

Abstract
DNA replication of the papillomaviruses is specified by cooperative binding of two proteins to the ori site: the enhancer E2 and the viral initiator E1, a distant member of the AAA+ family of proteins. Formation of this prereplication complex is an essential step toward the construction of a functional, multimeric E1 helicase and DNA melting. To understand how E2 interacts with E1 to regulate this process, we have solved the X-ray structure of a complex containing the HPV18 E2 activation domain bound to the helicase domain of E1. Modeling the monomers of E1 to a hexameric helicase shows that E2 blocks hexamerization of E1 by shielding a region of the E1 oligomerization surface and stabilizing a conformation of E1 that is incompatible with ATP binding. Further biochemical experiments and structural analysis show that ATP is an allosteric effector of the dissociation of E2 from E1. Our data provide the first molecular insights into how a protein can regulate the assembly of an oligomeric AAA+ complex and explain at a structural level why E2, after playing a matchmaker role by guiding E1 to the DNA, must dissociate for subsequent steps of initiation to occur. Building on previously proposed ideas, we discuss how our data advance current models for the conversion of E1 in the prereplication complex to a hexameric helicase assembly.
Keywords: Crystal structure E1E2 complex, papillomavirus, helicase structure, AAA+ protein, DNA replication
Papillomaviruses (PVs) are small DNA viruses that maintain latency in the stem cells of various epithelial tissues (for review, see Howley 1996). They are the etiological agents of a variety of benign lesions of the epithelium, and certain high-risk variants of the human papillomavirus (HPV), such as HPV-16, 18, and 31, are associated with cervical carcinomas (Bosch et al. 1995). The virus is maintained as an extrachromosomal nuclear plasmid that replicates in S phase with the host genome. Because PVs have few open reading frames (∼8 ORFs), the viral life cycle depends heavily on interaction with the host cell for enzymes and ancillary factors for its own gene expression and replication programs. The PVs have provided important novel insights into the regulation of cell proliferation and cancer etiology; for example, the targeted degradation of p53 directed by the HPV16 E6 protein first revealed how key cellular regulatory proteins could be specifically ubiquitinated by E3 ligases for destruction by the proteosome (Scheffner et al. 1993). The PVs also have provided models to understand how eukaryotic transcription factors coassociate to form loops between segments of DNA (Li et al. 1991) and to explore assembly of specific replication factories on defined origins of replication (Stenlund 2003b).
The first two universal steps in the formation of a prereplication complex are origin recognition and subsequent distortion and separation of duplex DNA strands. The simplicity of the PV prereplication complex offers advantages for mechanistic and structural dissection of these steps. Only two PV-encoded proteins, E1 and E2, are necessary for the initiation of viral DNA replication. The E1 protein (for review, see Sverdrup and Myers 1997) serves as the viral initiator protein that binds to the viral origin and, on DNA melting, converts to a helicase that functions during viral DNA replication. The N-terminal ∼200 amino acids of E1 are not well conserved between various papillomaviral types except for two boxes corresponding to the nuclear localization signal (Fig. 1A). The central portion E1 comprises the site-specific DNA-binding domain of E1 that associates with a series of inverted-repeat binding sites at the viral origin (Mendoza et al. 1995; Chen and Stenlund 1998; Enemark et al. 2002). The helicase domain of E1 is contained in the C-terminal half of the protein and contains motifs characteristic of Walker-type ATP-binding and hydrolysis proteins. Truncated versions of HPV E1 deleted for the entire site-specific DNA-binding domain are active helicases for templates containing forks or single strands (White et al. 2001; S.E. Moyer, E.A. Abbate, and M.R. Botchan, unpubl.) but are unable to load onto duplex DNA. The protein thus has one set of domains for non-specific DNA interactions that are critical for helicase activity and another domain for site-specific DNA recognition that is essential for origin targeting. The overall domain architecture of E1, including the spacing between the nuclear localization signals, distinct site-specific DNA recognition domain, and helicase motifs of E1 is similar to that of the SV40 T-antigen (Park et al. 1994; Mansky et al. 1997).
Figure 1.

Structure of the E1 · E2 complex. (A) The HPV18 E1 and E2 ORFs indicating the amino acid boundaries of key domains. (AD) Activation domain; (DBD) DNA-binding domain; (NLS) nuclear localization sequence. (B) Cartoon representation of the E1 · E2 structure. The N-terminal helical domain of E2 is colored green, the β-strand structural domain is colored red, and the linker segment between the two domains is yellow. E1 is depicted in blue. Secondary structural elements are labeled.
The E2 ORF contains a conserved N-terminal activation domain of ∼200 amino acids. This domain is essential for all viral DNA functions including transcription, replication, and viral DNA segregation. The modular activation domain is connected to the C-terminal DNA-binding/dimerization domain by a flexible proline-rich hinge domain (Fig. 1A). E2's role in PV replication is to help target monomers of E1 to the viral origin through cooperative DNA binding and to ensure the orderly conversion of the complex to an active helicase. In this respect, E2 is both an initiation protein and a helicase loader (Mohr et al. 1990; Yang et al. 1991a; Chiang et al. 1992; Seo et al. 1993; Sedman and Stenlund 1995). E2 is a homodimer and interacts with a well-characterized DNA recognition site that is conserved across the entire PV family.
The E2-binding sites lie adjacent to the E1-binding loci at the origin and are critical for formation of an E1 · E2 · ori ternary complex that consists of a dimer of E1 and a dimer of E2 (Chen and Stenlund 1998). The E1 · E2 assembly serves as the prereplication complex for the virus and is essential for replication in vivo, as E1 alone has low intrinsic DNA-binding specificity. Synergistic interactions between the two factors guides the specificity between the Scylla and Charybdis of tight binding to the viral DNA and nonspecific interactions with chromosomal DNA. Although both proteins bind to the origin through their respective DNA-binding domains, strong interactions between the C-terminal helicase domain of E1 and the activation domain of E2 create and stabilize an E1 · E2 assembly as well (Yang et al. 1991b; Li et al. 1993; Yasugi et al. 1997; Masterson et al. 1998).
For the bovine papillomavirus (BPV), additional contacts between the DNA-binding domains of each protein have been reported (Chen and Stenlund 1998). For BPV1, the interaction between the DNA-binding domains of E1 and E2 is believed to be an architectural one that produces a DNA shape that permits the stronger protein: protein interactions in the other domains of the partners. This situation may be restricted to BPV1 because of the especially close proximity of the E2-binding site to the adjacent E1-binding site (three base pairs separate the two) in the BPV origin. When E2 DNA-binding sites are displaced from the E1 assembly site, this particular interaction becomes dispensable (Gillitzer et al. 2000) and a hybrid E2 with a Gal4 DNA-binding domain can substitute for the DNA-binding tether if Gal4 sites are provided in cis (Winokur and McBride 1996). In contrast, the activation domain of E2 is specifically required for replication in vivo and that function cannot be replaced with other foreign activation sequences.
Following formation of the E1 · E2 complex at the viral origin, a series of poorly understood reactions ensue by which E1 molecules in the prereplication complex reorganize and convert into an active, doubly hexameric helicase (Fouts et al. 1999; Lin et al. 2002). Conversion requires the binding of additional E1 molecules to the origin and is accelerated by ATP (Sanders and Stenlund 1998). How E2 regulates E1 assembly in this conversion process, as well as its ultimate fate, is not clear. For BPV1, following the binding of additional E1 molecules, E2 is displaced from the viral origin (Lusky et al. 1994; Sanders and Stenlund 1998). For HPV-11, it has been reported that E2 remains associated with the DNA during the assembly of E1 molecules, even though E2 might serve as a potential roadblock for extensive unwinding (Lin et al. 2002). These differences raise several important questions. For example, if E2 leaves the BPV origin on the assembly of bound E1 molecules, is displacement of E2 necessary and, if so, why? Does ATP binding play a role in the assembly process or does nucleotide simply power the helicase to displace a proximal E2? Do differences between the viral systems reflect a fundamental divergence in how DNA replication is initiated?
To address some of these questions and better understand the interplay of E1 and E2 at the viral origin, we have determined the structure of a heterodimer containing the full-length activation domain of E2 in complex with the C-terminal helicase domain of E1. The structure shows that E2 regulates E1 assembly by sterically blocking key E1 oligomerization interfaces. This feature indicates that E2 functions not just as a “chaperone” that shuttles E1 to the origin, but it also acts as a molecular “matchmaker” between E1 and DNA to ensure the proper assembly of the E1 double hexamer. Our structural studies, coupled with biochemical experiments, demonstrate that ATP is an allosteric effector of this process and plays an important role in converting the site-specific DNA binding form of E1 in the prereplication complex to a nonsequence-specific DNA helicase that functions during replication.
