis Tools
Additional files
CEBP-1 expression in multiple tissues causes an abnormal pharyngeal morphology in nipi-3(0) cebp-1(0) animals. (a) Differential interference contrast (DIC) images of worms at 3 days post-hatching. Pharynx and intestine are denoted by a dotted red and yellow line, respectively. (b) Co-expression of cebp-1(+) in the epidermis and neurons in nipi-3(0) cebp-1(0) animals caused pharyngeal morphology defect. *P < 0.05; **P < 0.01; ns, not significant (two-tailed Fisher’s exact test). (PDF 3776 kb)
NIPI-3 interacts with CEBP-1 in yeast two-hybrid assay. CEBP-1 variants were fused to the Gal4 activation domain and tested for their interaction with full-length NIPI-3 fused to the LexA DNA-binding domain. A large CEBP-1 fragment (∆2, amino acids 1–115) was sufficient for the NIPI-3 interaction. – refers to no growth and + refers to growth in the absence of histidine. (PDF 433 kb)
Loss of cebp-1 rescues the nipi-3(fr4) phenotype. (a) Bright-field images and (b) body length of worms at 3 days post-hatching grown at 25 °C. (b) Each dot represents a single animal measured as shown; each red line represents the mean value. ***P < 0.001; ns, not significant (one-way ANOVA with Tukey’s post hoc tests). (PDF 1253 kb)
cebp-1 and nipi-3 are dispensable in immune response and axon regeneration, respectively. (a) qRT-PCR analysis of AMP gene, nlp-34. Relative abundance of nlp-34 mRNA normalized to actin mRNA. n = 3. Error bars represent SEM. ***P < 0.001; ns, not significant (one-way ANOVA with Tukey’s post hoc tests). Primary data are provided in Additional file 14. (b) Axotomy and axon regeneration analysis. Normalized PLM axon regrowth is shown in the bar graph. Error bars represent SEM. ***P < 0.001; ns, not significant (one-way ANOVA with Tukey’s post hoc tests). (PDF 472 kb)
Quantification of the body length of suppressor alleles identified from the screen and loss-of-function mutants of tpa-1, pmk-1, sta-2, dlk-1, pmk-3, mlk-1 and kgb-1. (a, b) Body length of worms at 3 days post-hatching. Each dot represents a single animal measured as shown; each red line represents the mean value; some data are replicated from Fig. 1 as shown with darker grey dots. ***P < 0.001 (one-way ANOVA with Tukey’s post hoc tests). (PDF 569 kb)
Phosphorylated PMK-1 levels are unchanged in nipi-3(0); mak-2(0) animals. (a) Western blot analysis on total protein lysate from various animal strains using the indicated antibodies, α-phospho-p38 MAPK antibody to detect a phosphorylated form of PMK-1 proteins (p-PMK-1) or α-actin antibody as a loading control. (b) Densitometric quantifications of immunoblot signals normalized to actin. n = 4; error bars represent SEM; ns, not significant (Student’s paired t test). Primary data are provided in Additional file 14. (PDF 595 kb)
cebp-1 shows a dosage sensitive effect in nipi-3(0) mutants. (a) Bright-field images and (b) body length of worms at 3 days post-hatching. (b) Each dot represents a single animal measured as shown; each red line represents the mean value. ***P < 0.001 (one-way ANOVA with Tukey’s post hoc tests). (PDF 1501 kb)
List of CEBP-1 target genes and hierarchical clustering of genes and functional classes (XLSX 64 kb)
The promoter of sek-1 contains a ChIP-seq peak of CEBP-1. The promoter region of sek-1 contains two consensus DNA-binding motifs for CEBP-1 (black triangles). Top, the sek-1 locus. Middle, sequencing reads from CEBP-1-IP. Bottom, sequencing reads from genomic DNA input. (PDF 253 kb)
Hierarchical clustering of genes and functional classes. The presence of a gene in a class is represented by a red rectangle, its absence in blue. See Additional file 8: Table S1 for class labels and full data. (PDF 680 kb)
List of strains and alleles. (XLSX 18 kb)
List of plasmids and primers used for qRT-PCR. (XLSX 13 kb)
FLAG-tagged CEBP-1 protein rescues PLM axon regeneration defects of cebp-1(0). Axotomy and axon regeneration analysis. Normalized PLM axon regrowth is shown in the bar graph. Error bars represent SEM. ***P < 0.001; ns, not significant (one-way ANOVA with Tukey’s post hoc tests). (PDF 432 kb)
Primary data for Figs. 3b, c, k and 5b and Figures S4a and S6b. (XLSX 20 kb)
Contributor Information
Kyung Won Kim, Email: k9kim@ucsd.edu.
