

Abstract
Insulin regulates skeletal muscle protein degradation, but the types of proteins being degraded in vivo remain to be determined due to methodological limitations. We present a method to assess the types of skeletal muscle proteins that are degraded by extracting their degradation products as low-molecular weight (LMW) peptides from muscle samples. High-resolution mass spectrometry was used to identify the original intact proteins that generated the LMW peptides, which we validated in rodents and then applied to humans. We deprived insulin from insulin-treated streptozotocin (STZ) diabetic mice for 6 and 96 h and for 8 h in type 1 diabetic humans (T1D) for comparison with insulin-treated conditions. Protein degradation was measured using activation of autophagy and proteasome pathways, stable isotope tracers, and LMW approaches. In mice, insulin deprivation activated proteasome pathways and autophagy in muscle homogenates and isolated mitochondria. Reproducibility analysis of LMW extracts revealed that ∼80% of proteins were detected consistently. As expected, insulin deprivation increased whole body protein turnover in T1D. Individual protein degradation increased with insulin deprivation, including those involved in mitochondrial function, proteome homeostasis, nDNA support, and contractile/cytoskeleton. Individual mitochondrial proteins that generated more LMW fragment with insulin deprivation included ATP synthase subunit-γ (+0.5-fold, P = 0.007) and cytochrome c oxidase subunit 6 (+0.305-fold, P = 0.03). In conclusion, identifying LMW peptide fragments offers an approach to determine the degradation of individual proteins. Insulin deprivation increases degradation of select proteins and provides insight into the regulatory role of insulin in maintaining proteome homeostasis, especially of mitochondria.
Keywords: peptidomics, isotope tracer, low molecular weight, method, autophagy
the anticatabolic effect of insulin was evident from early reports of humans with type 1 diabetes (T1D) showing dramatic atrophy of skeletal muscle without insulin and then restoration of muscle mass with insulin treatment (17). Skeletal muscle protein content represents the balance between protein synthesis and protein degradation. Previous studies demonstrated that skeletal muscle catabolism during insulin deprivation in T1D is due primarily to increased protein degradation, with little change to mixed muscle protein synthesis, representing the average of multiple protein pools (8, 16, 35). Separating specific muscle protein fractions revealed that insulin has a stimulatory effect on mitochondrial proteins when amino acids are provided (36, 40). In contrast, the synthesis rates of contractile proteins, such as myosin heavy chain, were not altered during insulin deprivation (7). Such studies indicate that insulin's effect on synthesis of specific proteins can vary, yet the lack of valid methodologies to measure degradation of specific proteins hampers research on understanding the regulation of degradation by insulin.
Current approaches to investigate protein degradation include assessing the activation state of major protein degradation pathways, such as the autophagy-lysosome and ubiquitin-proteasome pathways that are suppressed with insulin or amino acids (33, 38). However, measuring the activation status of associated pathways may not represent kinetics through the pathway. Kinetics can be measured in vivo using isotope tracer techniques that determine amino acid uptake into and release from skeletal muscle. Isotopic tracers are effective for measuring the synthesis rates of protein fractions such as mitochondria (37) or individual muscle proteins (23). Degradation of specific proteins is difficult to measure due to the requirement for longer prelabeling periods and potential for tracer reincorporation (recycling) into newly synthesized proteins (21). Isotope dilution approaches across tissue beds are used to measure global protein degradation (3, 5, 35) but cannot determine specific proteins or protein fractions that are degraded.
An approach that we consider valuable is to identify the specific muscle proteins that are degraded during insulin deficiency based on the peptide fragments that are released after protein degradation. Advances in high-resolution mass spectrometry provide an opportunity to identify low-molecular weight (LMW) peptides derived from protein degradation. The LMW peptides can be identified and compared against proteomic libraries to determine the original intact proteins. The original proteins are classified by their corresponding pathways and represent the pathways being actively degraded in vivo. This “peptidomics” approach is used to identify plasma peptide signatures as potential biomarkers (14, 26). Applying the approach to T1D humans and insulin-deficient mouse models provides an opportunity to understand the regulation of skeletal muscle protein degradation at the individual protein level with insulin deprivation.
We investigated the regulation of individual protein degradation by insulin using the LMW approach by first validating the approach in rodent models and then applying it to T1D humans. Here, we report the results based on this approach, demonstrating specific proteins and functional proteome pathways that are affected by insulin deprivation in T1D humans.
MATERIALS AND METHODS
Study overview.
We used insulin-deprived rodent models to develop and validate the LMW method for detecting protein degradation by-products. Reproducibility was determined using biological and technical replicates. We then applied the method on T1D humans to identify individual proteins that were degraded during insulin deprivation.
Mouse model.
The animal study protocol was approved by the Mayo Clinic Animal Care and Use Committee. Mice were caged individually (12-h light-dark cycle) and allowed ad libitum access to food and water except on the day of euthanasia, when the food was removed 4 h prior, to avoid acute effects of feeding on protein turnover. Eight-week-old C57BL/6J mice (Jackson Laboratories, Bar Harbor, ME) were experimentally induced with insulin deficiency using two intraperitoneal doses of streptozotocin (STZ) at 125 mg/kg body wt separated by 24 h. Glucose dysregulation was confirmed 5 days after the first STZ injection and subcutaneous insulin implants (LinBit; LinShin Canada, Toronto, ON, Canada) were inserted while mice were anaesthetized (0.090 mg/g body wt ketamine and 0.010 mg/g body wt xylazine). The erodible implants release ∼0.1 U/day of insulin. The mice were observed for 14 days for normalization of blood and urine glucose. On day 14, insulin implants were removed from mice for uncontrolled diabetes for 6 (D-6) and 96 h (D-96) compared with nondiabetic controls. All mice were euthanzied by decapitation and tissues harvested.
Human studies.
The Mayo Clinic Institutional Review Board approved the human study protocol, and the study design was described previously (25). The current report is on protein turnover measured during insulin treatment vs. insulin deprivation in T1D adults (n = 7) on two separate visits. Participants gave written, informed consent. Exclusionary criteria included circulating c-peptide, vascular disease, renal insufficiency, neuropathy, poor wound healing, or medications including β-blockers, anticoagulants, and tricyclic antidepressants. Fat-free mass (FFM) was determined by dual-energy X-ray absorptiometry (DPX-L; Lunar, Madison, WI).