Results
Structure determination
The E1 · E2 complexes used in crystallization trials were formed by expressing both proteins separately, and then mixing the extracts together. These complexes were stable throughout a three-column purification scheme (see Materials and Methods). The E2 portion of the complex contains the full-length activation domain of HPV18 E2 (amino acids 1-215), which is a longer variant of the HPV18 E2 protein whose uncomplexed structure (amino acids 66-215) was solved previously (Harris and Botchan 1999) and shown to be homologous to the HPV16 E2 activation domain (Antson et al. 2000). To find a minimal region of E1 that would be soluble and interact with E2, we constructed a series of N- and C-terminal deletions. A fragment of HPV18 E1 containing residues 428-631 in complex with the E2 activation domain yielded crystals suitable for structure determination. This E1 truncation showed affinities for E2 similar to longer fragments, as measured by glutathione-S-transferase (GST) pull-down experiments, and formed a stable heterodimer with E2 in solution, as measured by gel filtration (data not shown). Crystals of the complex were obtained by standard screening procedures, but extensive optimization of crystal harvesting and freezing conditions was necessary to produce high-resolution diffraction data (Materials and Methods). Phases were determined by using MAD from crystals soaked in erbium chloride. The final structure of the complex has been refined to 2.1 Å resolution (Table 1).
Table 1.
Data collection and refinement
| E1:E2 complex (Space group; P212121) | |||
|---|---|---|---|
| Cell parameters: a = 82.297 Å, b = 88.745 Å, c = 375.027 Å, α = β = γ = 90°
|
|
|
|
| Data collection | Native | Erb (peak) | Erb (remote) |
| Resolution (Å) | 50-2.1 | 50-3.0 | 50-3.0 |
| Wavelength (Å) | 1.0 | 1.483 | 1.501 |
| I/σ | 22.3 (2.6) | 13.4 (2.8) | 13 (2.4) |
| Rsym (%)a | 4.9 (37.9) | 9.2 (41.0) | 9.3 (41.6) |
| Completeness (%) | 99.0 (91.4) | 99.5 (99.1) | 99.1 (95.4) |
| Redundancy | 3.5 (2.5) | ||
| Phasing | |||
|---|---|---|---|
| No. of sites | 17 | ||
| Mean figures of merit (FOM)Solve | 0.40 | ||
| Mean figure of merit (FOM)Resolve | 0.58 |
| Refinement | |||
|---|---|---|---|
| Resolution (Å) | 30-2.1 | ||
| Rfree (%)b | 26.3 (33.1) | ||
| Rwork (%)c | 21.9 (26.6) | ||
| RMSDbond (Å) | 0.008 | ||
| RMSDangle (°) | 1.0 | ||
| Favored (%) | 92.0 | ||
| Additionally allowed (%) | 7.5 | ||
| Generously allowed (%) | 0.6 | ||
| Total atoms (protein) | 19218 | ||
| Total atoms (water) | 968 |
Values in parentheses are for the highest-resolution bin.
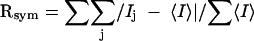 , where Ij is the recorded intensity of the reflection j and
, where Ij is the recorded intensity of the reflection j and 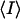 is the mean recorded intensity over multiple recordings.
is the mean recorded intensity over multiple recordings.
Rfree is the R value calculated for a test set of reflections, comprising a randomly selected 5% of the data that is not used during refinement.
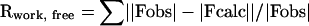 .
.
Structure of the E1 · E2 complex
The E1 · E2 complex crystallized with six heterodimers in the asymmetric unit, offering six independent views of the assembly. Few structural differences are evident between the heterodimers (average Cα RMSD < 0.9 Å). Potential E2 activation domain dimerization interfaces seen in different structures of uncomplexed E2 monomers (Harris and Botchan 1999; Antson et al. 2000) are not present in any of the E1 · E2 complexes observed here, even though the three E2 N-terminal helices that form various dimerization contacts in uncomplexed molecules are structurally superposable to those in the E1 · E2 heterodimer. The absence of E2-AD homodimer contacts is not due to E1 binding, because E1 associates with the inner surface of the helical domain of E2 (Fig. 1B), as opposed to the outer surface, which mediates E2 · E2 interactions in the free HPV16 and HPV18 E2 structures. The absence of dimerized E2 molecules in the E1 · E2 complex, as well as in the uncomplexed HPV11 E2 structure (Wang et al. 2004), supports the notion that the dimerization of E2-AD observed in some crystal forms may be fortuitous. For E2-dependent DNA replication in vivo, a single E2-binding site is sufficient in transient assays (Russell and Botchan 1995); similarly, an E2 dimer containing a single activation domain functions as wild type in vitro (Lim et al. 1998). These data all indicate that the E1 · E2 interaction does not require dimers of the E2 activation domain. For other E2 functions, such as transcriptional regulation or plasmid segregation, E2 · E2 activation domain interactions may be important, as suggested (Antson et al. 2000). No functionally significant E1 · E1 interactions are apparent in the crystal lattice.
The E1 · E2 complex is characterized by an overall “C”-shaped structure with dimensions of 75 × 60 × 40 Å. The top and side of the “C” are formed by the E2 activation domain, whereas the bottom is formed by E1 (Fig. 1B). E2 is formed by a largely α-helical N-terminal domain, which is in turn linked to the C-terminal β-strand domain by an extended linker segment (Fig. 1B). There is no significant change in either the location or organization of the major structural elements of E2 in the E1 complex as compared with its unbound forms. The Cα RMSD for E2 in the complex as compared with free E2 is 1.0 Å (over 128 amino acids) and 1.2Å (over 189 amino acids) for the HPV18 and HPV16 activation domains, respectively. The only noteworthy difference between the previously solved E2 structures and E2 in the complex is that there is no clear density for the last β-strand at the C terminus, an element that was visible in prior HPV18 and HPV16 structures. This C-terminal stretch of amino acids, which is distant from the E1 interaction surface, lies proximal to the E2 hinge region (see Fig. 1A). The differences observed here likely reflect a high degree of mobility in this end of the E2 activation domain.
The E1 helicase core is predominantly globular, though long loops extend out from the core at either end. The core contains a central parallel β-sheet comprising five strands that are sandwiched by several α-helices. The α/β topology of E1 is characteristic of Walker-type, P-loop ATPases, consistent with its membership in the AAA+ (ATPases associated with various cellular activities) superfamily of proteins (Neuwald et al. 1999) and the SF3 family of helicases (Koonin 1993). The SF3 helicase superfamily contains three characteristic motifs.
The first two motifs include the Walker-A (P-loop) and Walker-B (D(D/E)XX) elements, which are necessary for nucleotide binding and hydrolysis. The P-loop is located between strand β1 and helix α4, whereas the Walker-B motif is formed by two successive aspartate residues at the end of strand β3. Motif “C” of the SF3 family contains an invariant Asn residue, which in E1 maps to the C terminus of β6. This motif, often designated as the “sensor 1” region of AAA+ proteins, is thought to either respond to the hydrolytic state of bound nucleotide or to participate in positioning a water for nucleophilic attack on the γ-phosphate of ATP.
The last helix in E1 (α9) is also notable. This element runs through the center of a small, circular constellation of helices formed by helices α1-α3. This cluster appears to be important for the proper folding of this domain, because further N-terminal or C-terminal deletions resulted in E1 proteins that are poorly expressed and largely insoluble (E.A. Abbate and M.R. Botchan, unpubl.). Because the E1 helicase core domain is highly conserved between different variants of the virus (Fig. 2B), we expect the structure to be homologous in other E1 variants.
Figure 2.

Sequence/structure alignment of papillomavirus E1 and E2 proteins. (A) Sequence alignment of the E2 activation domain from some important papillomaviral types. Amino acid boundaries for each variant are indicated and amino acids are colored according to their amino acid type. The secondary structure from the HPV18 E2 structure is depicted above the alignment. Helices are represented by rectangles, strands by arrows, and coil regions by black lines. Residues making contact between E1 and E2 are indicated by black arrows. The rulers above each alignment delineate 10-amino acid increments. (B) Sequence alignment of the homologous regions of various E1 proteins with the amino acids of HPV18 E1. Various elements are labeled as in A. Walker-A and Walker-B motifs and the SF3 superfamily C motif are indicated, as are two arginines critical for either the E1 · E2 interaction or ATPase activity of the E1 hexamer.
