

Abstract
Muscle wasting resulting wholly or in part from disuse represents a serious medical complication that, when prolonged, can increase morbidity and mortality. Although much knowledge has been gained over the past half century, the underlying etiology by which disuse alters muscle proteostasis remains enigmatic. Multidisciplinary and novel methodologies are needed to fill gaps and overcome barriers to improved patient care. The present review highlights seminal concepts from a symposium at Experimental Biology 2016. These proceedings focus on 1) the role of insulin resistance in mediating disuse-induced changes in muscle protein synthesis (MPS) and breakdown (MPB), as well as cross-talk between carbohydrate and protein metabolism; 2) the relative importance of MPS/MPB in mediating involuntary muscle loss in humans and animals; 3) interpretative limitations associated with MPS/MPB “markers,” e.g., MuRF1/MAFbx mRNA; and finally, 4) how OMIC technologies can be leveraged to identify molecular pathways (e.g., ATF4, p53, p21) mediating disuse atrophy. This perspective deals primarily with “simple atrophy” due to unloading. Nonetheless, it is likely that disuse is a pervasive contributor to muscle wasting associated with catabolic disease-related atrophy (i.e., due to associated sedentary behaviour of disease burden). Key knowledge gaps and challenges are identified to stimulate discussion and identify opportunities for translational research. Data from animal and human studies highlight both similarities and differences. Integrated preclinical and clinical research is encouraged to better understand the metabolic and molecular underpinnings and translational relevance,for disuse atrophy. These approaches are crucial to clinically prevent or reverse muscle atrophy, thereby reestablishing homeostasis and recovery.
Keywords: disuse atrophy, protein synthesis, protein degradation, muscle ring finger 1, activating transcription factor 4
this article is a written representation of a symposium at Experimental Biology (San Diego, CA, 2016) that focused on the mechanisms of disuse atrophy in skeletal muscle. The rationale was to highlight the timeliness of this topic from a clinical perspective, to present what is known of the molecular underpinnings of disuse atrophy, and to highlight the multitude of varied experimental approaches being employed to scrutinize the mechanisms of disuse atrophy. Drs. Atherton and Lang were cochairs of the symposium, and Drs. Greenhaff, Phillips, Bodine, and Adams were speakers. Each of the speaker's contributions will follow this foreword.
Skeletal muscle atrophy, manifested as a loss of muscle mass, is a consequence of many noncommunicable and communicable diseases, e.g., cancers, AIDS, metabolic diseases, sepsis, burn injury, organ failures, and respiratory diseases. Nonetheless, even in the absence of disease the unloading of skeletal muscles causes muscle atrophy, as is observed due to cast immobilization, extended bed rest, or exposure to microgravity. This is termed “simple atrophy” in that it is intrinsic processes (e.g., atrophy limited to the affected limb) rather than extrinsic or systemic factors at work. That said, there is interplay between disuse atrophy and disease-related atrophy. For example, many atrophy-related diseases are associated with sedentary behaviors that likely act to exacerbate disuse-induced atrophy. Nonetheless, it would be churlish not to make this distinction at the outset; disuse is a process intrinsic to the muscles that are exposed to it.
The consequences of muscle atrophy are not trivial. Disuse atrophy is associated with morbidities such as functional decline, disability, metabolic disorders, and even premature death. Therefore, it is unsurprising that seeking understanding of the metabolic and mechanistic drivers of disuse atrophy and strategies to mitigate it remains a hotbed of research. Yet the regulatory underpinnings of disuse atrophy remain poorly defined. In this symposium overview, our coauthors present distinct contemporary approaches to addressing these issues. Dr. Greenhaff focuses upon the role of insulin resistance. The importance of this area is to better understand the established link between muscle insulin resistance and disuse atrophy in the context of protein metabolism, with the central question being, is there an insulin resistance of protein metabolism contributing to disuse atrophy? Dr. Phillips, the second speaker, poses the central question: What are the roles and contributions of muscle protein synthesis (MPS) and muscle protein breakdown (MPB) and associated molecular markers in the context of disuse atrophy in observational clinical studies in humans? Dr. Bodine, the third speaker, addresses this same question, albeit from the perspective of mechanistic and followup observational studies conducted in preclinical animal models. Finally, Dr. Adams discusses what can be understood by adopting systems (transcriptomics) approaches in preclinical models with this central question: How can untargeted OMICS be harnessed to construct molecular pathways regulating disuse atrophy?
Our coauthors each also touch on various other side issues, such as the impact of aging, muscle fiber type, and muscle function in addition to how “simple atrophy” compares with “nonsimple” forms of atrophy. This article should provide the reader a past, present, and future outlook of this area from leaders in the field.
Potential Role of Insulin Resistance in Driving Muscle Disuse Atrophy (Greenhaff)?
Mounting evidence suggests that physical inactivity is causative in the development of many noncommunicable diseases, including obesity, insulin resistance, type 2 diabetes, dyslipidemia, ischemic heart disease, and hypertension, among others. In 2012, ∼30% of adults worldwide failed to meet government physical activity guidelines, and it is estimated that the situation is worsening (25). Models of partial (reduced step count) and complete (bed rest, unilateral limb immobilization) physical inactivity have provided valuable information regarding the nature and extent of metabolic dysregulation associated with physical inactivity.
Immobilization-induced muscle atrophy.
Both reduced step count and immobilization result in muscle atrophy in healthy human volunteers, and under these conditions it is indisputable that postabsorptive state MPS is suppressed. Furthermore, immobilization is accompanied by a loss of muscle strength that is of a greater magnitude than the loss of muscle mass (27). Decline in the rate of MPS is of sufficient magnitude to wholly account for muscle mass losses in healthy, young volunteers (16). This, in combination with a lack of robust evidence for increased MPB during immobilization, has led some to suggest that a decline in the rate of MPS is solely responsible for the loss of muscle mass in humans. Nevertheless, animal hindlimb immobilization studies have reported MPS to be reduced and MPB to be increased and, moreover, essential for muscle loss to occur (30). This balance between MPS and MPB is covered further later in this review.
Although postabsorptive MPS is reduced in immobilization, Glover et al. (21) demonstrated that suppressed MPS also extends to the postprandial state in humans, even with high amino acid provision. Furthermore, under both postabsorptive and postprandial states, the phosphorylation state of signaling proteins in the mammalian target of rapamycin (mTOR) pathway could not explain these immobilization-induced deficits in MPS in humans (16, 21). Further studies conducted in immobilized rodents (30) and nonimmobilized healthy human volunteers (24, 46) question the established notion that mTOR signaling regulates MPS as routinely described. This does not preclude mTOR signaling from having an important function in the processes that govern human MPS during immobilization but does advocate that a single-point determination of protein phosphorylation does not necessarily reflect rates of MPS. Collectively, these observations highlight our lack of understanding of the mechanisms responsible for disuse-induced muscle mass loss.
Immobilization-induced insulin resistance.
Immobilization results in the development of whole body and muscle insulin resistance (MIR) in people within 3–5 days (26, 40, 43). Mikines et al. (34) were the first to demonstrate this phenomenon under euglycemic hyperinsulinemic clamp conditions in human volunteers, where it was found that MIR was present across a wide range of serum insulin concentrations (10–52 μU/ml). This seminal finding has since been independently confirmed on a number of occasions and in both arms and legs (34, 41, 42). Additionally, MIR develops after 3–14 days of reduced ambulatory activity (29, 31, 36). Overall, these observations suggest that a lack of muscle contraction per se is the physiological driver of dysregulation, but the mechanistic basis of this is far from clear. It has been proposed that immobilization-induced MIR is linked to reduced amounts and phosphorylation of key proteins involved in muscle glucose uptake, phosphorylation, and storage (3), but whether this is causative or a consequence of immobilization is not known. The substantive decline in limb glucose uptake within 24 h of immobilization speaks against a causative effect. The recently demonstrated inverse association between the increase in muscle pyruvate dehydrogenase complex (PDC) activation and leg lactate release during contraction following immobilization points to PDC also being a potential key regulator of immobilization-induced insulin resistance (44), much in the same way that it has been implicated with dietary- and obesity-induced MIR and type 2 diabetes (37). Indeed, an increase in intramuscular lipid content has been observed following unilateral limb immobilization and bed rest (13, 33).