Participants consumed a weight-maintaining diet (55% carbohydrate, 15% protein, and 30% fat) for 3 days prior to each inpatient study day and used short-acting insulin to maintain blood glucose at 80–120 mg/dl. Participants finished the meals by 1800 on the evening of admission and remained fasting (with the exception of water) until 1200 on the day of the study. A dorsal hand vein was catheterized in a retrograde direction for arterialized blood sampling (60°C), and a catheter was inserted into the opposite arm for intravenous infusions. For the insulin-treated protocol, insulin was infused to maintain euglycemia (80–120 mg/dl) between the evening meal and 1200 the next day. For the insulin deprivation protocol, insulin was stopped during the night from ∼0400 until 1200 the next day [average deprivation was 8.6 ± 0.6 h (25)].
T1D humans: protein synthesis and breakdown.
A primed, continuous infusion (1 mg/kg FFM prime and 1 mg/kg FFM infusion) of [ring-13C6]phenylalanine (Cambridge Isotopes, Andover, MA) was started at 0400, and percutaneous muscle biopsies were obtained under local anesthesia (2% lidocaine) at 0700 and 1200. The fractional synthesis rate (FSR) of mixed muscle proteins was calculated as FSR (%/h) = [(E1200 − E0700)/(Ep × t)] × 100, with E1200 and E0700 as the tracer enrichment at biopsy time points and Ep as the plasma enrichment at isotopic steady state during time (t) in hours between biopsies (2). The rate of appearance (Ra) of phenylalanine into circulation from endogenous protein breakdown was calculated as Ra = F[1/Ep − 1], using the infusion rate (F) of [ring-13C6]phenylalanine and Ep as the plasma enrichment at isotopic steady state. Plasma enrichment (molar %excess) was determined using gas chromatography (GC; Agilent Technologies, Santa Clara, CA) and mass spectrometry (MS; Waters, Milford, MA), with selective ion monitoring at m/z 399.2 and 403.2 for the [M-HF]− fragment of phenylalanine and [ring-13C6]phenylalanine, respectively. Muscle protein enrichment was determined by high-performance liquid chromatography (Cohesive Technologies, Franklin, MA) and MS/MS (Applied Biosystems, Boston, MA), with monitoring of transitions of m/z 222.4 > 121.6 and 226.4 > 125.6 for the m + 2 and m + 6 fragments of phenylalanine and [ring-13C6]phenylalanine, respectively, as described previously (44).
Mass spectrometric analysis of LMW peptides.
Mass spectrometry identified LMW peptide fragments from endogenous protein degradation. Muscle samples (∼10 mg) were homogenized in 9.8 M urea and passed through an Amicon Ultra-0.5, Ultracel-10-kDa molecular weight cutoff centrifugal filter. The “peptidome” present in the filtrates was desalted using a Nest Group (Southborough, MA) Macrospin C18 spin column concentrated to <5 μl on a spinning vacuum centrifuge and resuspended in 0.2% trifluoroacetic acid (TFA). The suspension (∼40%) was loaded to an OptiPak 250nl trap (Optimize Technologies, Oregon City, OR) packed with Michrom Magic C8 solid phase (Michrom Bioresources, Auburn, CA), using an Eskigent nano liquid chromatography (LC) system. The “peptidome” was separated on a 100 mm × 30 cm PicoFrit column (NewObjective, Woburn, MA) packed with Poroshell 120 EC-C18, 2.7 μm solid phase (Agilent, Santa Clara, CA), using a 68-min linear gradient of 4–40% mobile phase. Eluting peptides were analyzed with a QExactive MS (Thermo Fisher, Waltham, MA) configured to a resolution of 70K (at 200 m/z), and the top 20 peptide ions for fragmentation were isolated using a 2-Da window. MS/MS was acquired at a resolution of 17,500, with a repeat count of 1 and ion exclusion duration of 45 s. A total of 454,428 MS/MS were collected from all LC-MS/MS analyses.
MyriMatch (41) software matched the MS/MS against a composite protein sequence database containing the RefSeq human protein database (release 53) and common contaminants. Reversed sequence entries were appended to the database for estimating peptide identification false discovery rate (FDR). The software used 10 ppm m/z error for both precursor and fragments, derived nonenzymatic peptides from the sequence database, and searched for methionine oxidation and NH2-terminal pyroglutamic acid as variable modifications. IDPicker version 3.0 (32, 46) filtered the peptide identifications at 2% FDR using an optimal combination of Multivariate Hypergeometric, mzFidelity, and XCorr scores. IDPicker assembled the filtered peptides into protein identifications, following parsimony rules. Protein identifications with at least one distinct peptide identity and two MS/MS matches were considered present in the sample. Proteins that yielded LMW fragments were classified with Ingenuity Pathway Analysis (Qiagen, Valencia, CA) to determine the canonical pathways being degraded.
Our method of development included performing five extractions of LMW peptides (for reproducibility) from a single muscle sample from rat quadriceps and analyzing each extract five times (for mass spectrometry repeatability) for a total of 25 analyses.
Mass spectrometry of protein expression and posttranslational modifications.
Biopsy samples were suspended in homogenization buffer (50 mM Tris, 0.1% SDS) and then underwent reduction with 5 mM DTT and alkylation with 10 mM iodoacetamide. Proteins were digested overnight with trypsin (enzyme/substrate ratio of 1:50). Peptide digests were cleaned with SDS removal spin column (Thermo Fisher Scientific, Rockford, IL), evaporated to dryness, and stored at −20°C. Dried peptide digest was reconstituted (0.2% formic acid, 0.1% TFA, and 0.001% Zwittergent 3–16), and 250 ng of digest was loaded on an OptiPak Magic 200A°C8 trap (Phenomenex, Torrance, CA), using an Eskigent nano LC system. Peptides were separated on a 75-μm id fused silica column packed with 25 cm of Poroshell 120 EC-C18 stationary phase (2.7 mm, 120A°; Agilent Technologies), using a 90-min linear gradient of 2–40% mobile phase. Eluting peptides were analyzed with a QExactive MS (Thermo Fisher, Waltham, MA) configured as described above. A total of 573,983 MS/MS was collected from all LC-MS/MS analyses. MS/MS data were processed with a two-step bioinformatics protocol for detecting unanticipated posttranslational modifications (PTMs) (11).
First, proteins were detected using MyriMatch configured to match MS/MS spectra against a composite protein sequence containing RefSeq mouse proteome (version 58) and common contaminants. Reversed protein sequences were appended to the database for estimating peptide identification FDRs. MyriMatch used 10 ppm m/z error for both precursor and fragments derived semitryptic peptides from the database and identified carbamidomethylation of cysteine, oxidation of methionine, NH2-terminal pyroglutamic acid, and acetylation of lysine as variable modifications. IDPicker version 3.0 filtered the peptide identifications at 2% FDR and assembled protein identifications, as described above. Protein identifications with at least two distinct peptide identifications were included in a subset FASTA database.