The E1 · E2 interaction surface
The interaction surface between E1 and E2 requires residues from structural elements of both proteins that are spread out over an extended range of primary amino acid sequence (Figs. 2A,B, 3C,D). This may explain why it has been difficult to define the interaction domain by simple deletion analysis or scanning mutagenesis. In total, 940 Å2 of surface area is buried per protomer on formation of the E1 · E2 complex. The three helices of the N-terminal domain of E2 comprise the major contact point for E1. The helical linker segment connecting the N-terminal and C-terminal domains of E2, as well as the short anti-parallel β-strands between helices αB and αC, also make numerous contacts with E1 (Fig. 3). Interestingly, there is no interaction between the C-terminal β-strand domain structure of E2 and E1, creating an open cavity that gives the complex its overall “C” shape (Fig. 3A). The absence of contacts between the C-terminal domain of E2 and E1 allows this region of E2 to flex slightly with respect to the N-terminal domain, a characteristic reflected in the higher B-factors for residues contained in this region. In addition, superposition of the six dimers in the asymmetric unit reveals that the observed differences between the E2 molecules are due to subtle shifts in the relative orientation of the C-terminal domain of E2 with respect to the N-terminal helical domain. The main interaction surface on E1 is formed by a bundle of helices comprising α2, α3, and α9 (Fig. 3D). E1 also makes additional contacts to E2 through a long extended loop between β7 and α9 (loop-2; Fig. 3A,D).
Figure 3.

The E1 · E2 interaction surface. (A) Surface rendering of the E1 · E2 complex with underlying cartoon representation. The N-terminal helical domain of E2 is colored green, the C-terminal β-strand domain is in red, and the linker between the two is yellow. E1 is depicted in blue. (B) Exploded view of the E1 · E2 complex rotated to reveal the surface of interaction. The color scheme is as in A. The interaction surface between E1 and E2 (defined as atoms ≤4.5 Å apart) is depicted in orange. (C) Cα wire trace of E2 with residues involved in the E1 · E2 complex formation highlighted in orange. The color scheme is the same as in A. (D) Cα wire depiction of E1, with residues making contact between E1 and E2 depicted in orange.
No single type of chemical interaction predominates the contacts between E1 and E2. This may explain why the complex is stable to a variety of different conditions, including nonionic detergents; indeed, formation of the E1 · E2 complex studied here was performed in the presence of 1 M NaCl to eliminate spurious protein contamination. An inspection of some of the specific interactions reveals why several amino acids, such as E2 Glu 43 (Glu 39 for BPV1 E2), are critical for the replication function of E2 in both the human and bovine variants of the virus (Ferguson and Botchan 1996; Sakai et al. 1996; Harris and Botchan 1999). In the structure of the complex, this glutamate maps to the middle helix (αB) of the three N-terminal helices of E2 and forms a buried salt bridge with Arg 454 of E1 (Fig. 4A). Alignment of E1 sequences from HPV 6, 11, 16, and 18 showed that Arg 454 is absolutely conserved in the human variants of the virus (Fig. 2B). In BPV1, however, this arginine is replaced by an asparagine, a substitution that not only shortens the side chain, but also precludes any favorable ionic attraction with the corresponding glutamic acid. Interestingly, it appears that other interactions in this region have evolved to account for this variation in BPV1. For all HPV's, a hydrophobic residue points directly toward the salt bridge (EG, Ile 19 in HPV18), which may strengthen the ionic interaction, whereas in BPV the corresponding hydrophobic residue is replaced by a glutamine. Thus, intimate contact between E2 and E1 at this position in the complex appears to be preserved in BPV by creating a three-amino acid hydrogen bonding network (Asn:Gln: Arg) in place of the HPV ion pair (Glu:Arg).
Figure 4.

Mutation of a conserved, buried ion pair at the E1 · E2 interface affects complex formation. (A) The three helices of the N-terminal domain of E2 are in green and E1 is colored blue. The side chains of the Glu 43:Arg 454 ion pair is highlighted yellow. (B) Graph of E1 · E2 pull-down data. The amount of E1 pulled-down by GST-E2 (100 nM) is plotted on the Y-axis in arbitrary units. The concentration of E1 used in each pull-down experiment is plotted on the X-axis.
To test the predicted importance of the E2 · E1 Glu: Arg interaction, as well as other interfacial residues for the relative stability of the E1 · E2 complex, we performed GST pull-down assays with a variety of mutants (Fig. 4B). In these studies, an N-terminal GST-E2 fusion was used as bait for a C-terminal fragment of E1 (residues 358-631). Tests with the E2 E43A mutant produced a measurable but significantly reduced interaction with E1, and mutation of this residue's partner on E1, Arg 454, severely impaired formation of the E1 · E2 complex by >20-fold. These data substantiate the importance of this buried ion pair to the overall stability of the complex. In contrast, mutation of Glu 24 of E2, which forms surface interactions with E1 Arg 447 and Arg 622, only slightly reduces the stability of the complex. As a control, mutation of E1 residues not at interacting surfaces Lys 557 (in loop-1) and R589 (near the N terminus of β7), do not have an effect on formation of the complex.
E1 and the SV40 large T-antigen
E1 and SV-40 T-antigen are thought to be highly similar proteins in both structure and function. Both proteins bind site specifically to and assemble on their respective viral origins and distort the DNA in a manner that leads to origin melting. Following the melting event, both proteins function as doubly hexameric helicases (Bullock 1997; Simmons 2000). Unexpectedly, structural studies have revealed that the site-specific DNA-binding domains of BPV E1 and T-antigen possess the same fold, despite sharing <10% amino acid sequence identity (Luo et al. 1996; Enemark et al. 2000).
Li et al. (2003) have recently determined the crystal structure of the hexameric T-antigen helicase domain. To compare the C-terminal helicase modules of E1 and T-antigen, we aligned the primary and secondary structural features of these domains (Fig. 5). Sequence analysis of the fully functional helicase region, which extends beyond the residues included in our structure, reveals that the two proteins exhibit ∼20% pairwise identity (Fig. 5D). A set of helices N-terminal to the ATPase domain of T-antigen appear to have equivalent counterparts in E1 (Fig. 5A; Li et al. 2003). As in T-antigen, this N-terminal region of E1 is important for oligomerization of the protein; the E1 fragment crystallized here (residues 428-631) is monomeric in the absence of E2, as judged by gel filtration chromatography (data not shown), whereas a fragment that includes the N-terminal domain (amino acids 358-631) forms a stable hexamer (see following).
Figure 5.

Comparison of E1 and T antigen. (A) Side-by-side comparison of the SV40 T-antigen (PDB 1N25) and E1 helicase domain structures. The N-terminal domain in E1 that is critical for hexamerization was not contained in the fragment used for cocrystallization of the E1 · E2 complex. The N terminus of E1 is predicted to continue into a “linker helix” similar to that seen in T-antigen (see secondary structure alignments in D). The ATPase core for each protein is shown in gray. (B) Superposition of E1 and T-antigen ATPase core regions (colored gray in A), with E1 colored blue and T-antigen in orange. (C) Stereo view of the superposition of the entire E1 structure onto the analogous region of T-antigen. E1 is in blue and T-antigen in orange. The difference between E1 and T-antigen at the position of loop-2 in E1 is indicated. Analogous helices between E1 and T-antigen that form a knot-like structure are labeled. (D) Sequence and secondary structure alignments of E1 and T-antigen. The boundaries of the E1 structure are indicated. Helices and predicted helices are indicated by blue rectangles and red rectangles, respectively. Strands are represented by blue arrows and predicted strands are depicted as red arrows.
In the ATPase core itself, there is a high degree of secondary structural similarity between E1 and T-antigen (Fig. 5A-C). The location and organization of the various secondary structure elements of these two proteins is conserved in this module, with the region of highest structural overlap residing in the RecA/AAA+ subdomain (Fig. 5B; this portion of the domain is colored gray in Fig. 5A). The RMSD between the core regions of the two proteins is 1.6 Å over 146 amino acids. The ATP-binding module is connected to a series of helices (Fig. 5C) that are not found in other AAA+ proteins but that do appear to be present in other E1 and T-antigen variants. Significantly, a major divergence between the E1 and T-antigen structures occurs at the loop-2 position of E1 (Fig. 5C,D). Loop-2 contains many residues involved in E2 contacts (Fig. 3D) and in T-antigen is replaced by a helical segment. This divergence accounts for the unique ability of PV E1 proteins to interact with E2 and appears to be part of a mechanism by which ATP functions as an allosteric regulator of E1 · E2 complex stability (see following).