Yishi Jin, Email: yijin@ucsd.edu.
Nathalie Pujol, Email: pujol@ciml.univ-mrs.fr.
References
- 1.Murphy JM, Nakatani Y, Jamieson SA, Dai W, Lucet IS, Mace PD. Molecular mechanism of CCAAT-enhancer binding protein recruitment by the TRIB1 pseudokinase. Structure. 2015;23(11):2111–21. doi: 10.1016/j.str.2015.08.017. [DOI] [PubMed] [Google Scholar]
- 2.Lohan F, Keeshan K. The functionally diverse roles of tribbles. Biochem Soc Trans. 2013;41(4):1096–100. doi: 10.1042/BST20130105. [DOI] [PubMed] [Google Scholar]
- 3.Taylor SS, Shaw A, Hu J, Meharena HS, Kornev A. Pseudokinases from a structural perspective. Biochem Soc Trans. 2013;41(4):981–6. doi: 10.1042/BST20130120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dobens LL, Jr, Bouyain S. Developmental roles of tribbles protein family members. Dev Dyn. 2012;241(8):1239–48. doi: 10.1002/dvdy.23822. [DOI] [PubMed] [Google Scholar]
- 5.Seher TC, Leptin M. Tribbles, a cell-cycle brake that coordinates proliferation and morphogenesis during Drosophila gastrulation. Curr Biol. 2000;10(11):623–9. doi: 10.1016/S0960-9822(00)00502-9. [DOI] [PubMed] [Google Scholar]
- 6.Mata J, Curado S, Ephrussi A, Rørth P. Tribbles coordinates mitosis and morphogenesis in Drosophila by regulating string/CDC25 proteolysis. Cell. 2000;101(5):511–22. doi: 10.1016/S0092-8674(00)80861-2. [DOI] [PubMed] [Google Scholar]
- 7.Rørth P, Szabo K, Texido G. The level of C/EBP protein is critical for cell migration during Drosophila oogenesis and is tightly controlled by regulated degradation. Mol Cell. 2000;6(1):23–30. doi: 10.1016/S1097-2765(00)00004-6. [DOI] [PubMed] [Google Scholar]
- 8.Grosshans J, Wieschaus E. A genetic link between morphogenesis and cell division during formation of the ventral furrow in Drosophila. Cell. 2000;101(5):523–31. doi: 10.1016/S0092-8674(00)80862-4. [DOI] [PubMed] [Google Scholar]
- 9.Yokoyama T, Nakamura T. Tribbles in disease: signaling pathways important for cellular function and neoplastic transformation. Cancer Sci. 2011;102(6):1115–22. doi: 10.1111/j.1349-7006.2011.01914.x. [DOI] [PubMed] [Google Scholar]
- 10.Sung HY, Guan H, Czibula A, King AR, Eder K, Heath E, Suvarna SK, Dower SK, Wilson AG, Francis SE, et al. Human tribbles-1 controls proliferation and chemotaxis of smooth muscle cells via MAPK signaling pathways. J Biol Chem. 2007;282(25):18379–87. doi: 10.1074/jbc.M610792200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yokoyama T, Kanno Y, Yamazaki Y, Takahara T, Miyata S, Nakamura T. Trib1 links the MEK1/ERK pathway in myeloid leukemogenesis. Blood. 2010;116(15):2768–75. doi: 10.1182/blood-2009-10-246264. [DOI] [PubMed] [Google Scholar]
- 12.Eder K, Guan H, Sung HY, Ward J, Angyal A, Janas M, Sarmay G, Duda E, Turner M, Dower SK, et al. Tribbles-2 is a novel regulator of inflammatory activation of monocytes. Int Immunol. 2008;20(12):1543–50. doi: 10.1093/intimm/dxn116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Masoner V, Das R, Pence L, Anand G, LaFerriere H, Zars T, Bouyain S, Dobens LL. The kinase domain of Drosophila Tribbles is required for turnover of fly C/EBP during cell migration. Dev Biol. 2013;375(1):33–44. doi: 10.1016/j.ydbio.2012.12.016. [DOI] [PubMed] [Google Scholar]
- 14.Satoh T, Kidoya H, Naito H, Yamamoto M, Takemura N, Nakagawa K, Yoshioka Y, Morii E, Takakura N, Takeuchi O, et al. Critical role of Trib1 in differentiation of tissue-resident M2-like macrophages. Nature. 2013;495(7442):524–8. doi: 10.1038/nature11930. [DOI] [PubMed] [Google Scholar]