Second, DirecTag-TagRecon software interrogated the subset of FASTA proteins for unanticipated PTMs (11). DirecTag generated the 50 best tags of three amino acids from each MS/MS. TagRecon reconciled tags against the protein database with allowance for unanticipated mass shifts in peptides. TagRecon used the same variable modifications as MyriMatch and derived semitryptic peptides from the FASTA database. IDPicker version 3.0 filtered the resulting peptide identifications at 2% FDR, as described above. Detected PTMs were attested following previously published guidelines (11). Relative expression of protein PTMs (at sample level) between two groups was performed using peptide intensities as described previously (24) and then displayed as percent difference between study days.
Blood measurements.
Blood glucose concentrations were measured from tail vein puncture (∼5 μl) using a hand-held glucometer. Whole blood was collected into sodium-heparin tubes at euthanasia from trunk bleed following decapitation and then centrifuged and plasma stored at −80°C. Plasma insulin was measured using an enzyme-linked immunosorbent assay (Chrystal Chem, Downers Grove, IL).
Immunoblotting.
Mouse muscles were powdered in liquid N2 and diluted in RIPA buffer (20 μl/mg) with protease/phosphatase inhibitors, homogenized (Beadruptor, Omni International), and centrifuged (10,000 g for 20 min at 4°C). The supernatant was collected and analyzed for protein concentration (Pierce 660; Thermo Fisher Scientifi, Rockford, IL), which was normalized by diluting in loading buffer (LDS; Life Technologies, Carlsbad, CA). Thirty micrograms of protein was separated using 4–12% Bis-Tris NuPage gels (Life Technologies) and then transferred to nitrocellulose membranes, blocked in 5% bovine serum albumin in 0.05 M Tris-buffered saline, pH 7.5 (TBS), and incubated overnight with primary antibodies diluted in 5% nonfat dry milk in TBS with 0.05% Tween 20 (TBST). Membranes were washed for 3 × 10 min in TBST, incubated for 1 h with fluorescent secondary antibodies (Thermo Fisher Scientific) at 1:5,000 in blocking buffer, and imaged (LI-COR Biosciences, Lincoln, NE). Antibody dilutions were p62 (1:200, no. SC28359; Santa Cruz Biotechnology), Vinculin (1:1,000, Calbiochem; EMD Millipore, Billerica, MA), and OxPhos Cocktail (1:1,000, no. MS604; Mitosciences).
Autophagy.
Mitochondrial autophagy was measured in mouse quadriceps by immunoblotting of isolated mitochondria samples, similar to a previous approach (42). Mitochondria were isolated from quadriceps samples using differential centrifugation, as described previously (36). We examined mitochondrial purity using mass spectrometry and determined that the fraction was enriched in mitochondrial proteins but also included nonmitochondrial proteins, including contractile and lysosome proteins. For autophagy analysis, 20 μg of mitochondrial lysates was separated, following the above immunoblotting conditions, and membranes were probed with primary antibodies for p62 (1:200, no. SC28359; Santa Cruz Biotechnology) and normalized to mitochondrial respiratory complex content by OxPhos Cocktail (1:1,000, no. MS604; Mitosciences). A commercially available positive control (HeLa + Chloroquine extract) or negative control (untreated HeLa extract) for autophagy was included on every gel (no. 11972S; Cell Signaling Technology).
Quantitative real-time polymerase chain reaction.
Total mRNA was extracted from mouse quadriceps muscle using an RNeasy kit (Qiagen), and then purity and concentration were determined using a NanoDrop spectrophotometer (Thermo Fisher Scientific). Two micrograms of mRNA was reverse transcribed to cDNA (Life Technologies). Two hundred nanograms of cDNA was amplified using Taqman reagents and predesigned gene expression assay FAM-labeled probes (Life Technologies) for p62 (Mm00448091_m1), beclin-1 (Mm01265461m_1), and muscle RING finger 1 (MuRF1; Mm01185221_m1), with β2 microglobulin (VIC labeled) as reference gene (no. 4448486). Multiplex quantitative PCR was performed with target and reference genes amplified in 20 μl on 384-well plates with 40 cycles of 15 s of denaturing (95°C) and 60 s of annealing/extension (60°C). A no-template control and internal repeated control was included on every plate. Relative quantification was performed using a six-point relative standard curve spanning four log dilutions with the target gene normalized to the reference gene.
Statistical analysis.
Results were analyzed using Prism 6 (GraphPad Software) with significance at P ≤ 0.05. Paired human data were analyzed with paired t-test. Human plasma enrichment was analyzed using two-way (treatment × time) ANOVA with repeated measures (n = 6 due to 1 missing plasma value for 1 participant). Control and insulin-deprived mice were compared using unpaired t-test. Relative protein expression for proteomics data was performed using quasi-likelihood modeling of spectra counts (30). Ingenuity Pathway Analysis was used to identify canonical pathways from LMW peptide lists. Data are presented as means ± SD, with group numbers as indicated.
RESULTS
LMW method development.
First, we wanted to assess the contribution of ex vivo protein degradation during sample handling to the LMW peptidome. For this, we prepared muscle samples for LMW analysis in the presence and absence of protease inhibitors, using six mice in each condition. Omitting protease inhibitors led to generation of more peptide fragments (identified by more mass spectra) and subsequently more proteins being identified (Fig. 1). These results demonstrate that protease inhibitors are necessary to avoid ex vivo generation of LMW peptide fragments. Hence, all subsequent LMW preparations were performed in the presence of protease inhibitors.
Fig. 1.
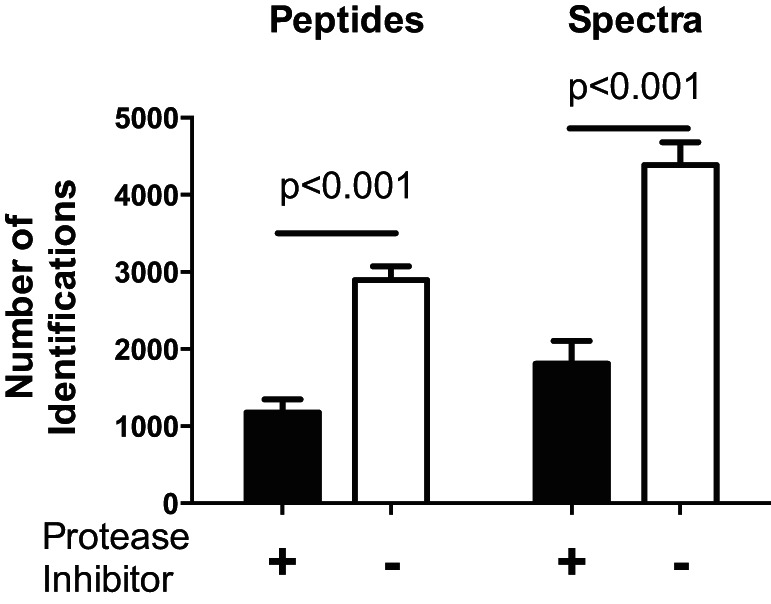
Minimizing ex vivo generation of low-molecular weight (LMW) peptides. LMW peptides were extracted with and without protease inhibitors from quadriceps from 6 mice. More LMW peptides and mass spectra and were generated without protease inhibitors, thus demonstrating that protease inhibitors are necessary to avoid ex vivo generation of LMW fragments. Data displayed as means ± SD and P values are from unpaired t-test between treatment conditions with and without protease inhibitors.