E1 hexamer organization
Given the close structural similarity between the helicase domains of T-antigen and E1, we used the hexameric structure of T-antigen as a guide to model a hexameric E1 state. Only those Cα positions in the core ATPase domain of E1 and T-antigen were included in the superposition (Fig. 6A). Many loop regions, as well as side chains in the Walker-A and -B regions overlapped, despite the fact that we were comparing the structure of E1 in a complex with E2 to that of a hexameric T-antigen structure. In the intact E1 molecule, the domain solved here is part of a multitiered structure, wherein the N-terminal extension of the helicase domain sits on top of the large ring depicted in our model. This upper tier is expected to contribute significantly to the total E1 · E1 interaction surface, which we expect to be similar to the 2500 Å2 reported for T-antigen (Li et al. 2003).
Figure 6.

A predicted model for the helicase domain of the E1 hexamer. (A) Cartoon representation of the E1 hexamer resulting from superposition on SV40 T-antigen. Modeling of nucleotide bound to E1 is depicted (see Fig. 7), as is the arginine finger. (B) Electrostatic potential surface representation (GRASP; Nicholls et al. 1991) of the E1 hexamer. Areas of positive potential are blue and negative potential red. (C) Kinetic parameters for various E1 ATPase mutants. See Materials and Methods for a description of the assay. (D) Superposition of the E1 · E2 complex onto one subunit of the E1 hexamer. The E2 activation domain is depicted as in Figure 1B. The steric clash is diagrammed by the overlaps between the C-terminal β-strand domain of E2 (red) and a monomer of E1 (blue). Approximately 10% of the volume of E1 is invaded by the β-strand domain of E2.
Several lines of evidence support a hexameric model for E1 on the basis of T-antigen. First, this organization positions loop-1 from each E1 protomer, a segment that contains a basic trio of amino acids (RKH) in the central hole of the hexamer. An analogous loop is seen in the structure of T-antigen that also contains a similar sequence of amino acids (KKH). The loop-1 region confers a positive charge to the central pore of the hexamer (Fig. 6B) and may play a role in binding DNA during helicase function. In addition, the channel in the E1 hexamer is ∼15 Å in diameter, a value that agrees with the reported size of the central channel of the analogous lower tier of T-antigen (Li et al. 2003).
A second feature consistent with our E1 hexamer model arises at interfaces between the protomers. One characteristic of many multimeric ATPases is the presence of a conserved “arginine finger”, a motif that stimulates ATPase activity in an oligomerized complex by extending from one subunit into the catalytic site of an adjacent protomer and interacting with the γ-phosphate of bound ATP. In AAA+ proteins, mutation of the arginine finger, which generally resides in the “Box VII” or “SRC” motifs (Neuwald et al. 1999; Davey et al. 2002) results in a protein significantly impaired for ATPase activity (Song et al. 2000; Putnam et al. 2001). Accordingly, on modeling the E1 hexamer, we noted that a conserved arginine, Arg 589 (Fig. 2), at the N terminus of β7 became positioned adjacent to the ATP-binding site of an adjacent monomer (Fig. 6A).
To test whether Arg 589 might serve as an arginine finger in E1, we replaced this residue with alanine and assayed the mutant for ATPase activity. The R589A mutation was made in a slightly longer E1 (358-631) construct than that used for crystallization because the shorter fragment (428-631) fails to stably oligomerize. Coupled ATPase assays showed that mutation of Arg 589 greatly impaired nucleotide hydrolysis activity compared with a wild-type E1 fragment of the same length (Fig. 6C). Indeed, the kinetic parameters for ATP hydrolysis for this mutant were equivalent to a double aspartate substitution of the E1 Walker-B motif (D529A/D530A). As controls, two other mutants were also made and tested for function: K557A, which maps to the predicted DNA-binding loop in the central cavity of E1, and R454A, which cripples E2 interaction. As expected, neither had any appreciable effect on E1 ATPase activity. Importantly, mutation of Arg 589 had no effect on the ability of the protein to oligomerize, as judged by gel filtration chromatography (data not shown) and was not defective for E2 interaction (Fig. 4B), indicating the R589A protein was properly folded. These data provide the first evidence that E1, like other AAA+ proteins, uses an arginine finger motif to construct a catalytically competent, bipartite ATP-binding site. Our data also indicate that a hexamer organization for E1 subunits predicated on the T-antigen model is likely to capture the salient features of this quaternary E1 state.
Regulation of E1 hexamerization by E2 and ATP
The E1 hexamer model also reveals the mechanism by which E2 blocks E1 oligomerization. As can be seen in Figure 6D, superposition of the E1 · E2 complex on the E1 hexamer shows that the C-terminal β-sheet domain of E2 shields the E1 oligomerization surface. This observation predicts that E2 can directly regulate hexamer assembly by physically preventing additional E1 subunits from associating with E1 protomers tethered at the origin. Consequently, disruption of the intimate E1:E2 complex on the DNA must occur before monomer E1:E1 interactions commence. Therefore, our results suggest that previous reports of HPV-11 E2 remaining bound to origin DNA during the assembly of E1 (Lin et al. 2002) is a consequence of E2 being tethered at a distance from the E1 sites following disruption of the E1 · E2 association. In contrast, the close proximity of the E2-binding sites to the location of E1 assembly in BPV likely necessitates physical displacement of E2 from the origin.
If E2 bound to E1 sterically occludes hexamerization, what serves as the switch to disengage E2? The gel filtration profiles of these proteins in the presence and absence of ATP provide a clue (Fig. 7A). Without ATP, the E1 · E2 complex elutes as a single peak (top trace), with an elution volume corresponding to a 1:1 complex of E1 and E2. When the ratio of E1 · E2 in each fraction eluted from the column is measured by SDS-PAGE, the complex appears to have a 1:1 stoichiometry (Fig. 7A, bottom panels). However, in the presence of ATP, a high-molecular-weight peak and a low-molecular-weight peak now appear. The elution volume of the high-molecular-weight peak is consistent with the size of an E1 hexamer and contains predominantly free E1. The later-eluting peak contains free E2 and, as judged by elution volume, is monomeric.
Figure 7.

Effect of ATP on the E1 · E2 complex. (A) Elution profile of the E1 · E2 complex from a gel filtration column in the presence and absence of ATP. These experiments were performed with the 358-631 construct of E1, which contains structural elements necessary for hexamerization (Fig. 5). An SDS-PAGE gel of fractions from the columns is shown below each chromatogram. A quantitative profile of the amount of E1 and E2 in each fraction (stained with SYPRO Orange and scanned with a Typhoon, Amersham) is given for each protein below the gels. (B) Superposition of RuvB (PDB 1IN4) bound to ADP onto E1 reveals the manner in which nucleotide would bind to E1. Overlay of the E1 · E2 surface shows the contacts between loop-2 of E1 and E2. E2 is colored green and E1 blue, with residues of loop-2 depicted in red. The view is the same as that of Figure 1B. The bottom panel depicts a surface representation of the E1 · E2 complex with modeled ADP showing the clash between the adenine ring and loop-2 of E1. (C) Structural comparison of E1 and the AAA+ protein RuvB. The ATPase modules are colored green. The C-terminal helical “lid” domain that clamps over the adenine of the bound nucleotide of RuvB is colored red.
The simplest interpretation of this finding is that, in the presence of ATP, the binding equilibrium between E1 and E2 becomes shifted toward dissociation of the complex and that this dissociation in turn allows E1 molecules to self-assemble to form a hexamer. This idea is substantiated by limited proteolysis data. In the E1 · E2 complex, E1 is susceptible to hydrolysis with trypsin, whereas the hexamer form of E1 is resistant to such cleavage. Incubation of the complex with ATP converts a fraction of the E1 to a proteolysis-resistant form, at a level consistent with the amount of hexamer detected by gel filtration (data not shown). Similar results were obtained when this experiment was performed with ATP-γS, indicating that ATP binding, rather than hydrolysis, was important for the release. Although ATP shifts the equilibrium toward dissociation, association of the complex is still favored. However, we speculate that in the context of the full-length proteins bound and positioned on origin DNA, there will be a hysteresis that irreversibly drives the formation of a hexameric E1 helicase locked onto melted DNA (see model following).