Does insulin resistance drive muscle disuse atrophy?.
Although a series of molecular pathways can be dovetailed to illuminate a sequence of coordinated events that explain immobilization-induced changes in MPS, MPB, and MIR (Fig. 1), in reality, in vivo evidence to substantiate such speculation is currently unavailable. Indeed, obese individuals presenting with marked blunting of leg glucose disposal under insulin clamp conditions do not have a lower lean tissue mass compared with their age-matched lean counterparts, which goes against such an idea. However, interpretation of data of this nature can be problematic because of several confounding variables. Certainly, in the scenario of sepsis-induced muscle inflammation, it does appear that the same cascade of events subsequent to cytokine-mediated dysregulation of Akt signaling (Fig. 1) can explain the decline in MPS and mass and increases in MPB and MIR seen under these conditions in rodents (14, 15) and humans (12).
Fig. 1.

Putative mechanism underpinning the observed changes in muscle protein synthesis, muscle protein breakdown, and muscle insulin resistance in immobilization (less likely) and sepsis-induced inflammation (highly likely). mTOR, mammalian target of rapamycin; FOXO, forkhead box O; MAFbx, muscle atrophy F-box; MuRF1, muscle RING finger 1; PDK4, pyruvate dehydrognase kinase 4; PDC, pyruvate dehydrogenase complex.
In conclusion, it is clear that a number of gaps exist in our understanding of the mechanisms controlling the dysregulation of protein and carbohydrate metabolism during immobilization and their integration (Table 1). However, although the unfavorable metabolic effects of immobilization are often indirectly inferred from the positive effects of exercise training in sedentary populations, the mechanisms underlying inactivity-induced metabolic dysregulation are far from resolved. Furthermore, it is highly likely that they are not the direct opposite of those mechanisms routinely associated with the beneficial metabolic effects of exercise training (9).
Table 1.
Research gaps in immobilization research
| • Explanation for the apparent lack of commonality between rodent and human-based immobilization research with regard to the relative contribution of MPS and MPB to muscle mass loss |
| • Lack of data from humans on temporal changes in MPS, MPB, and MIR from the onset of immobilization and whether they follow the same pattern of change |
| • Lack of mechanistic understanding for disuse-induced muscle mass loss and MIR |
| • Unknown whether the widespread metabolic dysregulation observed in muscle during immobilization is underpinned by a group of coordinated common molecular events |
| • Data pertaining to longer-term integrated rates of muscle protein synthesis |
| • Lack of direct measurements of MPB |
| • Impact of aging and sex difference on altering the atrophic response |
| • Investigations on therapeutic interventions (nutritional, nutraceutical, pharmacological) |
| • Identification of specific proteins that are increased or decreased in translation under disuse conditions |
| • Identification of specific substrates for MuRF1 and MAFbx under conditions of unloading and inactivity and whether the ubiquitination of these proteins alters their degradation, activity, or cellular location |
| • Discovery effort to identify mTORC1-independent pathways regulating protein synthesis under conditions that modify muscle size and metabolic capacity |
| • Mechanism by which immobilization increases activity of ATF4 and p53 in skeletal muscle |
| • Mechanism for p21 repression of the spermine oxidase gene |
| • Mechanism by which spermine oxidase protects against skeletal muscle atrophy |
| • More detailed investigation of the hundreds of mRNAs that are increased or decreased in immobilized skeletal muscle |
MPS, muscle protein synthesis; MPB, muscle protein breakdown; MIR, muscle insulin resistance; MuRF1, muscle RING finger 1; MAFbx, muscle atrophy F-box; mTORC1, mammalian target of rapamycin; ATF4, activating transcription factor 4.
Regulation of Protein Metabolism in Human Disuse Atrophy: Now and the Future (Phillips)
As indicated above, disuse-induced atrophy occurs in a variety of situations in humans, including bed rest, limb immobilization, and abrupt reductions in physical activity (15). The loss of load bearing in skeletal muscle is translated via currently undefined signaling pathway(s) (8) and results in a reduction in the rate of MPS, an increase in MPB, or most likely some degree of both (8, 15). A recent exchange of ideas on whether it is a reduction in MPS or an increase in MPB that predominates (and is most effectively treated) resulting in disuse-induced atrophy has been published (14, 16). However, the answer to this question remains unresolved (14, 16), and yet it is an important question since a clinically relevant target for amelioration, synthesis or proteolysis, would depend on the solution. An important point to make about distinguishing possibly distinct differences in the predominant process leading to disuse atrophy is that the discussion in the present review revolves around simple unloading-mediated disuse atrophy in nondiseased states. There is also a rapid and pronounced decline in muscle in disease states such as sepsis, cancer cachexia, AIDS wasting, uncontrolled diabetes, and renal failure that may have a hypodynamic component, and clinical states are distinct from simple disuse atrophy (14, 15).A hallmark distinct feature of the former disease states is a pronounced hypercortisolemia, hypercytokinemia (14), and markedly elevated oxidative stress, all of which are procatabolic triggers that would increase rates of MPB. Thus, a relevant question is how does disuse atrophy affect the processes of MPS and MPB and the resultant protein balance (MPS minus MPB) to produce atrophy? Importantly, much of the data from this field show that in multiple disuse models muscular atrophy is a process that proceeds rapidly and then slows, reaching a plateau (Fig. 2), even in the case of “complete” unloading with spinal cord injury (15).
Fig. 2.

Schematic representation of the known and hypothesized changes in rates of muscle protein synthesis (MPS) and muscle protein breakdown (MPB) leading to net protein accretion or net protein loss in early (10 days or less) and later (beyond 10 days) disuse. The total daily integrated protein turnover response is represented in the bar graphs showing the daily integrated fractional synthesis rates (FSR) and fractional breakdown rates (FBR) of proteins.
In humans, a highly reproducible finding with disuse is a reduction in the resting fasted rate of MPS (14, 15), which has been seen in multiple models of disuse in humans. The amount of contractile activity required to offset this decline is decidedly minimal, whether undertaken involuntarily or voluntarily (15). Another hallmark of disuse atrophy is the reduction in mitochondrial gene expression, protein content (1), and likely mitochondrial function (20). In addition to the reduction in resting, fasted-state MPS, numerous studies have reported that the highly reproducible hyperaminoacidemia-induced rise in MPS is blunted in humans during amino acid infusion (6) or following the ingestion of protein (21). Thus, the stereotypical finding in simple disuse-induced atrophy in humans is that there are reductions in both the fasted-state and fed-state MPS, which would suggest a global daily reduction in the rate of MPS (Fig. 2). The magnitude of the daily reduction in MPS has been estimated, based on acute measurements, to be ∼40–50% (15). However, we currently lack data on the integrated daily rates of MPS, which would be useful to obtain to confirm that acute measurements have been extrapolated correctly to give daily rates (15).
Multiple lines of evidence from preclinical models show elevated expression of proteolytic genes and proteins, which lends some support to the thesis that proteolysis is elevated with disuse (16). One study in humans reported that interstitial 3-methylhistidine, a proxy marker of myofibrillar proteolysis, was elevated following only 3 days of immobilization (19), with concordant elevations in proteolytic gene biomarkers (7). However, direct measurement following 21 days of bed rest showed no elevation in MPB (18). Clearly, the measurement of MPB is an area that requires more experimentation, and better attempts are needed to measure this variable in humans and with some attention paid to temporal changes with different durations of disuse. It would also be advantageous if molecular methods were combined with in vivo dynamic measurements of MPB to gain mechanistic insight. This is particularly relevant as transcriptomic profiles of early (2-day) and later (14-day) muscle from limb immobilization do not show the pronounced increases in proteolytic transcripts seen in preclinical models (1). In addition, measurements of proteolytic complex activity, as opposed to gene or protein abundances, would also be useful outcomes to include in future studies.