We next investigated the reproducibility of the LMW method in detecting degraded protein fragments. For this, we performed five repeated LMW extracts of a single muscle sample (from a rat) and analyzed each extract five times using mass spectrometry analysis. We identified a mean of 1,796 proteins across all biological and technical replicates. We considered the extent to which the LMW method could consistently identify proteins across biological replicates and determined that an average of 12 ± 2 proteins (<1% of total) were detected in only one sample, whereas ∼1,418 ± 70 proteins (∼80% of total) were consistently detected in all five samples (Table 1). We identified that the LMW peptide abundance is the source of the 20% variability; i.e., high abundance peptides will have higher intensity in mass spectrometry analysis and hence, are reproducibly detectable when compared with low-abundance peptides. To verify this, we separated the LMW peptides detected in all replicates by their MS1 intensity quartile. We found that 93% of the peptides in the high-intensity quartile (4th) were detected in all replicates compared with only 58% of the peptides in the lowest-intensity quartile (1st), which were detected in all replicates (Table 1). This suggested that the LMW method could reproducibly identify full-length proteins that are increasingly degraded upon stimulus. In contrast, the LMW method has a potential blind spot when detecting proteins that decrease degradation upon stimulus.
Table 1.
Reproducibility analysis
| LMW Peptides Detected in 5 Extractions of 5 Samples |
|||||
|---|---|---|---|---|---|
| MS Intensity Quartiles | Detected in 1 sample | Detected in 2 sample | Detected in 3 sample | Detected in 4 sample | Detected in all 5 samples |
| 1 (low) | 6 ± 3 | 20 ± 4 | 45 ± 10 | 117 ± 12 | 260 ± 9 |
| 2 | 3 ± 3 | 5 ± 1 | 17 ± 4 | 78 ± 11 | 346 ± 5 |
| 3 | 2 | 3 ± 1 | 10 ± 1 | 42 ± 5 | 392 ± 5 |
| 4 (high) | 1 | 3 ± 1 | 5 ± 1 | 21 ± 2 | 420 ± 7 |
| Sum of all intensities | 12 ± 2 | 31 ± 8 | 77 ± 18 | 258 ± 42 | 258 ± 42 |
| %Total LMW (total = 1,796) | 0.7% | 1.7% | 4.3% | 14.4% | 79.0% |
Data are means ± SD number of peptides Reproducibility analysis from 5 extracts from the same muscle sample (rat quadriceps) were analyzed 5 times and revealed that low-molecular-weight (LMW) peptides were consistently detected between biological replicates (e.g., overlapping between samples), with greater detection of peptides at higher intensities. Mass spectrometry (MS) intensities were separated into quartiles from 1 (low) to 4 (high).
STZ mouse model.
The LMW approach was applied to an insulin-deprived mouse model. Mice were injected with STZ to experimentally induce insulin deficiency and then treated with subcutaneous insulin implants for 14 days (Fig. 2A), as described previously (45). The implants were removed to induce 6 (D-6) and 96 h (D-96) of insulin deprivation compared with nondiabetic control (CON). By design, plasma glucose was higher at euthanasia in D-6 and D-96 mice compared with control (Fig. 2B). Mice were fasted for 4 h before euthanasia, leading to low plasma insulin in all groups (CON: 0.7 ± 0.1; D-6: 1.1 ± 0.95; D-96: 0.6 ± 0.9 ng/ml; ANOVA, P = 0.17) despite marked hyperglycemia in D-6 and D-96 mice. Body weight was lower in D-6 and D-96 mice (Fig. 2C).
Fig. 2.
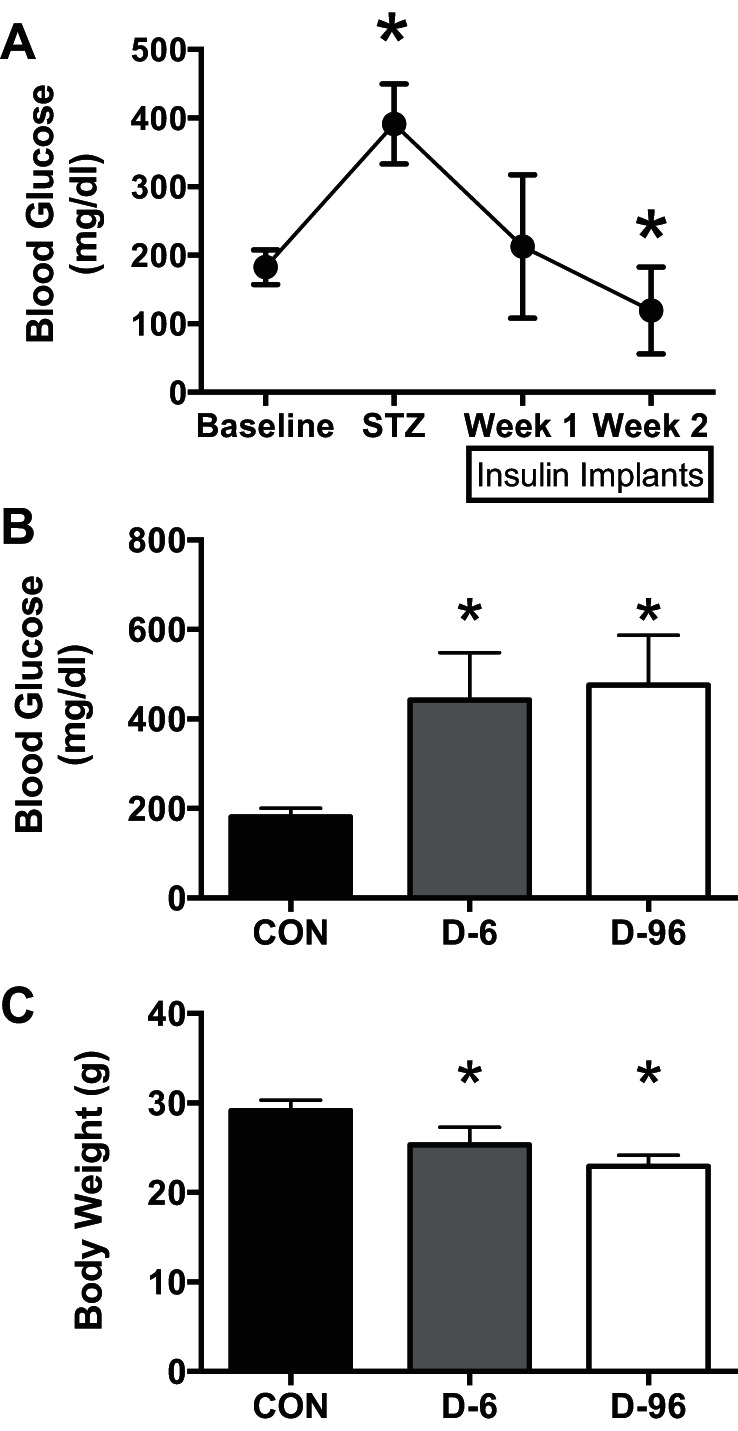