The E1 · E2 structure provides a molecular explanation for the ability of ATP to shift the equilibrium of the complex toward dissociation. Juxtaposed to the P-loop of the ATP-binding site is a long extension that provides one of the major interaction surfaces in contact with E2 (Fig. 7B, loop-2). Loop-2 is unusual in that it substitutes for the helical “lid” domains typically found in AAA+ proteins (Fig. 7C); in this regard both E1 (and its cousin T-antigen) lack the sensor-II regions of AAA+ proteins and actually represent a distinct subclass of AAA+ ATPases. Fitting nucleotide into the E1 ATP-binding site by superposition with the structure of the RuvB AAA+ protein (Putnam et al. 2001) shows how this PV-specific reconfiguration of the AAA+ fold influences E1 · E2 association/dissociation (Fig. 7B). There is good steric and chemical complementarity of the sugar and phosphate groups of the modeled nucleotide with the surface of E1 in the complex. The fit of the adenine moiety, however, is another matter. The position of the base of the modeled nucleotide is such that it points at a number of solvent-exposed aromatic and aliphatic residues contained in loop-2. The conformation of loop-2, however, is maintained by its contacts with E2, such that these residues sterically block the space expected to be occupied by the adenine moiety (Fig. 7B, bottom). This observation indicates that a conformational change must occur in loop-2 in order to accommodate binding of the base of ATP.
Together, these findings suggest a mechanism by which ATP might help remodel the E1 · E2 complex and promote assembly of the replication complex during initiation. In one model, loop-2 would undergo an induced fit on ATP binding that would reconfigure its structure. This, in turn, would disrupt numerous interactions that bond loop-2 to the N-terminal domain of E2, thereby decreasing the stability of the complex. Alternatively, it is possible that E2 might be poised to respond to the presence of an exogenous E1 subunit that is appropriately positioned with respect to the E1 protein of the E1 · E2 complex to correctly oligomerize. In this scheme, the β-domain of E2 might act as a trigger to disengage E2 from E1, thereby allowing ATP to bind to E1. Once bound, ATP would reconfigure the loop-2 region, preventing E2 from reassociating with E1.
Discussion
Our data provide a structural basis for understanding specific interactions between E1 and E2. Although the E1 molecule has strong structural homologies with T-antigen and other members of this superfamily, including the Rep40 helicase (James et al. 2003), specific motifs, including loop-2, have evolved in unique ways to yield such interaction interfaces. Significantly, the E2 molecule binds to E1 in a manner that may be relevant for the ordered regulation of assembly of more distantly related AAA+ proteins. We have solved the structures of domains of both E1 and E2. Several lines of reasoning secure the point that the structures of these regions are sufficient for the subsequent discussion. First, the specific DNA-binding domain of E2 is not critical for the replication process in cells and can be swapped with other DNA-binding motifs, provided the appropriate cis-acting sites are present in the replication reporter. In addition, we believe that our structure captures a view of the entire E1 · E2 interaction surface. Using the T-antigen structure as a guide, the E1 N-terminal and C-terminal extensions would continue in directions away from the E1 · E2 interaction surface and the E2 β-sheet domain, respectively. Furthermore, our binding studies reveal no appreciable difference in the affinity between various extended E1 fragments and E2.
Taken together, the results reported here have a number of implications for constructing a model that accounts for the progression of PV replication initiation. The pathway begins when a complex consisting of E2 and a dimer of E1 binds origin DNA sites through their respective recognition domains (Fig. 8, step 1). Here, the N-terminal activation domains of E2 would be complexed with the C-terminal helicase domains of E1, preventing inadvertent E1 oligomerization. The E1 · E2 interaction would also increase the overall specificity of the complex for the PV origin by combining the DNA-binding affinities of both of the E1 and E2 DNA-binding domains and could serve to suppress the nonspecific DNA-binding activity of the E1 helicase domain (Stenlund 2003a). We suspect that the basic residues of loop-1 (labeled in Fig. 8 as the “DNA-binding loop”) contribute to the nonspecific DNA-binding surface, because they appear to form the positively charged inner surface of the hexamer (Fig. 6C) that is expected to interact with nucleic acid during helicase progression.
Figure 8.

Model for the assembly of E1 molecules at the viral origin. A dimer of E1 and a dimer of E2 bind to the viral origin (step 1). The E2 DNA-binding domains are depicted in red and the activation domains in green. The E1 DNA-binding domains are colored yellow and the helicase domains blue. The strands of DNA contacted by the E1 DNA-binding domains are shown by the red and blue extensions. In step 2, a second E1 dimer binds the PV origin. The inset (middle) view depicts how the second set of E1 molecules is predicted to bind to the DNA relative to the first (Enemark et al. 2002). Competition between the helicase domains of the second set of E1 molecules and the activation domain of E2, coupled with ATP binding, drives the conversion of intermediate 2 to 3. The helicase domains interact with partners across the helix, and the nonspecific DNA-binding loops of the E1 helicase domains are expected to engage DNA prior to or during oligomerization. Following this association, the recruitment of additional helicase domains leads to the assembly of the E1 double hexamer and may induce the melting of the viral origin (step 4).
The next intermediate to form in the assembly process may rely on the recruitment of an additional E1 dimer to the origin, forming an E1 tetramer. DNA cocrystal studies using E1 fragments comprising the origin-binding domains have revealed the presence of such assemblies and that each of the two dimers touches DNA recognition motifs in the PV origin with the strand biases, as indicated in Figure 8 (Enemark et al. 2002). In particular, the structure of Enemark et al. shows that the DNA-binding domains of the E1 dimers are offset by a rotation of ∼100° about the central DNA axis when bound to the PV origin (Fig. 8, inset). When four molecules of E1 bind to the origin in vivo, we expect that the E2 activation domains would initially block interactions between their partner E1 subunits and the newly bound monomers (Fig. 8, step 2). Although this 4:2 E1:E2 intermediate has not been detected, it may have a short half-life, because the second pair of E1 monomers could dissociate, reversing the complex back to step 1.
Because the β-sheet domain of E2 shields the oligomerization surface on its associated E1 partner from the newly bound E1 subunits, E2 must be disengaged to progress to the next stage of the initiation reaction. ATP could directly drive this process by binding to E1, thereby reconfiguring the loop-2 region and disrupting the E2 interaction interface. It is also possible that helicase domains of the E2-free E1 monomers, which are positioned approximately a quarter of a turn about the DNA helix away from the E1 · E2 complex, might lie close enough to engage the E2 β-sheet “shield” domain and help dislodge E2. Thus, the switch that leads to E2 release might rely on a combination of ATP binding and an appropriately docked E1 dimer, which would serve as the nucleus for assembly of the hexameric helicase.
In this regard, as first proposed by Enemark et al. (2002), we suggest that the oligomerization of E1 helicase domains may commence not between two E1 proteins that reside within a dimer, but instead between two E1 proteins that are part of different dimers (Fig. 8, step 3). The primary determinant for this organization derives from the fact that E1-binding sites exist in the PV origin as inverted repeats. As a consequence, E1 proteins first bind their target regions in a twofold rotationally symmetric, or head-to-head, manner (Fig. 8, step 1; Enemark et al. 2002). This configuration presents a problem for the formation of a hexameric helicase, which forms a cyclically symmetric (head-to-tail) assembly of oligomers. Thus, for E1 monomers within a dimer to assemble properly into a hexamer, one protomer would need to rotationally reorient its position with respect to the other by as much as 180°. In contrast, a pair of E1 monomers from different dimers that are offset from each other about the DNA helix, as is seen in structures of the E1 DBD tetramer, are pre-positioned such that the “head” of one partner points in the general direction of the “tail” of the other (Fig. 8).
There are several lines of reasoning that support this oligomerization model. In the Enemark et al. (2002) co-crystal structure of the tetrameric E1 DNA-binding domain · DNA complex, it was noted that the two DNA-binding domains within a dimer each interacted predominately with one of the two DNA strands, and the strand contacted by one protomer was the complement of the one contacted by its dimer mate. Curiously, this organization was reflected in both dimers, such that two domains in different dimers, but related to one another about the DNA axis, actually contact the same DNA strand (Fig. 8, step 2; Enemark et al. 2002). This configuration is significant, because it provides a means for E1 helicase domains within a hexamer to jointly coordinate the same DNA strand as they begin to associate. Moreover, each of the two hexamers would assemble around a different DNA, potentially providing the energy needed to distort and melt the DNA duplex.