Mathematical estimates suggest that it appears unlikely that rates of MPB contribute appreciably to disuse-induced muscle atrophy in humans (15). Nonetheless, it appears likely that MPB, if it is appreciably elevated in humans during disuse, happens early and then is likely adaptively reduced with longer periods of disuse. If MPB were not downregulated, as MPS is (see above), then rates of reduction in muscle/muscle fiber loss in disuse would be far greater than those observed in multiple ground-based models of disuse (2). However, these mathematical estimates are based on assumptions, some of which have not been tested, and thus integrated measurements of MPS and MPB would be a valuable advance in this area. It is also possible that MPB acts to downregulate MPS. That is, disuse-induced increments in MPB do not result in bulk myofibrillar proteolysis but instead act to degrade elements of the protein-synthetic machinery that result in a reduction in the rate of MPS. For example, eukaryotic initiation factor (eIF)-3f, a protein that is critical for the commencement of translation initiation, is degraded by the ubiquitin E3 ligase MAFbx/atrogin-1 (4, 5). It has been shown that the prevention of polyubiquitination of eIF3f reduces starvation-induced muscle atrophy (9, 10). Moreover, myostatin-induced myotube atrophy is associated with the upregulation of components of the ubiquitin-proteasome pathway but also degradation of proteins associated with ribosomal biogenesis, translation initiation, and elongation as well as a reduction in MPS (11, 12).
The time course of disuse atrophy bears some consideration, as it is clear that much of the muscle loss occurs early; for a review of the time course of changes in muscle fiber and muscle cross-sectional area from a number of ground-based models of disuse, see Ref. 2. Importantly, the data from a number of disuse models in humans indicate a reduction in muscle mass/muscle fiber size that has a half-life of about 10 days. Importantly, when the mathematical functions that can be fit to these data are examined (2), there are no marked differences between human muscle fiber types with respect to magnitude or time course of reductions in muscle fiber cross-sectional area (Phillips SM, unpublished observations). However, that there are not marked differences between fiber types with disuse atrophy in humans is not unexpected, as the magnitude of reported differences in protein turnover between fibers is only 10–15% (13). However, as is presented next, fiber type differences are seen in some rodent models of disuse. Although it may be impractical to repeatedly measure differences in protein turnover between fibers in human skeletal muscle, it would be interesting to examine differences between rates of atrophy and recovery in differing muscles, for example, the soleus, the vastus lateralis, and the elbow extensors (triceps), as examples of muscles being composed of predominantly slow, mixed, and faster muscle fibers.
An area that has received less attention is the impact of disuse atrophy in the elderly (17). From what little we know in this area it appears that older people are not able to recover as fully, or perhaps not at all, as younger persons (17). Thus, periods of disuse superimposed on the decline in muscle mass resulting from simple and disease-related sarcopenia are likely to have dire consequences for older persons related to the loss of muscle mass, muscle strength, and functional abilities (3). Thus, it would be beneficial to obtain more data from the elderly undergoing disuse atrophy and, importantly, look for nutritional and or minimal loading prescriptions that might help offset the atrophy occurring during disuse in this vulnerable group.
Future work on disuse in humans would benefit from a number of experimental models that focus on answering the types of questions laid out in Table 1. Some of the solutions are a simple matter of study design, but others await development of methods and/or the application of existing methods in the appropriate models.
Regulation of Protein Metabolism in Preclinical Models of Disuse Atrophy: Now and the Future (Bodine)
Skeletal muscle is an adaptable tissue that responds to multiple stressors during a lifetime to modify/maintain muscle mass. During adulthood, maintenance of muscle mass and adaptive growth are dependent on two primary factors: external loading and neural activation. Decreases in the amount of external loading and/or neural activation of a muscle will lead to a loss of muscle mass and are often referred to as “disuse atrophy.” The severity of disuse atrophy is dependent on the extent to which both load and neural activity are affected. For example, denervation leads to severe disuse atrophy due to a dramatic decrease in both neural activation and external loading of a muscle. In contrast, a decrease in external loading with less involvement of neural activity, as occurs during bedrest, microgravity, and joint immobilization, yields less overall muscle loss. However, it is noteworthy that joint immobilization can lead to varying amounts of muscle atrophy, depending on the degree of joint restriction that can affect neural activity and the position of the joint that will affect muscle length (4).
The mechanisms responsible for disuse atrophy have been debated recently and likely involve variable changes in both protein synthesis and protein degradation (38). To address the question of whether alterations in protein synthesis or protein degradation contribute to muscle atrophy under conditions of disuse, we have utilized the tail suspension model, which results in unloading of the hindlimb muscle. Hindlimb suspension is an excellent disuse model because there is minimal impact on neural activity and one can examine the impact of unloading on muscles of variable physiological function (i.e., flexors vs. extensors) and fiber type composition. We have purposely chosen to study the rat, as opposed to the mouse, because of the greater fiber type variability in hindlimb muscles of the rat. In addition, we have compared disuse atrophy in adult (9 mo old) and old (28 mo old) male Fisher Brown Norway rats (National Institute of Aging) and focused our analysis on three lower-limb muscles: the soleus (SOL), a predominately slow extensor muscle; the medial gastrocnemius (MG), a predominately fast oxidative extensor muscle; and the tibialis anterior (TA), a predominately fast nonoxidative flexor muscle.
Disuse atrophy: protein synthesis or protein degradation?
Disuse atrophy was measured after 14 days of unloading, and loss of mass occurred in all muscles, being greatest in the ankle extensor muscles (SOL, −38%; MG, −31%) and least in the ankle flexor muscle (TA, −20%) (2). To examine the mechanisms responsible for the differential loss of muscle mass, we measured protein synthesis using the SUnSET method (23) and protein degradation by 20S and 26S proteasome subunit activities and cathepsin L activity (22). Although protein synthesis significantly decreased in all three muscles with unloading, the time course and extent of the decrease varied across the muscles. After 3 days of unloading, protein synthesis was reduced in all muscles, but the decrease was greater in the SOL and MG than in the TA. In the SOL, protein synthesis continued to decrease at 7 days of unloading and remained depressed at 14 days of unloading. In the MG, synthesis was down at 7 days postunloading but was increasing toward control levels, which were achieved by 14 days of unloading. In the TA, synthesis was no longer suppressed at 7 or 14 days of unloading.
With respect to protein degradation following unloading, the 20S and 26S β5-proteasome activities increased steadily from 3 to 14 days of unloading in the soleus and were elevated at 7 and 14 days of unloading. In contrast, no increase in the activities of the 20S and 26S β5-proteasome subunits was observed in the MG or TA upon unloading, in fact, proteasome activity tended to decrease below resting levels. Activity of cathepsin L, a lysosomal enzyme, showed the same trend as the β5 proteasome activities, increasing in the soleus at 7 and 14 days but showing no change in the MG or TA (Fig. 3).
Fig. 3.

Summary of the relative changes in multiple variables to 3, 7, and 14 days of tail suspension (hindlimb unloading) in adult male Fisher Brown Norway rats. The %loss of muscle mass at 14 days of hindlimb unloading is given for the soleus (SOL), medial gastrocnemius (MG), and tibialis anterior (TA) muscles. The variables measured were protein synthesis (PS), 26S β5 proteasome activity (β5), cathepsin L activity (Cath L), MuRF1 mRNA (MuRF1), MAFbx mRNA (MAFbx), FOXO1 mRNA (FOXO1), and FOXO3a mRNA (FOXO3a).
In summary, changes in muscle mass following unloading in rats are a consequence of the net changes in both protein synthesis and degradation. The response to unloading differs across muscles of varying physiological function (extensors vs. flexors) and fiber type composition (oxidative vs. glycolytic), with oxidative extensor muscles such as the soleus atrophying to a greater extent than nonoxidative flexor muscles such as the TA. Although all muscles experienced some degree of unloading-induced decreased protein synthesis, the greatest loss of muscle mass occurred in those muscles that experienced an increase in protein degradation. Predominantly slow oxidative antigravity muscles appear to be the most susceptible to atrophy in response to unloading, exhibiting decreases in protein synthesis and increases in multiple degradation pathways. These data highlight the importance of the muscle under study and the time points chosen for analysis when making conclusions about the role of protein synthesis and degradation in disuse-induced atrophy.