Streptozotocin (STZ) mouse model. A: glucose dysregulation was confirmed in mice following STZ injection and normalized for 2 wk by subcutaneous insulin implants. Implants were removed from the mice for 6 (D-6) and 96 h (D-96) of untreated diabetes (n = 7 each) compared with nondiabetic controls (CON; n = 5). B and C: at euthanasia compared with control mice, D-6 and D-96 mice had higher blood glucose (B) and lower body weight (C). *P < 0.05 vs. baseline (A) or CON (B and C) for multiple comparisons following significant ANOVA. Data are displayed as means ± SD.
The activation of protein degradation pathways was determined using real-time PCR and immunoblotting of whole muscle lysates and isolated mitochondria. A rigorous investigation of autophagy flux would require the use of proteolytic inhibitors, which would hinder our attempts to measure protein degradation products. Therefore, we chose a representative panel of autophagy and proteasome markers as indicative but not definitive markers to assess activation with insulin deprivation. Trim63 mRNA content, representing the proteasome pathway, increased in D-6 and D-96 mice (Fig. 3A), and beclin mRNA, representing autophagy, increased in D-96 (Fig. 3B). There was a trend for increased p62 mRNA content in D-96 mice (CON: 1.1 ± 0.4 vs. D-96: 1.45 ± 0.6, P = 0.056). There was decline in protein content for the autophagy receptor protein p62 in whole muscle lysates (Fig. 3C) and isolated mitochondria (Fig. 3D), suggesting increased degradation through the autophagy-lysosome pathway during insulin deprivation. Collectively, these results are consistent with activation of protein degradation with insulin deprivation.
Fig. 3.

Activation of protein degradation in STZ mice. A and B: activation of protein degradation pathways was determined by real-time PCR for proteasome pathway (E3 ubiquitin ligase; A) and autophagy (beclin; B). Immunoblotting revealed insulin deprivation decreased accumulation of the autophagy receptor protein p62 in quadriceps lysates (C) and isolated mitochondria fractions (D), indicating activation of autophagy. Autophagy negative (HeLa cells untreated) and positive controls (HeLa cells + chloroquine) were included in lanes 1 and 2. Loading controls were vinculin (Vinc) for muscle lysates and complexes III and V for isolated mitochondria. Representative images are from a single blot, with vertical bars representing discontinuous lanes. Data are means ± SD with n = 5–7 mice/group. *P < 0.05 vs. control. AU, arbitrary units.
Our overall aim of the project was to identify the individual proteins that were being degraded using the LMW method, particularly from the mitochondrial fraction. The analysis of LMW peptide fragments showed a total of 429 proteins consistently detected between groups. A total of 89 detected peptides belonged to mitochondrial proteins. Proteins identified from LMW fragments are listed in Supplemental File S1 (Supplemental Material for this article can be found on the AJP-Endocrinology and Metabolism web site). The detected LMW peptides were mapped to their corresponding proteins and compared between controls and D-6 (Fig. 4A) and D-96 mice (Fig. 4B). These results show that seven peptides derived from mitochondrial proteins were higher (defined as >0.5 fold change and FDR ≤ 0.05) following 6 h of insulin deprivation and 14 are higher following 96 h of insulin deprivation, consistent with activation of autophagy during insulin deprivation. Of these, four were exclusive to D-6 (ATP5K, MRPL21, COX5A, and RPL10A), and three were detected in both D-6 and D-96 (OGDH, GOT2, and COX6C), whereas 11 were exclusive to D-96 (ABCF2, MRPS18C, NUDFB10, ATP5A1, MP68, UQCRQ, MRPL48, NDUFS6, PHB2, COX5B, and HADHB).
Fig. 4.