The interior translocation loop of E1 (loop-1) might be expected to be the element of the helicase that interacts with the phosphate backbone of DNA during this process. In support of this, BPV E1 does form an extended footprint at the viral origin in the absence of the E2 activation domain, presumably through contacts between the helicase domain of E1 and DNA (Stenlund 2003a). Moreover, motifs in the region of T-antigen equivalent to the E1 loop-1 segment make contact with flanking DNA sequences at the SV-40 origin that are important for viral DNA replication (Reese et al. 2004). The interaction between the helicase domain of E1 and flanking DNA sequences may be important for restricting flexibility between the helicase domain and the DNA-binding domain and could help the protein manifest an unwinding torque on the DNA during E1 oligomerization.
A salient feature of this model is that assembly of the hexamer of E1 at the origin is coupled both to origin melting and creation of a functional helicase (step 4). Once assembled, the helicase is poised to processively unwind the melted strands. There is evidence that T-antigen and E1 (Stahl and Knippers 1983; Fouts et al. 1999; Sanders and Stenlund 2000; Alexandrov et al. 2002) actually function as double hexamers during this process. It is interesting to note that in the assembly scheme proposed earlier (Fig. 8), the dimer interfaces formed initially between the site-specific DNA-binding domains might serve as the contact points for the two hexamers, a model that has been similarly proposed for the N-terminal domain of the MCM helicase (Fletcher et al. 2003). In this regard, the combination of activities observed for E1 and T-antigen, which act both as replication initiators and as DNA melting and helicase proteins, may presage the coordinated action of a set of related AAA+ proteins that initiate DNA replication: ORC, Cdc6, and MCM.
Materials and methods
Protein expression and purification
The activation domain of the HPV18 E2 ORF was cloned into pGEX2T (Amersham) for expression as an N-terminal GST fusion. E1 fragments were cloned into either a modified N-terminal maltose-binding protein (MBP) fusion vector, pMAL-c2X (NEB), which replaced the factor Xa cleavage site with a thrombin cleavage site, or into the same vector with an added N-terminal hexa-histidine tag, for expression as a His-MBP fusion. Mutants were made using the QuikChange (Stratagene) method, according to the manufacturer's protocol. Vectors were transformed into the Escherichia coli strain XA90, and proteins were overexpressed in 2XYT media by induction at an O.D.600 of ∼1.2 with 0.5 mM IPTG for 16 h at 20°C.
For purification of the E1(maltose-binding protein) E2 complex, cells expressing MBP-E1 and GST-E2 were lysed separately by sonication in 50 mM Tris-HCl (pH 7.5), 10% glycerol, 1 M NaCl, 10 mM EDTA, 1 mM PMSF, and 1 mM Tris(2-carboxyethyl)phosphine hydrochloride (TCEP). The E2 extract was incubated with glutathione Sepharose (Amersham) with mixing for 2 h at 4°C. The E2 extract was then removed and the resin-bound E2 was incubated with the E1 extract for ∼16 h at 4°C with mixing. The resin was washed with lysis buffer, followed by extensive washing with 10% Q buffer (25 mM Tris-HCl at pH 8.2), 10% glycerol, 100 mM NaCl, 1 mM EDTA, 1 mM TCEP). Proteins were then eluted with 10% Q buffer containing 20 mM glutathione. GST and MPB were cleaved by incubation with thrombin and separated from the complex by passage over a Mono-Q column (Amersham). Further purification on a Superdex75 (Amersham) sizing column (10% Q buffer) separated the E1 · E2 complex from any remaining free E2. The purified complex was >95% pure as judged by SDS-PAGE and Coomassie staining. Proteins used for biochemical studies were used directly from gel filtration fractions or were concentrated by ultrafiltration (Centriprep10, Amicon) to ∼10 mg/mL, for crystallization.
For pull-down experiments and ATPase assays, GST-tagged E2 and a His-MBP tagged E1 were expressed as described earlier. Purification of E2 was the same as for the complex, except no incubation with E1 was performed. Following elution from the glutathione resin, E2 was bound to a Q-Sepharose (Amersham) column and washed with 10% Q buffer to remove free glutathione. GST-E2 was then eluted with 10% Q buffer containing 400 mM NaCl. His-MBP-E1 extract was made as described earlier, except that all buffers lacked EDTA and TCEP and contained 5 mM β-mercaptoethanol. The extract was incubated with Ni-NTA agarose (QIAGEN) for 2 h at 4°C. The resin was washed with lysis buffer followed by 10% Q buffer (plus 5 mM imidazole), and eluted with 10% Q buffer containing 250 mM imidazole. The eluate was bound to a Q-Sepharose column and washed with 10% Q buffer to remove free imidazole and then eluted with 10% Q buffer containing 400 mM NaCl.
Crystallization
The purified and concentrated E1(428-631) · E2(1-215) complex was dialyzed against 10 mM Tris-HCl (pH 8.0), 50 mM NaCl, and 1 mM TCEP for crystallization. Crystals were grown in microbatch, under oil, by mixing the protein solution with crystallization buffer (50 mM MES at pH 5.9-6.1, 7%-10% PEG 3,350, 20-40 mM MgCl2) in a 1:1 ratio at 4°C. Crystals appeared overnight and grew to average dimensions of 75 × 75 × 125 μm in 3 d. Cryoprotectant was introduced slowly by a 20-step, sequential transfer at 4°C into the final cryo solution (1:2 dilution of the crystallization buffer plus 30% MPD), with 10-min incubations between steps. Crystals were then flash frozen in liquid nitrogen. Heavy atom derivatives were obtained by soaking crystals in a solution containing a 1:2 dilution of crystallization buffer (without MgCl2) and 1 mM erbium (III) chloride overnight. Cryoprotectant was added in a similar manner as with the native crystals, except MgCl2 was replaced by 1 mM ErCl3, which was included in all steps and the final cryoprotectant.
Data collection, structure determination, and refinement
The native data set was collected on Beamline 8.2.2 and derivative data sets on Beamline 8.3.1 at the Advanced Light Source of Lawrence Berkeley National Laboratory, using ADSC Quantum-Q315 CCD detectors. Diffraction data were processed using DENZO and SCALEPAK (Otwinowski and Minor 1997). The crystals are in the orthorhombic space group P212121 with cell parameters a = 82.297 Å, b = 88.745 Å, c = 375.027 Å. There are six E1 · E2 heterodimers in the asymmetric unit. Heavy atom (erbium) sites were identified with SOLVE (Terwilliger and Berendzen 1999). Density modification of experimental maps was performed with RESOLVE (Terwilliger and Berendzen 1999) and DM (Cowtan 1994). Three of the six E2 activation domains were located in the density maps by the program FFFEAR (Cowtan 1998) using the solved HPV18 E2 structure. The remaining three E2 activation domains were manually positioned and E1 and the first two helices of E2 were manually built using O (Jones et al. 1991). Refinement was carried out with REFMAC5 (Murshudov et al. 1999) using ARP (Lamzin and Wilson 1997) for placement of water. The model was refined to a final Rwork of 21.9% and an Rfree of 26.3%. Amino acids 3-198 of E2 and amino acids 428-629 are accounted for in the best E1 · E2 dimer in the asymmetric unit. Geometric analysis was carried out with Procheck (Laskowski et al. 1993).
GST pull-down assays
GST-E2(1-215) and His-MBP-E1(358-631) were used to perform GST pull-down experiments. E2 (100 nM) was incubated with various concentrations of E1 (25 nM, 100 nM, 200 nM) in pull-down buffer (25 mM Tris-HCl at pH 8.2, 10% glycerol, 150 mM NaCl, 1 mM EDTA, 1 mM DTT) containing 100 μg/mL BSA for 1 h at 4°C with mixing. This mixture was then transferred to a GST MicroSpin Purification Module (Amersham) and mixed with the resin for 1 h at 4°C. The resin was washed three times with pull-down buffer and bound proteins were eluted with pull-down buffer containing 20 mM glutathione. Samples were resolved by SDS-PAGE and gels were stained with the fluorescent dye SYPRO Orange (Amersham) and scanned (Typhoon 9400, Amersham) for quantitation.