Aging and disuse atrophy.
Our laboratory and others have shown that disuse-induced atrophy is less in old muscle relative to adult muscle (2). We investigated whether the difference was related to alterations in protein synthesis vs. protein degradation. We found that for the soleus suppression of protein synthesis was similar for adult and old rats in response to the unloading. In contrast, there was no increase in protein degradation, as measured by proteasome subunit activities, in the soleus of the old rats that could explain the difference in percent muscle loss (−38 vs. −30%, adult vs. old). For the MG, the major difference between adult and old rats was that protein synthesis was suppressed to a lesser extent in the old compared with the adult, resulting in slightly less atrophy in the old (31 vs. 28%, adult vs. old). The unloading response of the old TA differed greatly from that of the adult TA. There was no decrease in protein synthesis in the old TA and a significant increase in 20S and 26S β5-proteasome subunit activities in response to unloading. These data show that muscle from old rats responds differently from adult muscle to unloading and that changes to both protein synthesis and degradation occur in a muscle-specific manner.
Disuse and atrophy-associated genes.
A number of genes whose transcription is upregulated in response to signals that induce atrophy have been identified, including unloading and inactivity (6, 32). Four of the most commonly examined atrophy-associated genes are muscle RING finger 1 (MuRF1), muscle atrophy F-box (MAFbx)/atrogin-1, forkhead box O1 (FOXO1), and forkhead box O3a (FOXO3a). The increased expression of all four genes, especially the ubiquitin ligases MuRF1 and MAFbx, has routinely been thought to be an indicator of an increase in protein degradation (5). We measured the expression of these four genes in SOL, MG, and TA in response to unloading using quantitative PCR. The expression of MuRF1 and MAFbx increased in all muscles in response to unloading, although the time course varied across muscles. In the SOL and MG, MuRF1 and MAFbx expression rises quickly, peaking at 7 days and returning to baseline by 14 days. Moreover, the fold increase of MuRF1 and MAFbx mRNA expression was higher in the MG than SOL. In contrast, MuRF1 and MAFbx mRNA in TA was elevated at 3 days of unloading and then returned to baseline by 7 days. It is noteworthy that although MuRF1 and MAFbx mRNA increased in all muscles following unloading, proteasome activity was selectively increased only in the SOL. Consequently, although MuRF1 and MAFbx are clearly atrophy-associated genes, their upregulation was not uniformly associated with an increase in protein degradation (as measured by proteasome subunit activity and cathepsin L activity). Moreover, increased MuRF1 and MAFbx mRNA content is highly dependent on the time point being measured and the muscle under study.
The FOXO transcription factors FOXO1 and FOXO3a translocate from the cytoplasm to the nucleus under atrophy-inducing conditions such as unloading and regulate the increased transcription of the E3 ubiquitin ligases MuRF1 and MAFbx, leading to increased protein degradation. Examination of the expression patterns revealed that FOXO1 is upregulated in all muscles upon unloading, with the greatest increase in expression occurring in the MG, followed by the TA and then the SOL. The pattern of FOXO1 expression over the 14 days of unloading resembled that observed for MuRF1 and MAFbx. In contrast to FOXO1, the expression of FOXO3a changed very little over the 14 days of unloading, highlighting the fact that FOXO1 and FOXO3a should not be used interchangeably. These data reveal that changes in the expression patterns of FOXO1 and FOXO3a mRNA are not always linked and that changes in the expression levels of both genes are dependent on the muscle and the time after unloading (Fig. 3).
Denervation: a more severe disuse model.
The loss of neural innervation to a muscle, i.e., denervation, results in severe muscle atrophy. As with unloading, denervation alters both protein synthesis and degradation. We have demonstrated that denervation increases protein degradation, especially 20S and 26S proteasome subunit activities (22). Interestingly, the loss of MuRF1 does not inhibit proteasome activity but actually increases the activity of all of the 20S and 26S proteasome subunits (22). With respect to protein synthesis, we and others have shown that protein synthesis actually increases with denervation (1, 22). Moreover, the increase in protein synthesis is associated with an increase in Akt/mTORC1 signaling. Thus, muscle atrophy is not always associated with decreases in protein synthesis, as is often described in the literature. The increase in protein synthesis following denervation appears to be mTORC1 mediated, and interestingly, blocking this activity with rapamycin does not lead to an increase in muscle loss (28). Thus, this increase in protein synthesis may not be a compensatory mechanism, but it may be related to the increased translation of the hundreds of atrophy-associated genes whose transcription is upregulated in response to denervation.
In summary, it is too simplistic to envision disuse atrophy as simply the result of a decrease in protein synthesis and an increase in protein degradation. In disuse atrophy caused by unloading, a decrease in protein synthesis was apparent in all muscles; however, the extent to which suppression of protein synthesis occurred was dependent on the muscle and the time following unloading. In this regard, the data highlight the danger in using a single time point to assess changes in either protein synthesis or degradation. Our data show that increases in protein degradation can occur in response to unloading and lead to greater muscle atrophy in rats. However, increases in protein degradation in response to unloading appear to be most prevalent in predominantly slow-twitch antigravity muscle such as the soleus. Our data also highlight the error in using mRNA expression of the ubiquitin ligases MuRF1 and MAFbx as markers of protein degradation. Sustained increases in the expression of MuRF1 and MAFbx for 7 days or more appear to be a clear indicator of muscle atrophy; however, it is does not always lead to a concomitant increase in protein degradation, at least as measured by proteasome subunit activity and cathepsin L activity. Disuse atrophy is a complex process, and many gaps remain in our understanding of the process as outlined in Table 1.
Insights into Disuse Muscle Atrophy Using Systems Approaches: Now and the Future (Adams)
Severe illnesses and injuries typically impose a period of immobilization that, depending on the clinical context, may be temporary or permanent and either localized (e.g., limb casting) or generalized (e.g., bed rest). Although immobilization can sometimes be of benefit to a patient (e.g., allowing a fracture to heal), it always has a negative side effect: skeletal muscle atrophy. Immobilization-induced skeletal muscle atrophy often creates new problems for patients, including weakness, falls, and impaired recovery from the primary illness or injury; it may also create a vicious cycle of atrophy and weakness leading to even more muscle disuse and thus more atrophy and weakness, etc., culminating in debilitation and loss of independent living.
Although immobilization-induced skeletal muscle atrophy is a relatively common and serious clinical problem, it lacks a medical therapy, and it remains poorly understood and largely unexplored at the molecular level. To illustrate the gaps in our mechanistic understanding of this condition, let us briefly consider what is currently known about a well-established and reliable mouse model of immobilization-induced muscle atrophy. In this model, one tibialis anterior muscle in each mouse is immobilized with a metal clip, which, similar to a cast, induces progressive muscle atrophy that is localized to the immobilized muscle (10). Skeletal muscle atrophy in this model, like other forms of skeletal muscle atrophy in humans or mice, is highly complex at the molecular level. For example, using genome-wide mRNA expression arrays, we found that a short, 3-day period of immobilization significantly alters the levels of >600 mRNAs in the immobilized muscle (>200 mRNAs increased and >400 mRNAs decreased) while inducing a degree of atrophy (≈10% loss of mean muscle fiber diameter) similar to the degree of atrophy that occurs in human patients with orthopedic injuries (20).
All of the mRNAs that are increased or decreased in immobilized skeletal muscle represent potentially important molecular mediators of immobilization-induced skeletal muscle atrophy. Moreover, a number of readily available molecular and genetic approaches can be used to investigate the functional significance of individual mRNAs that are differentially expressed in immobilized mouse skeletal muscle. However, up to now, <1% of immobilization-induced changes in skeletal muscle mRNA expression have been investigated at a functional level. The best-studied mRNAs encode MuRF1 and MAFbx, muscle-specific E3 ubiquitin ligases that are highly induced at the transcriptional level by immobilization (20) and other atrophy stimuli (5). And yet, although MuRF1 and MAFbx have been studied in great detail over the past 15 years (more than 500 publications in PubMed) and are clearly important to the pathogenesis of skeletal muscle atrophy (5), many other mRNAs that are just as highly induced in immobilized skeletal muscle have not been examined at all. Furthermore, virtually nothing is known about the skeletal muscle mRNAs that are repressed by immobilization. This knowledge gap represents a rich opportunity for new discoveries in an important and understudied area of biomedical research.