- 15.Dedhia PH, Keeshan K, Uljon S, Xu L, Vega ME, Shestova O, Zaks-Zilberman M, Romany C, Blacklow SC, Pear WS. Differential ability of Tribbles family members to promote degradation of C/EBPalpha and induce acute myelogenous leukemia. Blood. 2010;116(8):1321–8. doi: 10.1182/blood-2009-07-229450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ohoka N, Hattori T, Kitagawa M, Onozaki K, Hayashi H. Critical and functional regulation of CHOP (C/EBP homologous protein) through the N-terminal portion. J Biol Chem. 2007;282(49):35687–94. doi: 10.1074/jbc.M703735200. [DOI] [PubMed] [Google Scholar]
- 17.Ohoka N, Yoshii S, Hattori T, Onozaki K, Hayashi H. TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J. 2005;24(6):1243–55. doi: 10.1038/sj.emboj.7600596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ord D, Meerits K, Ord T. TRB3 protects cells against the growth inhibitory and cytotoxic effect of ATF4. Exp Cell Res. 2007;313(16):3556–67. doi: 10.1016/j.yexcr.2007.07.017. [DOI] [PubMed] [Google Scholar]
- 19.Jousse C, Deval C, Maurin AC, Parry L, Cherasse Y, Chaveroux C, Lefloch R, Lenormand P, Bruhat A, Fafournoux P. TRB3 inhibits the transcriptional activation of stress-regulated genes by a negative feedback on the ATF4 pathway. J Biol Chem. 2007;282(21):15851–61. doi: 10.1074/jbc.M611723200. [DOI] [PubMed] [Google Scholar]
- 20.Pujol N, Cypowyj S, Ziegler K, Millet A, Astrain A, Goncharov A, Jin Y, Chisholm AD, Ewbank JJ. Distinct innate immune responses to infection and wounding in the C. elegans epidermis. Curr Biol. 2008;18(7):481–9. doi: 10.1016/j.cub.2008.02.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ziegler K, Kurz CL, Cypowyj S, Couillault C, Pophillat M, Pujol N, Ewbank JJ. Antifungal innate immunity in C. elegans: PKCdelta links G protein signaling and a conserved p38 MAPK cascade. Cell Host Microbe. 2009;5(4):341–52. doi: 10.1016/j.chom.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 22.Yan D, Wu Z, Chisholm AD, Jin Y. The DLK-1 kinase promotes mRNA stability and local translation in C. elegans synapses and axon regeneration. Cell. 2009;138(5):1005–18. doi: 10.1016/j.cell.2009.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bounoutas A, Kratz J, Emtage L, Ma C, Nguyen KC, Chalfie M. Microtubule depolymerization in Caenorhabditis elegans touch receptor neurons reduces gene expression through a p38 MAPK pathway. Proc Natl Acad Sci U S A. 2011;108(10):3982–7. doi: 10.1073/pnas.1101360108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339(6121):819–23. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Friedland AE, Tzur YB, Esvelt KM, Colaiacovo MP, Church GM, Calarco JA. Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nat Methods. 2013;10(8):741–3. doi: 10.1038/nmeth.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineering via Cas9. Science. 2013;339(6121):823–6. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu S. The application of CRISPR-Cas9 genome editing in Caenorhabditis elegans. J Genet Genomics. 2015;42(8):413–21. doi: 10.1016/j.jgg.2015.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kelly WG, Xu S, Montgomery MK, Fire A. Distinct requirements for somatic and germline expression of a generally expressed Caernorhabditis elegans gene. Genetics. 1997;146(1):227–38. doi: 10.1093/genetics/146.1.