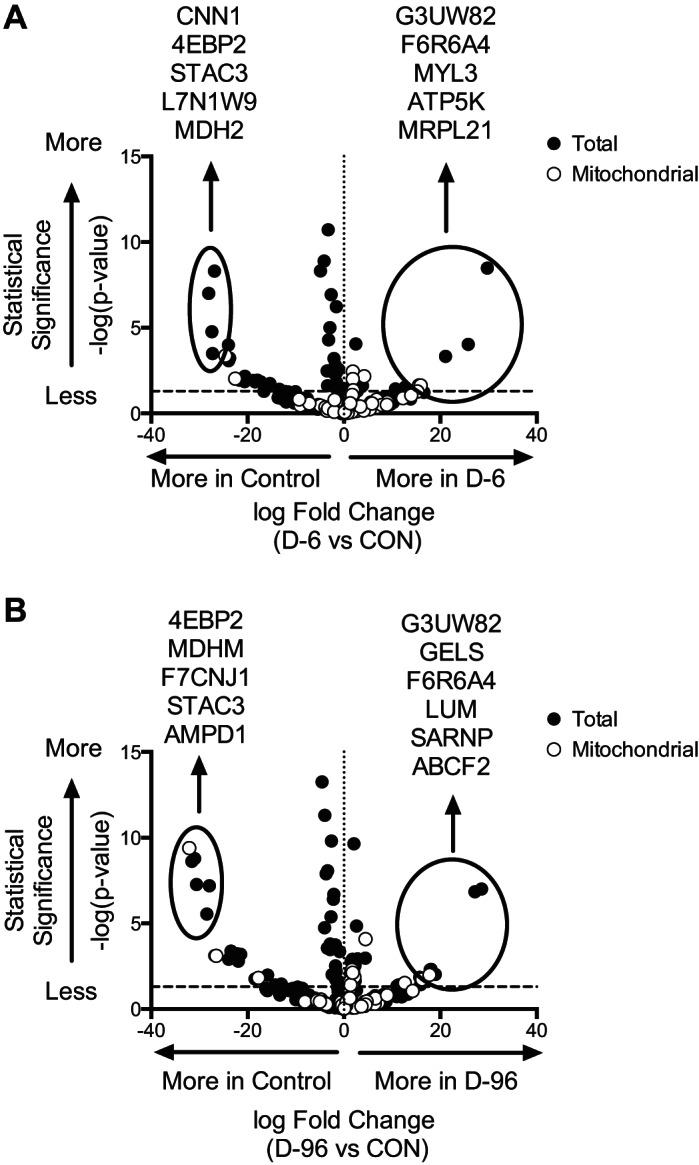
Mitochondrial protein degradation in STZ mice identified by LMW. Volcano plots represent total and mitochondrial proteins identified in LMW peptide fragments from quadriceps during 6 (A) and 96 h (B) of insulin deprivation in STZ diabetes mice compared with control mice. Data are presented with statistical significance on vertical axis and fold change on horizontal axis relative to control. The horizontal dashed line at y = 1.3 corresponds to significance at 0.05 for false discovery rate. Proteins with the greatest change in each condition are identified and circled.
T1D humans.
We next applied the LMW approach to T1D humans to investigate mitochondrial protein degradation during insulin deprivation. Humans with T1D (n = 7) were studied on 2 separate inpatient days (Fig. 5) to investigate skeletal muscle protein turnover during insulin treatment (INS+) or insulin deprivation (INS−). Protein turnover kinetics were measured using infusion of [ring-13C6]phenylalanine, providing a measure of whole body protein degradation (by dilution of plasma isotope from release of unlabeled amino acids). Plasma [ring-13C6]phenylalanine isotopic enrichment at steady state was lower during INS− than during INS+ due to a higher rate of appearance of unlabeled phenylalanine from whole body protein degradation (Fig. 5, A and B). Mixed-muscle FSR was not different between INS+ and INS− (Fig. 5C). We considered whether the changes in protein damage as measured by irreversible posttranslational modifications to proteins would increase degradation. Muscle proteins in the insulin deprivation state had greater oxidative damage but lower deamination (Fig. 5D). These results are consistent with oxidative damage leading to increased degradation of proteins.
Fig. 5.

Protein degradation increased with insulin deprivation in type 1 diabetic (T1D) humans. T1D humans (n = 7) were studied on 2 visits during insulin treatment (INS+) or insulin deprivation (INS−) with primed, continuous infusion of [ring-13C6]phenylalanine and biopsies of the vastus lateralis. A: plasma tracer enrichment in molar %excess (MPE) at steady state was lower during INS− study day. B: whole body protein breakdown was increased during insulin deprivation conditions and is displayed as the rate of appearance (Ra) of phenylalanine (Phe) into circulation. C: fractional synthesis rate (FSR) of mixed-muscle proteins was not different between insulin-treated and deprivation conditions. D: posttranslational modifications to proteins were determined by mass spectrometry and revealed that oxidative damage was increased, whereas deamidation was decreased during insulin deprivation conditions. Posttranslational modifications to proteins were determined by mass spectrometry and differential expression based on peptide intensities made by paired comparison and then presented as %difference between INS− and INS+. *P < 0.05 and **P < 0.01 for paired comparison between INS+ and INS−. Data are means ± SD. FFM, fat-free mass.
Analyzing LMW peptides revealed differences between insulin-treated and -deprived conditions (full list is in Supplemental File S2). In T1D humans, similar to the mouse model, individual mitochondrial proteins were detected by LMW peptide fragments, and specific proteins generated more (P ≤ 0.05) LMW peptide fragments during insulin deprivation, including individual proteins involved with oxidative phosphorylation (Fig. 6). Additionally, degraded proteins were detected across major regulatory pathways, including ribosomal proteins (representing protein translation), histones (regulating DNA structure and transcription), and myosin isoforms (contractile proteins).
Fig. 6.

Proteins identified by LMW peptides released during INS+ and INS− in humans. LMW peptide fragments identified individual proteins that were differentially generated (P < 0.05) during insulin treatment (INS+) and insulin deprivation (INS−). Each original full-length protein is identified along with P value (paired t-test) and fold change between INS+ and INS−, with negative values indicating more fragments generated during INS+ and positive values indicating more fragments generated during INS−.
DISCUSSION
The current investigation describes a mass spectrometry-based approach for identifying individual skeletal muscle proteins that are degraded in vivo. We applied the approach, along with stable isotope measurements of protein degradation, to short-term insulin deprivation in T1D humans. The stable isotope based approach, as expected, showed increased whole body protein degradation, with no effect on muscle protein synthesis. The STZ mouse model suggests that both proteasome and lysosome-autophagy pathways were activated by insulin deprivation. Using high-resolution mass spectrometry, we identified several LMW peptides representing many individual proteins that were differentially degraded in skeletal muscle during insulin deprivation, including proteins from pathways of mitochondrial physiology, proteome homeostasis, nDNA support, glucose metabolism, and contractile/cytoskeleton. The LMW approach provides an effective and novel method to determine the degradation of specific proteins from muscle biopsy and animal tissues.
The proteins or proteomic pathways that we have identified represent some of the important functions of insulin. Recently, an important role of insulin in regulating mitochondrial biogenesis (36, 40) and function (1) was recognized. We identified several LMW peptide fragments released from the degradation of individual mitochondrial proteins, including subunits of ATP synthase and cytochrome c oxidase complexes, which are involved in oxidative phosphorylation. These results are consistent with the decrease in mitochondrial proteins and ATP production rate in insulin-deprived STZ mice (45) and decreased muscle ATP production in insulin-deprived T1D individuals (25). Previous studies in STZ mice showed that insulin deprivation reduced mitochondrial protein content involved in ATP production and β-oxidation (45). Insulin deficiency can affect mitochondrial protein synthesis since insulin administration with amino acid supplementation increased mitochondrial protein synthesis (36, 40). Collectively, the LMW approach indicates that mitochondrial protein degradation is increased during insulin deprivation, which may contribute to reduced mitochondrial protein content (45) and mitochondrial function.