From this perspective, our research uses genome-wide mRNA expression data, coupled with mechanistic investigations in mice, to search for previously unrecognized mediators of immobilization-induced skeletal muscle atrophy. In conjunction with those studies, we have been working to discover transcriptional regulators that mediate critical effects of immobilization on skeletal muscle gene expression. As illustrated below, those transcriptional regulators can provide new insights into the pathogenesis of immobilization-induced skeletal muscle atrophy while also serving as useful tools in the subsequent identification of additional mediators of skeletal muscle atrophy. Finally, because some molecular mechanisms of muscle atrophy are conserved across diverse causes of skeletal muscle atrophy, our investigations of immobilization-induced skeletal muscle atrophy are also guided by data from other causes of muscle atrophy, including fasting, aging, systemic illness, and other types of muscle disuse. In general, we have been most interested in identifying proteins that mediate muscle atrophy in several different contexts that include but are not limited to immobilization. Some examples of these approaches are described below.
Our initial studies of immobilization-induced skeletal muscle atrophy focused on a basic leucine zipper transcription factor called activating transcription factor 4 (ATF4). A series of unbiased genome-wide mRNA expression analyses demonstrated that ATF4 is one of many mRNAs that increase during muscle disuse and several other conditions that cause skeletal muscle atrophy (39). Subsequently, we found that increased ATF4 expression is sufficient to induce skeletal muscle fiber atrophy (18), and ATF4 is required for skeletal muscle atrophy in some but not all contexts. For example, ATF4 is required for skeletal muscle atrophy caused by fasting or aging (18, 19), but it is not required for muscle atrophy caused by muscle denervation (7). To test the hypothesis that ATF4 might be required for immobilization-induced skeletal muscle atrophy, we generated and studied muscle-specific ATF4 knockout (ATF4 mKO) mice, which lack ATF4 in skeletal muscle fibers from birth but develop normally and exhibit no apparent phenotype under basal conditions (17). We found that ATF4 mKO mice are partially (≈50%) resistant to muscle atrophy induced by 3 days of limb immobilization (17, 20). This finding indicated that ATF4 plays an essential role in immobilization-induced skeletal muscle atrophy. In addition, because ATF4 mKO mice were not completely resistant to immobilization-induced skeletal muscle atrophy, this finding indicated the existence of at least one other essential factor that promotes atrophy in an ATF4-independent manner.
As we considered potential ATF4-independent factors, we saw p53 as an attractive candidate. Similar to ATF4, p53 is a transcription factor that mediates cellular adaptations to metabolic stress (45). Moreover, a wide variety of conditions producing skeletal muscle atrophy, including immobilization, increase p53 expression in skeletal muscle (shown and reviewed in Ref. 20). Thus, to test the hypothesis that p53 might be required for immobilization-induced skeletal muscle atrophy, we generated and studied mice that lack p53 expression in skeletal muscle fibers (p53 mKO mice). In parallel, to determine whether loss of p53 might provide additional protection to ATF4 mKO mice, we generated and studied ATF4/p53 double-mKO mice. We found that p53 mKO mice, much like ATF4 mKO mice, are partially resistant to immobilization-induced skeletal muscle atrophy (20). Furthermore, ATF4/p53 double-mKO mice are almost completely resistant to immobilization-induced skeletal muscle atrophy, suggesting that the effects of ATF4 and p53 are additive (20). To further investigate the potentially additive effects of ATF4 and p53, we used in vivo plasmid transfection to force expression of ATF4 and/or p53 in skeletal muscle fibers of wild-type, fully active mice. We found that both ATF4 and p53 are sufficient to induce muscle fiber atrophy even in the absence of immobilization, and their effects are additive (20). Furthermore, using ATF4 mKO and p53 mKO mice, we found that ATF4 is capable of causing atrophy in the absence of p53, and conversely, p53 is capable of causing atrophy in the absence of ATF4, indicating that ATF4 and p53 do not require each other to cause atrophy. Collectively, these data identified ATF4 and p53 as essential transcription factors that can account for much of the atrophy in immobilized skeletal muscle (20).
The finding that ATF4 and p53 promote immobilization-induced skeletal muscle atrophy led us to investigate the downstream mechanisms. As transcription factors, ATF4 and p53 regulate many genes and thus have many potential downstream mechanisms. As a starting point for investigation, we tested the possibility that ATF4 and p53 might coinduce at least one critical proatrophy gene. To identify such a gene, we took an unbiased approach, using genome-wide mRNA expression arrays to search for mRNAs that met the following criteria: 1) increased by limb immobilization in wild-type muscle; 2) increased by ATF4 overexpression in wild-type muscle; 3) increased by p53 overexpression in wild-type muscle; 4) decreased in ATF4 mKO muscle; and 5) decreased in p53 mKO muscle. Through these analyses, one mRNA, p21, emerged as the strongest candidate, meeting all five criteria (17, 18, 20). Moreover, the effects of ATF4 and p53 on p21 mRNA were nearly additive, and the induction of p21 mRNA by ATF4 and p53 led to an increased level of p21 protein (20). In contrast to p21, MuRF1 and MAFbx mRNAs were not induced by either ATF4 or p53 (20). Thus, in healthy, active muscle fibers, p21 gene expression is low, but it rises dramatically during immobilization due to the combined actions of ATF4 and p53, leading to an increased level of p21 protein.
p21 (a.k.a. Cdkn1a, Waf1, and Cip1) is a well-characterized inhibitor of cyclin-dependent kinases, and numerous studies (reviewed in Ref. 20) have demonstrated that p21 is highly induced in adult skeletal muscle fibers during a wide variety of atrophy conditions, including muscle disuse, fasting, aging, and systemic illness. However, the potential role of p21 in skeletal muscle atrophy had not been explored. To test the hypothesis that p21 might promote skeletal muscle atrophy, we overexpressed p21 in nonimmobilized, wild-type mouse skeletal muscle fibers, which normally contain low levels of p21. We found that p21 reduces muscle fiber size, indicating that increased p21 expression is sufficient to induce muscle fiber atrophy (20). We then performed similar experiments in ATF4/p53 double-mKO mice and found that p21-mediated muscle fiber atrophy does not require ATF4 or p53, consistent with the earlier finding that p21 lies downstream, not upstream, of ATF4 and p53 (20). To determine whether p21 is required for muscle atrophy, we used in vivo RNA interference (RNAi) to knock down p21 expression in wild-type skeletal muscle fibers during immobilization, ATF4 overexpression, and p53 overexpression. Importantly, in each of these conditions, p21 knockdown reduces skeletal muscle fiber atrophy, indicating that p21 is a crucial downstream mediator of immobilization, ATF4, and p53 (20).
Since p21 caused skeletal muscle fiber atrophy, we investigated the mechanism downstream of p21. Surprisingly, a genome-wide analysis of the effects of p21 on mRNA expression in mouse skeletal muscle revealed that p21 strongly represses the mRNA encoding spermine oxidase, which was also the most strongly repressed mRNA in immobilized skeletal muscle (8). In addition, we found that 1) the reduction of spermine oxidase mRNA by immobilization or p21 expression was accompanied by reduction in spermine oxidase protein, 2) p21 knockdown in immobilized skeletal muscle not only reduced atrophy but also restored spermine oxidase expression, and 3) other muscle atrophy stimuli that increase skeletal muscle p21 expression (aging, fasting, and denervation) also decrease spermine oxidase mRNA and spermine oxidase protein (8).