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamamoto M, Uematsu S, Okamoto T, Matsuura Y, Sato S, Kumar H, Satoh T, Saitoh T, Takeda K, Ishii KJ, et al. Enhanced TLR-mediated NF-IL6 dependent gene expression by Trib1 deficiency. J Exp Med. 2007;204(9):2233–9. doi: 10.1084/jem.20070183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dierking K, Polanowska J, Omi S, Engelmann I, Gut M, Lembo F, Ewbank JJ, Pujol N. Unusual regulation of a STAT protein by an SLC6 family transporter in C. elegans epidermal innate immunity. Cell Host Microbe. 2011;9(5):425–35. doi: 10.1016/j.chom.2011.04.011. [DOI] [PubMed] [Google Scholar]
- 31.Hammarlund M, Nix P, Hauth L, Jorgensen EM, Bastiani M. Axon regeneration requires a conserved MAP kinase pathway. Science. 2009;323(5915):802–6. doi: 10.1126/science.1165527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nix P, Hisamoto N, Matsumoto K, Bastiani M. Axon regeneration requires coordinate activation of p38 and JNK MAPK pathways. Proc Natl Acad Sci U S A. 2011;108(26):10738–43. doi: 10.1073/pnas.1104830108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37(Web Server issue):W202–8. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Medina-Rivera A, Defrance M, Sand O, Herrmann C, Castro-Mondragon JA, Delerce J, Jaeger S, Blanchet C, Vincens P, Caron C, et al. RSAT 2015: Regulatory Sequence Analysis Tools. Nucleic Acids Res. 2015;43(W1):W50–6. doi: 10.1093/nar/gkv362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gupta S, Stamatoyannopoulos JA, Bailey TL, Noble WS. Quantifying similarity between motifs. Genome Biol. 2007;8(2):R24. doi: 10.1186/gb-2007-8-2-r24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vinson CR, Sigler PB, McKnight SL. Scissors-grip model for DNA recognition by a family of leucine zipper proteins. Science. 1989;246(4932):911–6. doi: 10.1126/science.2683088. [DOI] [PubMed] [Google Scholar]
- 37.Hosack DA, Dennis G, Jr, Sherman BT, Lane HC, Lempicki RA. Identifying biological themes within lists of genes with EASE. Genome Biol. 2003;4(10):R70. doi: 10.1186/gb-2003-4-10-r70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Engelmann I, Griffon A, Tichit L, Montanana-Sanchis F, Wang G, Reinke V, Waterston RH, Hillier LW, Ewbank JJ. A comprehensive analysis of gene expression changes provoked by bacterial and fungal infection in C. elegans. PLoS One. 2011;6(5):e19055. doi: 10.1371/journal.pone.0019055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zugasti O, Thakur N, Belougne J, Squiban B, Kurz CL, Soule J, Omi S, Tichit L, Pujol N, Ewbank JJ. A quantitative genome-wide RNAi screen in C. elegans for antifungal innate immunity genes. BMC Biol. 2016;14(1):35. doi: 10.1186/s12915-016-0256-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Andrusiak MG, Jin Y. Context specificity of stress-activated mitogen-activated protein (MAP) kinase signaling: the story as told by Caenorhabditis elegans. J Biol Chem. 2016;291(15):7796–804. doi: 10.1074/jbc.R115.711101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim DH, Ewbank JJ. Signaling in the innate immune response. WormBook. 2015 doi: 10.1895/wormbook.1.83.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Richardson CE, Kinkel S, Kim DH. Physiological IRE-1-XBP-1 and PEK-1 signaling in Caenorhabditis elegans larval development and immunity. PLoS Genet. 