We also identified many peptide fragments derived from proteins involved in proteome homeostasis, especially of ribosomal units that are critical protein synthetic machinery. Insulin plays a key role in protein turnover at the level of both protein synthetic machinery (27) and protein degradation (16, 19). The current study demonstrated that degradation of many ribosomal proteins (regulating protein synthesis) and a calpain 3 isoform (regulating protein degradation) was at a lower rate during insulin deprivation. Calpain 3 is a proteolytic enzyme, and its reduced degradation is potentially important for increased muscle protein degradation (20, 39). Insulin is a key hormone involved in glucose metabolism, and we found increased degradation of phosphoglucomutase 1 isoform 1, a key enzyme involved in glycogenolysis. We also found that many isoforms of myosin, a protein family involved in muscle contractile function, are degraded at higher level consistent with the muscle wasting that has been observed in insulin-deficient T1D (17). Nebulin is an actin-binding protein that exists in different isoforms and appears to be essential to maintain myofibrillar integrity (43). Higher LMW fragments from this protein isoform indicate a potential detrimental impact of insulin deprivation on myofibrillar integrity. The SR/ER Ca ATPase plays an important role in calcium transport from cytosol to the sarcoplasmic reticulum, and the increased degradation of this protein during insulin deprivation potentially affects its important function of the muscle in diabetes (13).
Current approaches to measure protein degradation include amino acid tracers for whole body assessment, which can be combined with arterial-venous balance studies across a specific tissue bed (35) but cannot measure degradation of individual proteins or organelles such as mitochondria. Other approaches to measure mitochondrial protein degradation include enzymatic activity, such as the Lon protease located within the mitochondria (15), but cannot investigate the organelle as a whole or as individual proteins. In the absence of any valid approach, we applied the measurement of LMW peptides that are derived from mitochondrial proteins and found them to be increased during insulin deprivation, showing that mitochondrial protein degradation is increased.
The LMW approach is an important tool not only for assessing tissue degradation of mitochondrial proteins but also for nonmitochondrial proteins. Indeed, although mitochondrial-specific proteins were detected, proteins representing a variety of other pathways were also observed and demonstrate the extensive effect of insulin deprivation on protein degradation. Of interest is that the degradation of ribosomal proteins was noted, raising a paradoxical situation in which the ribosome, the site of protein translation, was degraded without mixed-muscle protein synthesis rates being affected. Potential explanations include that degradation of damaged ribosomal proteins may have preserved the protein quality and that there is no evidence showing that protein synthesis is directly proportional to the ribosomal protein content. Previous work has shown that the availability of amino acids, which are released following protein degradation, is critical to maintain protein synthesis (2, 36). However, as we have reported previously, insulin deprivation increases net protein degradation (35), and longer periods of insulin deprivation would significantly reduce the content of muscle proteins whose degradation continues to exceed synthesis. We propose that during the 8-h study period, the decline of ribosomal protein content was not large enough to negatively impact protein synthesis. Degradation products of myosin isoforms were also increased with insulin deprivation, consistent with muscle atrophy that is observed with untreated T1D. The current study demonstrated that the LMW peptide approach showed not only the degradation of mitochondrial proteins during insulin deprivation but also that many other protein pools are also affected by insulin deprivation. However, it is possible that the current approach is not comprehensive enough to detect degradation of all proteins that occurred during insulin deprivation muscle. Even so, a variety of protein pathways were represented in the LMW approach. A main strength of the LMW approach is that this provides insight into the dynamics of specific proteins during disease progression.
A number of proteins degraded were higher in control mice, suggesting that greater degradation of these proteins occur, whereas insulin is sufficient to maintain glucose homeostasis. For example, LMW peptides derived from proteins involved with the cytoskeleton (CNN1, DEST, and SGCB) or neuromuscular transmission (Stac3) were higher in nondiabetic mice. There are potential explanations for this observation. We speculate that proteins that have undergone greater degradation in controls may belong to classes of proteins that are spared during insulin deprivation. Sparing of proteins during nutrient stress, such as during caloric restriction (29), is suggested to be protective against the high-energy cost of synthesizing new proteins. It is also possible that increased amino acids during insulin deprivation may have selective effect on inhibiting degradation of certain classes of proteins.
A small but significant increase in oxidatively damaged proteins was observed in the current study. These small differences in the short term may cause buildup of damaged and dysfunctional proteins over time, such as occurs with aging (10). Of interest, we observed a significant decrease of deamidation of proteins that may suggest that in the absence of insulin these modified proteins are degraded via the ubiquitin-dependent degradation pathway (12). The increased protein degradation during insulin deprivation may help remove proteins that are damaged with irreversible posttranslational modifications (18). Previous studies using leucine tracers showed that amino acids derived from protein degradation are preferentially acetylated to tRNA for protein synthesis (31). Therefore, protein degradation is usually followed by synthesis and allows replacement of damaged proteins by nascent proteins.
We investigated major pathways regulating protein degradation, including the autophagy-lysosome and ubiquitin-proteasome pathways, during insulin deprivation using an STZ mouse model that was not possible in humans due to biopsy sample size limitations. The autophagy adapter protein p62 is degraded in the lysosome, and therefore, decreased p62 content is representative of increased autophagy (6). p62 content was decreased in mitochondria fractions at 6 and 96 h of insulin deprivation and indicates that mitophagy was activated, although this finding requires definitive confirmation by flux measurement. Indeed, lysosomes can copurify with mitochondria, and autophagy could be increased independently of mitophagy, and fluorescent labeling of mitochondrial and autophagy proteins is an approach to measure mitophagy specifically (28). Similarly, the ubiquitin-proteasome pathway is highly regulated, and deubiquitination ezymes can be induced with insulin deprivation (9), and selective inhibitors help improve interpretation of immunoblotting. Autophagy is also a highly dynamic process, and rigorous investigation requires autophagy flux analysis using autophagy inhibitors (34). However, this was not feasible in the current analysis because using proteolytic inhibitors would impair the release of LMW fragments, thereby confounding our results. Instead, we were limited to using a representative panel of autophagy and proteasome markers that, in conjunction with the current tracer data in humans and historical data, indicate activation of protein degradation pathways. This is consistent with our previous report showing increased degradation of mitochondrial proteins with the STZ mouse model, which were identified as semitryptic peptides (45).
Previously, we used the semitryptic peptide analysis approach to determine muscle protein degradation at the tissue level in caloric restriction and diabetic mouse models (29, 45). This approach looks for in vivo cleaved nontryptic peptides in a sea of in vitro tryptic digest. On average, only ∼2% of the peptides in a tryptic digest would come from in vivo degradation. As such, this method can measure the in vivo degradation only at the whole sample level, and it is not sensitive enough to measure individual protein degradation. In contrast, the LMW approach begins by isolating and enriching for in vivo degraded peptide fragments. Hence, this method is more sensitive than the semitryptic approach and can reliably identify individual proteins and the extent to which they are being degraded.