Interestingly, spermine oxidase is an enzyme in polyamine metabolism that catabolizes spermine to spermidine, 3-aminopropanol, and hydrogen peroxide (11). Its role in the control of skeletal muscle mass was unknown; however, the finding that spermine oxidase expression was strongly repressed by p21 suggested that spermine oxidase might be an antiatrophy factor essential for the maintenance of skeletal muscle mass. To determine whether a reduction in spermine oxidase might be sufficient to induce muscle atrophy, we used in vivo RNAi to knoc kdown spermine oxidase expression in healthy, nonimmobilized, wild-type skeletal muscle fibers. We found that spermine oxidase knockdown decreased skeletal muscle fiber size, indicating that a reduction in spermine oxidase is sufficient to induce muscle fiber atrophy (8). To test whether a reduction in spermine oxidase expression is required for muscle atrophy, we transfected mouse muscle fibers in vivo with plasmid encoding spermine oxidase under control of a constitutively active promoter, and then, in the presence of constitutive spermine oxidase expression, we applied muscle atrophy stimuli (immobilization, p21 overexpression, fasting, or denervation). We found that forced expression of spermine oxidase reduced skeletal muscle fiber atrophy caused by immobilization, p21 overexpression, fasting, and denervation, indicating that a reduction in spermine oxidase expression is at least partially required for muscle atrophy during these conditions (8). Taken together, these data identified spermine oxidase as an important factor in the maintenance of skeletal muscle mass and a key downstream target of p21 during immobilization and other atrophy conditions.
Together, these results suggest a model that is illustrated in Fig. 4. Immobilization induces skeletal muscle atrophy via the actions within skeletal muscle fibers of two transcription factors: ATF4 and p53. Like all transcription factors, ATF4 and p53 have numerous downstream effects, most of which have not yet been investigated in detail. However, one particularly important overlapping effect of ATF4 and p53 in immobilized muscle fibers is activation of the p21 gene, which increases the level of p21 mRNA and the level of p21 protein. p21 causes muscle fiber atrophy, and it does so at least in part by inhibiting the expression of spermine oxidase. This information may help to guide future therapeutic development, and at least some aspects of these findings may also be relevant to other causes of skeletal muscle atrophy, such as aging, fasting, and muscle denervation. Yet it is also important to emphasize that this represents a very oversimplified model of immobilization-induced skeletal muscle atrophy, with substantial gaps that still need to be completed. In addition, we examined only a few of the hundreds of changes that occur as a muscle undergoes atrophy, so much work remains, and this field of research should remain, for a long time, a rich area for discoveries, which can be facilitated by unbiased systems-based approaches (Table 1).
Fig. 4.
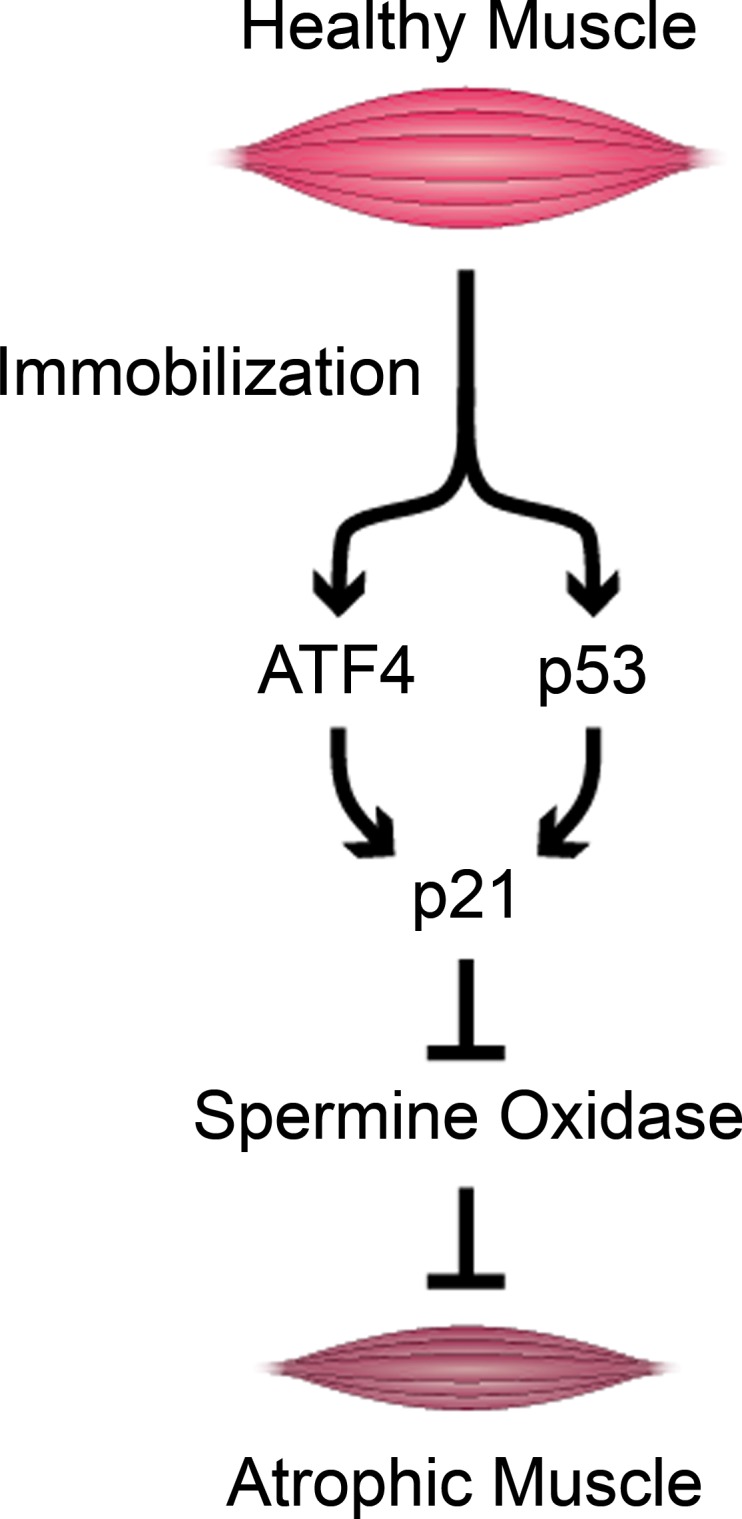
Schematic illustration of a molecular signaling pathway to skeletal muscle atrophy during limb immobilization. ATF4, activating transcription factor 4.
Concluding Remarks
The four authors present distinct experimental and methodological approaches to understanding the molecular regulation of disuse atrophy; all staunchly agree that this is a crucial research problem where research inroads must be made. From this collection of articles, we are led to make some general conclusions.
First, muscle insulin resistance is an established side effect of disuse, and its anabolic effects on muscle are well known. Nonetheless, the mechanistic role of muscle insulin resistance in driving atrophy in response to “simple disuse” is poorly defined. More research is needed in this area. Second, although common proteostatic processes regulating muscle disuse atrophy inevitably exist across disuse and other catabolic conditions, simple disuse atrophy does differ from where systemic drivers are also present (e.g., inflammation, such as in cachexia). Moreover, it is likely that sedentary behavior associated with many catabolic diseases and illnesses contributes to what researchers might in instances term “disease-related muscle atrophy.” On this basis we contend that disease-related atrophy and disuse atrophy can rarely be seen as being mutually exclusive. This is not a matter to be taken lightly; going forward, research needs to be conducted to dissect out these potentially confounding inputs and to define the mechanisms and contributions of inactivity in relation to disease-related muscle atrophy.