2011;7(11) doi: 10.1371/journal.pgen.1002391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pukkila-Worley R, Feinbaum R, Kirienko NV, Larkins-Ford J, Conery AL, Ausubel FM. Stimulation of host immune defenses by a small molecule protects C. elegans from bacterial infection. PLoS Genet. 2012;8(6) doi: 10.1371/journal.pgen.1002733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cheesman HK, Feinbaum RL, Thekkiniath J, Dowen RH, Conery AL, Pukkila-Worley R. Aberrant activation of p38 MAP kinase-dependent innate immune responses is toxic to Caenorhabditis elegans. G3 (Bethesda) 2016;6(3):541–9. doi: 10.1534/g3.115.025650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim KW, Jin Y. Neuronal responses to stress and injury in C. elegans. FEBS Lett. 2015;589(14):1644–52. doi: 10.1016/j.febslet.2015.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77(1):71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mello CC, Kramer JM, Stinchcomb D, Ambros V. Efficient gene transfer in C. elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 1991;10(12):3959–70. doi: 10.1002/j.1460-2075.1991.tb04966.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arribere JA, Bell RT, Fu BX, Artiles KL, Hartman PS, Fire AZ. Efficient marker-free recovery of custom genetic modifications with CRISPR/Cas9 in Caenorhabditis elegans. Genetics. 2014;198(3):837–46. doi: 10.1534/genetics.114.169730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pujol N, Zugasti O, Wong D, Couillault C, Kurz CL, Schulenburg H, Ewbank JJ. Anti-fungal innate immunity in C. elegans is enhanced by evolutionary diversification of antimicrobial peptides. PLoS Pathog. 2008;4(7):e1000105. doi: 10.1371/journal.ppat.1000105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dickinson DJ, Pani AM, Heppert JK, Higgins CD, Goldstein B. Streamlined genome engineering with a self-excising drug selection cassette. Genetics. 2015;200(4):1035–49. doi: 10.1534/genetics.115.178335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sarin S, Prabhu S, O’Meara MM, Pe’er I, Hobert O. Caenorhabditis elegans mutant allele identification by whole-genome sequencing. Nat Methods. 2008;5(10):865–7. doi: 10.1038/nmeth.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Giardine B, Riemer C, Hardison RC, Burhans R, Elnitski L, Shah P, Zhang Y, Blankenberg D, Albert I, Taylor J, et al. Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 2005;15(10):1451–5. doi: 10.1101/gr.4086505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rouger V, Bordet G, Couillault C, Monneret S, Mailfert S, Ewbank JJ, Pujol N, Marguet D. Independent synchronized control and visualization of interactions between living cells and organisms. Biophys J. 2014;106(10):2096–104. doi: 10.1016/j.bpj.2014.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu Z, Ghosh-Roy A, Yanik MF, Zhang JZ, Jin Y, Chisholm AD. Caenorhabditis elegans neuronal regeneration is influenced by life stage, ephrin signaling, and synaptic branching. Proc Natl Acad Sci U S A. 2007;104(38):15132–7. doi: 10.1073/pnas.0707001104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mukhopadhyay A, Deplancke B, Walhout AJ, Tissenbaum HA. Chromatin immunoprecipitation (ChIP) coupled to detection by quantitative real-time PCR to study transcription factor binding to DNA in Caenorhabditis elegans. Nat Protoc. 2008;3(4):698–709. doi: 10.1038/nprot.2008.38. [DOI] [PMC free article] [PubMed] [Google Scholar]