Sizing column with ATP
The E1(358-631) · E2(1-215) complex was incubated with MgCl2 (5 mM) or with ATP (1 mM) and MgCl2 (5 mM) in 10% Q buffer (see earlier) lacking EDTA for 30 min at room temp. Following incubation, samples were run on a Superdex75 (Amersham) sizing column. Fractions were resolved by SDS-PAGE and stained as described earlier for quantitation.
ATPase assays
ATPase activity was measured by coupling the hydrolysis of ATP to the oxidation of NADH through the enzymes pyruvate kinase and lactate dehydrogenase. Activity was monitored spectrophotometrically as a decrease in absorbance at 340 nm. The assay was performed essentially as described (Rocque et al. 2000). The reaction was initiated with E1 at a final concentration of 1.23 μM.
Acknowledgments
E. Abbate is grateful to Dr. Emmanuel Skordalakes and other members of the Berger lab for crystallography assistance and advice. We also thank James Holton at Beamline 8.3.1 of the Advanced Light Source for assistance with data acquisition and Jackie Wong for the construction of several E1 vectors. This research was supported by funds from NIH grant CA42414 to M.R.B. J.M.B. gratefully acknowledges support from the G. Harold and Leila Y. Mathers Charitable Foundation for this work. The coordinates have been deposited with the Protein Data Bank under accession number 1TUE.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Corresponding authors.
Article published online ahead of print. Article and publication date are at http://www.genesdev.org/cgi/doi/10.1101/gad.1220104.
References
- Alexandrov A.I., Botchan, M.R., and Cozzarelli, N.R. 2002. Characterization of simian virus 40 T-antigen double hexamers bound to a replication fork. The active form of the helicase. J. Biol. Chem. 277: 44886-44897. [DOI] [PubMed] [Google Scholar]
- Antson A.A., Burns, J.E., Moroz, O.V., Scott, D.J., Sanders, C.M., Bronstein, I.B., Dodson, G.G., Wilson, K.S., and Maitland, N.J. 2000. Structure of the intact transactivation domain of the human papillomavirus E2 protein. Nature 403: 805-809. [DOI] [PubMed] [Google Scholar]
- Bosch F.X., Manos, M.M., Munoz, N., Sherman, M., Jansen, A.M., Peto, J., Schiffman, M.H., Moreno, V., Kurman, R., and Shah, K.V. 1995. Prevalence of human papillomavirus in cervical cancer: A worldwide perspective. International biological study on cervical cancer (IBSCC) Study Group. J. Natl. Cancer Inst. 87: 796-802. [DOI] [PubMed] [Google Scholar]
- Bullock P.A. 1997. The initiation of simian virus 40 DNA replication in vitro. Crit. Rev. Biochem. Mol. Biol. 32: 503-568. [DOI] [PubMed] [Google Scholar]
- Chen G. and Stenlund, A. 1998. Characterization of the DNA-binding domain of the bovine papillomavirus replication initiator E1. J. Virol. 72: 2567-2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang C.M., Ustav, M., Stenlund, A., Ho, T.F., Broker, T.R., and Chow, L.T. 1992. Viral E1 and E2 proteins support replication of homologous and heterologous papillomaviral origins. Proc. Natl. Acad. Sci. 89: 5799-5803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowtan K. 1994. A CCP4 density modification package. Joint CCP4 ESF-EACBM. Newslett. Prot. Crystallogr. 31: 34-38. [Google Scholar]
- ____. 1998. Modified phased translation functions and their application to molecular-fragment location. Acta Crystallogr. D Biol. Crystallogr. 54 (Pt. 5): 750-756. [DOI] [PubMed] [Google Scholar]
- Davey M.J., Jeruzalmi, D., Kuriyan, J., and O'Donnell, M. 2002. Motors and switches: AAA+ machines within the replisome. Nat. Rev. Mol. Cell Biol. 3: 826-835. [DOI] [PubMed] [Google Scholar]
- Enemark E.J., Chen, G., Vaughn, D.E., Stenlund, A., and Joshua-Tor, L. 2000. Crystal structure of the DNA binding domain of the replication initiation protein E1 from papillomavirus. Mol. Cell 6: 149-158. [PubMed] [Google Scholar]
- Enemark E.J., Stenlund, A., and Joshua-Tor, L. 2002. Crystal structures of two intermediates in the assembly of the papillomavirus replication initiation complex. EMBO J. 21: 1487-1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson M.K. and Botchan, M.R. 1996. Genetic analysis of the activation domain of bovine papillomavirus protein E2: Its role in transcription and replication. J. Virol. 70: 4193-4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher R.J., Bishop, B.E., Leon, R.P., Sclafani, R.A., Ogata, C.M., and Chen, X.S. 2003. The structure and function of MCM from archaeal M. thermoautotrophicum. Nat. Struct. Biol. 10: 160-167. [DOI] [PubMed] [Google Scholar]
- Fouts E.T., Yu, X., Egelman, E.H., and Botchan, M.R. 1999. Biochemical and electron microscopic image analysis of the hexameric E1 helicase. J. Biol. Chem. 274: 4447-4458. [DOI] [PubMed] [Google Scholar]
- Gillitzer E., Chen, G., and Stenlund, A. 2000. Separate domains in E1 and E2 proteins serve architectural and productive roles for cooperative DNA binding. EMBO J. 19: 3069-3079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris S.F. and Botchan, M.R. 1999. Crystal structure of the human papillomavirus type 18 E2 activation domain. Science 284: 1673-1677. [DOI] [PubMed] [Google Scholar]
- Howley P.M. 1996. Papillomavirinae: The viruses and their replication. In Fields virology (eds. B.N. Fields et al.), pp. 2045-2076. Lippincott-Raven, Philadelphia, PA.
- James J.A., Escalante, C.R., Yoon-Robarts, M., Edwards, T.A., Linden, R.M., and Aggarwal, A.K. 2003. Crystal structure of the SF3 helicase from adeno-associated virus type 2. Structure (Camb.) 11: 1025-1035. [DOI] [PubMed] [Google Scholar]
- Jones T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47 (Pt. 2): 110-119. [DOI] [PubMed] [Google Scholar]
- Koonin E.V. 1993. A common set of conserved motifs in a vast variety of putative nucleic acid-dependent ATPases including MCM proteins involved in the initiation of eukaryotic DNA replication. Nucleic Acids Res. 21: 2541-2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamzin V.S. and Wilson, K.S. 1997. Automated refinement for protein crystallography. In Methods enzymology (eds. C.W. Carter Jr. et al.), pp. 269-305. Academic Press, San Diego, CA. [DOI] [PubMed]
- Laskowski A.R., MacArthur, W.M., Moss, S.D., and Thornton, M.J. 1993. PROCHECK: A program to check the stereo-chemical quality of protein structures. J. Appl. Cryst. 26: 283-291. [Google Scholar]
- Li R., Knight, J.D., Jackson, S.P., Tjian, R., and Botchan, M.R. 1991. Direct interaction between Sp1 and the BPV enhancer E2 protein mediates synergistic activation of transcription. Cell 65: 493-505. [DOI] [PubMed] [Google Scholar]
- Li R., Yang, L., Fouts, E., and Botchan, M.R. 1993. Site-specific DNA-binding proteins important for replication and transcription have multiple activities. Cold Spring Harb. Symp. Quant. Biol. 58: 403-413. [DOI] [PubMed] [Google Scholar]
- Li D., Zhao, R., Lilyestrom, W., Gai, D., Zhang, R., DeCaprio, J.A., Fanning, E., Jochimiak, A., Szakonyi, G., and Chen, X.S. 2003. Structure of the replicative helicase of the oncoprotein SV40 large tumour antigen. Nature 423: 512-518. [DOI] [PubMed] [Google Scholar]
- Lim D.A., Gossen, M., Lehman, C.W., and Botchan, M.R. 1998. Competition for DNA binding sites between the short and long forms of E2 dimers underlies repression in bovine papillomavirus type 1 DNA replication control. J. Virol. 72: 1931-1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin B.Y., Makhov, A.M., Griffith, J.D., Broker, T.R., and Chow, L.T. 2002. Chaperone proteins abrogate inhibition of the human papillomavirus (HPV) E1 replicative helicase by the HPV E2 protein. Mol. Cell. Biol. 22: 6592-6604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X., Sanford, D.G., Bullock, P.A., and Bachovchin, W.W. 1996. Solution structure of the origin DNA-binding domain of SV40 T-antigen. Nat. Struct. Biol. 3: 1034-1039. [DOI] [PubMed] [Google Scholar]