Next, it is clear that static molecular markers cannot be relied upon as proxies for MPS and MPB pathways. This includes both clinical and preclinical studies. These are snapshot measures at best. They may indeed be indicative of MPS/MPB, although they could also be acutely regulating subsets of mRNA/proteins rather than “bulk” or global rates of MPS or MPB. Moreover, although apparent up- or downregulation of certain molecular pathways may indeed be indicative of the dynamic metabolic processes at play, these are rarely looked at in sufficient depth to even confer such conclusions, i.e., gene and protein levels and robust pathway coverage. It is now feasible to quantify MPS and MPB dynamically in both humans and animals using a variety of techniques (e.g., arterio-venous balance techniques, stable or radioisotopic tracers, and release of nonreincorporating amino acids such as tyrosine). Crucially, there exist undeniable differences in allometry, metabolism, genetics, and epigenetic backgrounds between rodents and humans, not to mention any stress factors in animals subjected to unloading. Thus one cannot simply refer to overlap of the DNA sequence as a reason why rodents represent humans. This also relates to developmental knockouts with compensatory mechanisms at play. Either tissue-specific postnatal knockouts or acute shRNA approaches in vivo (Dr. Adams's work) can overcome this, with the latter permitting experimental rapidity.
Having brought together leading clinical and preclinical researchers, it is clear that MPS is suppressed, whereas the role of MPB and insulin resistance in simple disuse atrophy remain poorly defined, as is the clinical relevance of pathway constructions. Too few preclinical researchers are forward-translating findings, whereas too few targets derived from clinical studies (with OMICS) are being generated, followed up, or back translated. Integrating clinical and preclinical research in the future will surely represent the most likely route to understanding and mitigating the consequences of disuse and other atrophy in a clinical context.
GRANTS
This work was partially funded by Veteran Affairs RR & D Merit Grant 1I01RX000673 and National Institute on Aging Award No. R01-AG-045375. This work was also funded by the National Institute on Aging (Grant R01-AR-059115) and the US Department of Veterans Affairs (Grants IBX000976A and 1I01RX001477). C. H. Lang was partially supported by National Institute of General Medical Sciences Grant R01-GM-38032.
DISCLOSURES
C. M. Adams is a founder, shareholder, and officer of Emmyon, Inc.
The symposium on which this article is based was supported by travel funds from Abbott.
AUTHOR CONTRIBUTIONS
P.J.A. and C.H.L. conception and design of research; P.J.A., P.L.G., S.M.P., S.C.B., C.M.A., and C.H.L. analyzed data; P.J.A., P.L.G., S.M.P., S.C.B., C.M.A., and C.H.L. interpreted results of experiments; P.J.A., P.L.G., S.M.P., S.C.B., C.M.A., and C.H.L. prepared figures; P.J.A., P.L.G., S.M.P., S.C.B., C.M.A., and C.H.L. drafted manuscript; P.J.A., P.L.G., S.M.P., S.C.B., C.M.A., and C.H.L. edited and revised manuscript; P.J.A., P.L.G., S.M.P., S.C.B., C.M.A., and C.H.L. approved final version of manuscript.
ACKNOWLEDGMENTS
Dr. Greenhaff acknowledges the contributions of Ph.D students and academic colleagues who collaborated in studies cited in this short overview. Dr. Phillips thanks the National Science and Engineering Research Council of Canada, the Canadian Institutes for Health Research, and the Canada Research Chairs Program for their support. Dr. Bodine thanks the many colleagues and students that have contributed to this work. In particular, Dr. Bodine thanks fellow colleagues Drs. Keith Baar and Aldrin Gomes at the University of California Davis for their contributions and acknowledges the work done by many students and postdoctoral fellows over the years, especially, Leslie Baehr, Darren Hwee, George Marcotte, and Daniel West. Dr. Adams thanks all colleagues who helped conduct the studies of immobilization-induced skeletal muscle atrophy, including Scott Ebert, Michael Dyle, Daniel Fox, Steven Bullard, Jason Dierdorff, and Steven Kunkel.
REFERENCES
- 1.Argadine HM, Hellyer NJ, Mantilla CB, Zhan WZ, Sieck GC. The effect of denervation on protein synthesis and degradation in adult rat diaphragm muscle. J Appl Physiol 107: 438–444, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baehr LM, West DW, Marcotte G, Marshall AG, De Sousa LG, Baar K, Bodine SC. Age-related deficits in skeletal muscle recovery following disuse are associated with neuromuscular junction instability and ER stress, not impaired protein synthesis. Aging (Albany NY) 8: 127–146, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bienso RS, Ringholm S, Kiilerich K, Aachmann-Andersen NJ, Krogh-Madsen R, Guerra B, Plomgaard P, van Hall G, Treebak JT, Saltin B, Lundby C, Calbet JA, Pilegaard H, Wojtaszewski JF. GLUT4 and glycogen synthase are key players in bed rest-induced insulin resistance. Diabetes 61: 1090–1099, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bodine SC. Disuse-induced muscle wasting. Int J Biochem Cell Biol 45: 2200–2208, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bodine SC, Baehr LM. Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1. Am J Physiol Endocrinol Metab 307: E469–E484, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, Pan ZQ, Valenzuela DM, DeChiara TM, Stitt TN, Yancopoulos GD, Glass DJ. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 294: 1704–1708, 2001. [DOI] [PubMed] [Google Scholar]
- 7.Bongers KS, Fox DK, Ebert SM, Kunkel SD, Dyle MC, Bullard SA, Dierdorff JM, Adams CM. Skeletal muscle denervation causes skeletal muscle atrophy through a pathway that involves both Gadd45a and HDAC4. Am J Physiol Endocrinol Metab 305: E907–E915, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bongers KS, Fox DK, Kunkel SD, Stebounova LV, Murry DJ, Pufall MA, Ebert SM, Dyle MC, Bullard SA, Dierdorff JM, Adams CM. Spermine oxidase maintains basal skeletal muscle gene expression and fiber size and is strongly repressed by conditions that cause skeletal muscle atrophy. Am J Physiol Endocrinol Metab 308: E144–E158, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Booth FW, Chakravarthy MV, Gordon SE, Spangenburg EE. Waging war on physical inactivity: using modern molecular ammunition against an ancient enemy. J Appl Physiol 93: 3–30, 2002. [DOI] [PubMed] [Google Scholar]
- 10.Caron AZ, Drouin G, Desrosiers J, Trensz F, Grenier G. A novel hindlimb immobilization procedure for studying skeletal muscle atrophy and recovery in mouse. J Appl Physiol 106: 2049–2059, 2009. [DOI] [PubMed] [Google Scholar]
- 11.Casero RA, Pegg AE. Polyamine catabolism and disease. Biochem J 421: 323–338, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Constantin D, McCullough J, Mahajan RP, Greenhaff PL. Novel events in the molecular regulation of muscle mass in critically ill patients. J Physiol 589: 3883–3895, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cree MG, Paddon-Jones D, Newcomer BR, Ronsen O, Aarsland A, Wolfe RR, Ferrando A. Twenty-eight-day bed rest with hypercortisolemia induces peripheral insulin resistance and increases intramuscular triglycerides. Metabolism 59: 703–710, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crossland H, Constantin-Teodosiu D, Gardiner SM, Constantin D, Greenhaff PL. A potential role for Akt/FOXO signalling in both protein loss and the impairment of muscle carbohydrate oxidation during sepsis in rodent skeletal muscle. J Physiol 586: 5589–5600, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Crossland H, Constantin-Teodosiu D, Greenhaff PL, Gardiner SM. Low-dose dexamethasone prevents endotoxaemia-induced muscle protein loss and impairment of carbohydrate oxidation in rat skeletal muscle. J Physiol 588: 1333–1347, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Boer MD, Selby A, Atherton P, Smith K, Seynnes OR, Maganaris CN, Maffulli N, Movin T, Narici MV, Rennie MJ. The temporal responses of protein synthesis, gene expression and cell signalling in human quadriceps muscle and patellar tendon to disuse. J Physiol 585: 241–251, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ebert SM, Dyle MC, Kunkel SD, Bullard SA, Bongers KS, Fox DK, Dierdorff JM, Foster ED, Adams CM. Stress-induced skeletal muscle Gadd45a expression reprograms myonuclei and causes muscle atrophy. J Biol Chem 287: 27290–27301, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ebert SM, Monteys AM, Fox DK, Bongers KS, Shields BE, Malmberg SE, Davidson BL, Suneja M, Adams CM. The transcription factor ATF4 promotes skeletal myofiber atrophy during fasting. Mol Endocrinol 24: 790–799, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ebert SM, Dyle MC, Bullard SA, Dierdorff JM, Murry DJ, Fox DK, Bongers KS, Lira VA, Meyerholz DK, Talley JJ, Adams CM. Identification and Small Molecule Inhibition of an Activating Transcription Factor 4 (ATF4)-dependent Pathway to Age-related Skeletal Muscle Weakness and Atrophy. J Biol Chem 290: 25497–25511, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fox DK, Ebert SM, Bongers KS, Dyle MC, Bullard SA, Dierdorff JM, Kunkel SD, Adams CM. p53 and ATF4 mediate distinct and additive pathways to skeletal muscle atrophy during limb immobilization. Am J Physiol Endocrinol Metab 307: E245–E261, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Glover EI, Phillips SM, Oates BR, Tang JE, Tarnopolsky MA, Selby A, Smith K, Rennie MJ. Immobilization induces anabolic resistance in human myofibrillar protein synthesis with low and high dose amino acid infusion. J Physiol 586: 6049–6061, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gomes AV, Waddell DS, Siu R, Stein M, Dewey S, Furlow JD, Bodine SC. Upregulation of proteasome activity in muscle RING finger 1-null mice following denervation. FASEB J 26: 2986–2999, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goodman CA, Mabrey DM, Frey JW, Miu MH, Schmidt EK, Pierre P, Hornberger TA. Novel insights into the regulation of skeletal muscle protein synthesis as revealed by a new nonradioactive in vivo technique. FASEB J 25: 1028–1039, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Greenhaff PL, Karagounis LG, Peirce N, Simpson EJ, Hazell M, Layfield R, Wackerhage H, Smith K, Atherton P, Selby A, Rennie MJ. Disassociation between the effects of amino acids and insulin on signaling, ubiquitin ligases, and protein turnover in human muscle. Am J Physiol Endocrinol Metab 295: E595–E604, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hallal PC, Andersen LB, Bull FC, Guthold R, Haskell W, Ekelund U; Lancet Physical Activity Series Working Group. Global physical activity levels: surveillance progress, pitfalls, and prospects. Lancet 380: 247–257, 2012. [DOI] [PubMed] [Google Scholar]
- 26.Hamburg NM, McMackin CJ, Huang AL, Shenouda SM, Widlansky ME, Schulz E, Gokce N, Ruderman NB, Keaney JF Jr, Vita JA. Physical inactivity rapidly induces insulin resistance and microvascular dysfunction in healthy volunteers. Arterioscler Thromb Vasc Biol 27: 2650–2656, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jones SW, Hill RJ, Krasney PA, O'Conner B, Peirce N, Greenhaff PL. Disuse atrophy and exercise rehabilitation in humans profoundly affects the expression of genes associated with the regulation of skeletal muscle mass. FASEB J 18: 1025–1027, 2004. [DOI] [PubMed] [Google Scholar]
- 28.Kline WO, Panaro FJ, Yang H, Bodine SC. Rapamycin inhibits the growth and muscle-sparing effects of clenbuterol. J Appl Physiol 102: 740–747, 2007. [DOI] [PubMed] [Google Scholar]
- 29.Knudsen SH, Hansen LS, Pedersen M, Dejgaard T, Hansen J, Hall GV, Thomsen C, Solomon TP, Pedersen BK, Krogh-Madsen R. Changes in insulin sensitivity precede changes in body composition during 14 days of step reduction combined with overfeeding in healthy young men. J Appl Physiol 113: 7–15, 2012. [DOI] [PubMed] [Google Scholar]
- 30.Krawiec BJ, Frost RA, Vary TC, Jefferson LS, Lang CH. Hindlimb casting decreases muscle mass in part by proteasome-dependent proteolysis but independent of protein synthesis. Am J Physiol Endocrinol Metab 289: E969–E980, 2005. [DOI] [PubMed] [Google Scholar]
- 31.Krogh-Madsen R, Thyfault JP, Broholm C, Mortensen OH, Olsen RH, Mounier R, Plomgaard P, van Hall G, Booth FW, Pedersen BK. A 2-wk reduction of ambulatory activity attenuates peripheral insulin sensitivity. J Appl Physiol 108: 1034–1040, 2010. [DOI] [PubMed] [Google Scholar]
- 32.Lecker SH, Jagoe RT, Gilbert A, Gomes M, Baracos V, Bailey J, Price SR, Mitch WE, Goldberg AL. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J 18: 39–51, 2004. [DOI] [PubMed] [Google Scholar]
- 33.Manini TM, Clark BC, Nalls MA, Goodpaster BH, Ploutz-Snyder LL, Harris TB. Reduced physical activity increases intermuscular adipose tissue in healthy young adults. Am J Clin Nutr 85: 377–384, 2007. [DOI] [PubMed] [Google Scholar]
- 34.Mikines KJ, Richter EA, Dela F, Galbo H. Seven days of bed rest decrease insulin action on glucose uptake in leg and whole body. J Appl Physiol 70: 1245–1254, 1991. [DOI] [PubMed] [Google Scholar]
- 36.Olsen RH, Krogh-Madsen R, Thomsen C, Booth FW, Pedersen BK. Metabolic responses to reduced daily steps in healthy nonexercising men. JAMA 299: 1261–1263, 2008. [DOI] [PubMed] [Google Scholar]
- 37.Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1: 785–789, 1963. [DOI] [PubMed] [Google Scholar]
- 38.Reid MB, Judge AR, Bodine SC. CrossTalk opposing view: The dominant mechanism causing disuse muscle atrophy is proteolysis. J Physiol 592: 5345–5347, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sacheck JM, Hyatt JP, Raffaello A, Jagoe RT, Roy RR, Edgerton VR, Lecker SH, Goldberg AL. Rapid disuse and denervation atrophy involve transcriptional changes similar to those of muscle wasting during systemic diseases. FASEB J 21: 140–155, 2007. [DOI] [PubMed] [Google Scholar]
- 40.Smorawinski J, Kaciuba-Uscilko H, Nazar K, Kubala P, Kaminska E, Ziemba AW, Adrian J, Greenleaf JE. Effects of three-day bed rest on metabolic, hormonal and circulatory responses to an oral glucose load in endurance or strength trained athletes and untrained subjects. J Physiol Pharmacol 51: 279–289, 2000. [PubMed] [Google Scholar]
- 41.Sonne MP, Alibegovic AC, Hojbjerre L, Vaag A, Stallknecht B, Dela F. Effect of 10 days of bedrest on metabolic and vascular insulin action: a study in individuals at risk for type 2 diabetes. J Appl Physiol 108: 830–837, 2010. [DOI] [PubMed] [Google Scholar]
- 42.Sonne MP, Hojbjerre L, Alibegovic AC, Nielsen LB, Stallknecht B, Vaag AA, Dela F. Endothelial function after 10 days of bed rest in individuals at risk for type 2 diabetes and cardiovascular disease. Exp Physiol 96: 1000–1009, 2011. [DOI] [PubMed] [Google Scholar]
- 43.Stuart CA, Shangraw RE, Prince MJ, Peters EJ, Wolfe RR. Bed-rest-induced insulin resistance occurs primarily in muscle. Metabolism 37: 802–806, 1988. [DOI] [PubMed] [Google Scholar]
- 44.Vigelso A, Gram M, Dybboe R, Kuhlman AB, Prats C, Greenhaff PL, Constantin-Teodosiu D, Birk JB, Wojtaszewski JF, Dela F, Helge JW. The effect of age and unilateral leg immobilization for 2 weeks on substrate utilization during moderate-intensity exercise in human skeletal muscle. J Physiol 594: 2339–2358, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vousden KH, Ryan KM. p53 and metabolism. Nat Rev Cancer 9: 691–700, 2009. [DOI] [PubMed] [Google Scholar]
- 46.Wilkinson SB, Phillips SM, Atherton PJ, Patel R, Yarasheski KE, Tarnopolsky MA, Rennie MJ. Differential effects of resistance and endurance exercise in the fed state on signalling molecule phosphorylation and protein synthesis in human muscle. J Physiol 586: 3701–3717, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]