- Lusky M., Hurwitz, J., and Seo, Y.S. 1994. The bovine papillomavirus E2 protein modulates the assembly of but is not stably maintained in a replication-competent multimeric E1-replication origin complex. Proc. Natl. Acad. Sci. 91: 8895-8899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansky K.C., Batiza, A., and Lambert, P.F. 1997. Bovine papillomavirus type 1 E1 and simian virus 40 large T antigen share regions of sequence similarity required for multiple functions. J. Virol. 71: 7600-7608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masterson P.J., Stanley, M.A., Lewis, A.P., and Romanos, M.A. 1998. A C-terminal helicase domain of the human papillomavirus E1 protein binds E2 and the DNA polymerase α-primase p68 subunit. J. Virol. 72: 7407-7419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza R., Gandhi, L., and Botchan, M.R. 1995. E1 recognition sequences in the bovine papillomavirus type 1 origin of DNA replication: Interaction between half sites of the inverted repeats. J. Virol. 69: 3789-3798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr I.J., Clark, R., Sun, S., Androphy, E.J., MacPherson, P., and Botchan, M.R. 1990. Targeting the E1 replication protein to the papillomavirus origin of replication by complex formation with the E2 transactivator. Science 250: 1694-1699. [DOI] [PubMed] [Google Scholar]
- Murshudov G.N., Vagin, A.A., Lebedev, A., Wilson, K.S., and Dodson, E.J. 1999. Efficient anisotropic refinement of macromolecular structures using FFT. Acta Crystallogr. D Biol. Crystallogr. 55 (Pt. 1): 247-255. [DOI] [PubMed] [Google Scholar]
- Neuwald A.F., Aravind, L., Spouge, J.L., and Koonin, E.V. 1999. AAA+: A class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 9: 27-43. [PubMed] [Google Scholar]
- Nicholls A., Sharp, K.A., and Honig, B. 1991. Protein folding and association: Insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins 11: 281-296. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode. In Methods enzymology (eds. C.W. Carter Jr. et al.), pp. 307-326. Academic Press, San Diego, CA. [DOI] [PubMed]
- Park P., Copeland, W., Yang, L., Wang, T., Botchan, M.R., and Mohr, I.J. 1994. The cellular DNA polymerase α-primase is required for papillomavirus DNA replication and associates with the viral E1 helicase. Proc. Natl. Acad. Sci. 91: 8700-8704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putnam C.D., Clancy, S.B., Tsuruta, H., Gonzalez, S., Wetmur, J.G., and Tainer, J.A. 2001. Structure and mechanism of the RuvB Holliday junction branch migration motor. J. Mol. Biol. 311: 297-310. [DOI] [PubMed] [Google Scholar]
- Reese D.K., Sreekumar, K.R., and Bullock, P.A. 2004. Interactions required for binding of simian virus 40 T antigen to the viral origin and molecular modeling of initial assembly events. J. Virol. 78: 2921-2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocque W.J., Porter, D.J., Barnes, J.A., Dixon, E.P., Lobe, D.C., Su, J.L., Willard, D.H., Gaillard, R., Condreay, J.P., Clay, W.C., et al. 2000. Replication-associated activities of purified human papillomavirus type 11 E1 helicase. Protein Expr. Purif. 18: 148-159. [DOI] [PubMed] [Google Scholar]
- Russell J. and Botchan, M.R. 1995. cis-acting components of human papillomavirus (HPV) DNA replication: Linker substitution analysis of the HPV type 11 origin. J. Virol. 69: 651-660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai H., Yasugi, T., Benson, J.D., Dowhanick, J.J., and Howley, P.M. 1996. Targeted mutagenesis of the human papillomavirus type 16 E2 transactivation domain reveals separable transcriptional activation and DNA replication functions. J. Virol. 70: 1602-1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders C.M. and Stenlund, A. 1998. Recruitment and loading of the E1 initiator protein: An ATP-dependent process catalysed by a transcription factor. EMBO J. 17: 7044-7055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ____. 2000. Transcription factor-dependent loading of the E1 initiator reveals modular assembly of the papillomavirus origin melting complex. J. Biol. Chem. 275: 3522-3534. [DOI] [PubMed] [Google Scholar]
- Scheffner M., Huibregtse, J.M., Vierstra, R.D., and Howley, P.M. 1993. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 75: 495-505. [DOI] [PubMed] [Google Scholar]
- Sedman J. and Stenlund, A. 1995. Co-operative interaction between the initiator E1 and the transcriptional activator E2 is required for replicator specific DNA replication of bovine papillomavirus in vivo and in vitro. EMBO J. 14: 6218-6228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo Y.S., Muller, F., Lusky, M., Gibbs, E., Kim, H.Y., Phillips, B., and Hurwitz, J. 1993. Bovine papilloma virus (BPV)-encoded E2 protein enhances binding of E1 protein to the BPV replication origin. Proc. Natl. Acad. Sci. 90: 2865-2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons D.T. 2000. SV40 large T antigen functions in DNA replication and transformation. Adv. Virus Res. 55: 75-134. [DOI] [PubMed] [Google Scholar]
- Song H.K., Hartmann, C., Ramachandran, R., Bochtler, M., Behrendt, R., Moroder, L., and Huber, R. 2000. Mutational studies on HslU and its docking mode with HslV. Proc. Natl. Acad. Sci. 97: 14103-14108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl H. and Knippers, R. 1983. Simian virus 40 large tumor antigen on replicating viral chromatin: Tight binding and localization on the viral genome. J. Virol. 47: 65-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenlund A. 2003a. E1 initiator DNA binding specificity is unmasked by selective inhibition of non-specific DNA binding. EMBO J. 22: 954-963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ____. 2003b. Initiation of DNA replication: Lessons from viral initiator proteins. Nat. Rev. Mol. Cell Biol. 4: 777-785. [DOI] [PubMed] [Google Scholar]
- Sverdrup F. and Myers, G. 1997. The E1 proteins. In Human papillomaviruses 1997 (ed. G. Myers), pp. 37-53. Theoretical Division, Los Alamos National Laboratory, Los Alamos, NM.
- Terwilliger T.C. and Berendzen, J. 1999. Automated MAD and MIR structure solution. Acta Crystallogr. D Biol. Crystallogr. 55 (Pt. 4): 849-861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Coulombe, R., Cameron, D.R., Thauvette, L., Massariol, M.J., Amon, L.M., Fink, D., Titolo, S., Welchner, E., Yoakim, C., et al. 2004. Crystal structure of the E2 transactivation domain of human papillomavirus type 11 bound to a protein interaction inhibitor. J. Biol. Chem. 279: 6976-6985. [DOI] [PubMed] [Google Scholar]
- White P.W., Pelletier, A., Brault, K., Titolo, S., Welchner, E., Thauvette, L., Fazekas, M., Cordingley, M.G., and Archambault, J. 2001. Characterization of recombinant HPV6 and 11 E1 helicases: Effect of ATP on the interaction of E1 with E2 and mapping of a minimal helicase domain. J. Biol. Chem. 276: 22426-22438. [DOI] [PubMed] [Google Scholar]
- Winokur P.L. and McBride, A.A. 1996. The transactivation and DNA binding domains of the BPV-1 E2 protein have different roles in cooperative origin binding with the E1 protein. Virology 221: 44-53. [DOI] [PubMed] [Google Scholar]
- Yang L., Li, R., Mohr, I.J., Clark, R., and Botchan, M.R. 1991a. Activation of BPV-1 replication in vitro by the transcription factor E2. Nature 353: 628-632. [DOI] [PubMed] [Google Scholar]
- Yang L., Mohr, I., Li, R., Nottoli, T., Sun, S., and Botchan, M. 1991b. Transcription factor E2 regulates BPV-1 DNA replication in vitro by direct protein-protein interaction. Cold Spring Harb. Symp. Quant. Biol. 56: 335-346. [DOI] [PubMed] [Google Scholar]
- Yasugi T., Benson, J.D., Sakai, H., Vidal, M., and Howley, P.M. 1997. Mapping and characterization of the interaction domains of human papillomavirus type 16 E1 and E2 proteins. J. Virol. 71: 891-899. [DOI] [PMC free article] [PubMed] [Google Scholar]