There are key practical considerations for using the LMW approach. As we demonstrated, peptide fragments can be generated during sample preparation, and peptide fragments generated during sample preparation cannot be differentiated from endogenously degraded proteins. Hence, protease inhibitors must be included in homogenization buffers. The 10-kDa filter might exclude degraded peptide fragments from larger proteins because large proteins when degraded might produce peptides that are >10 kDa in size. Hence, the size of the filter must be chosen appropriately for the underlying biology and study design. We tested both 5- and 10-kDa filters and chose the latter due to increased yield (data not shown).
A further limitation is inherent for all current approaches (including the LMW method) that measure protein degradation; the intended signal (a degraded protein) is continually decreasing, and detecting a fragment from the protein means it has not yet been fully degraded. The LMW approach identifies proteins that have initiated degradation but are not yet fully degraded to their amino acid constituents. A fully degraded protein released as amino acids can be detected using isotopic tracer approaches, but not with the LMW method. However, amino acids do not represent any fingerprint of the degraded proteins. Amino acid tracer-based kinetic measurements can provide an accurate measurement of protein degradation in a tissue bed or at a whole body level. Hence, combining the LMW approach with isotope trace-based static and kinetic measures provides a comprehensive insight into protein degradation, including activation, overall kinetics, and identification of specific proteins.
The current study identified degradation of only a fraction of the total mitochondrial proteins. It is possible that small increases in protein degradation accumulate over time to alter protein content, possibly contributing to declines in specific pools like mitochondria with aging (22) or diabetes (4). Since only a limited number of proteins were identified, insulin deprivation per se may not wither away all mitochondrial proteins in a short time span. The current approach may not comprehensively detect all mitochondrial proteins that are degraded. The LMW approach identifies a relatively small portion of proteins compared with the entire protein pool. Although the LMW approach provides insight into individual protein degradation, additional methods for simultaneously measuring individual protein synthesis are necessary for net effects of individual protein turnover on protein expression.
In conclusion, we used a peptidomics approach to identify individual proteins that are degraded in vivo based on the release of LMW peptide fragments. This approach reproducibly detected peptides generated during insulin deprivation in T1D humans, with additional validation using STZ-treated mouse models. Insulin deprivation in human and mouse models leads to increased degradation of distinct skeletal muscle proteins, including mitochondrial proteins and those involved in proteome homeostasis. Assessing individual protein degradation using LMW peptide fragments provides important insight into protein turnover.
GRANTS
The publication of this article was made possible by Grant No. 5-R01-DK-041973 (K. S. Nair) and Clinical and Translational Science Award Grant No. UL1-TR-000135 from the National Center for Advancing Translational Sciences, a component of the National Institutes of Health (NIH), and U24-DK-100469, originating from the NIH Director's Common Fund. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of NIH. Additional funding was provided by Grant No. T32-D-K007352 (M. M. Robinson) and the Center for Individualized Medicine at the Mayo Clinic (SD).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.M.R. and K.S.N. conception and design of research; M.M.R. and H.K. performed experiments; M.M.R., S.D., H.R.B., and K.S.N. analyzed data; M.M.R., S.D., and K.S.N. interpreted results of experiments; M.M.R. and S.D. prepared figures; M.M.R. drafted manuscript; M.M.R., S.D., H.K., H.R.B., and K.S.N. edited and revised manuscript; M.M.R., S.D., H.K., H.R.B., and K.S.N. approved final version of manuscript.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge the skilled assistance of Dawn Morse, Dan Jakaitis, Katherine Klaus, Gabriel Marrero, Jill Schimke, and the Proteomics Core and the nursing staff of the Clinical Research Unit at the Mayo Clinic.
REFERENCES
- 1.Asmann YW, Stump CS, Short KR, Coenen-Schimke JM, Guo Z, Bigelow ML, Nair KS. Skeletal muscle mitochondrial functions, mitochondrial DNA copy numbers, and gene transcript profiles in type 2 diabetic and nondiabetic subjects at equal levels of low or high insulin and euglycemia. Diabetes 55: 3309–3319, 2006. [DOI] [PubMed] [Google Scholar]
- 2.Barazzoni R, Short KR, Asmann Y, Coenen-Schimke JM, Robinson MM, Nair KS. Insulin fails to enhance mTOR phosphorylation, mitochondrial protein synthesis, and ATP production in human skeletal muscle without amino acid replacement. Am J Physiol Endocrinol Metab 303: E1117–E1125, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barrett EJ, Revkin JH, Young LH, Zaret BL, Jacob R, Gelfand RA. An isotopic method for measurement of muscle protein synthesis and degradation in vivo. Biochem J 245: 223–228, 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Befroy DE, Petersen KF, Dufour S, Mason GF, de Graaf RA, Rothman DL, Shulman GI. Impaired mitochondrial substrate oxidation in muscle of insulin-resistant offspring of type 2 diabetic patients. Diabetes 56: 1376–1381, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Biolo G, Fleming RY, Maggi SP, Wolfe RR. Transmembrane transport and intracellular kinetics of amino acids in human skeletal muscle. Am J Physiol Endocrinol Metab 268: E75–E84, 1995. [DOI] [PubMed] [Google Scholar]
- 6.Bjørkøy G, Lamark T, Pankiv S, Øvervatn A, Brech A, Johansen T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol 452: 181–197, 2009. [DOI] [PubMed] [Google Scholar]
- 7.Charlton MR, Balagopal P, Nair KS. Skeletal muscle myosin heavy chain synthesis in type 1 diabetes. Diabetes 46: 1336–1340, 1997. [DOI] [PubMed] [Google Scholar]
- 8.Chow LS, Albright RC, Bigelow ML, Toffolo G, Cobelli C, Nair KS. Mechanism of insulin's anabolic effect on muscle: measurements of muscle protein synthesis and breakdown using aminoacyl-tRNA and other surrogate measures. Am J Physiol Endocrinol Metab 291: E729–E736, 2006. [DOI] [PubMed] [Google Scholar]
- 9.Combaret L, Adegoke OA, Bedard N, Baracos V, Attaix D, Wing SS. USP19 is a ubiquitin-specific protease regulated in rat skeletal muscle during catabolic states. Am J Physiol Endocrinol Metab 288: E693–E700, 2005. [DOI] [PubMed] [Google Scholar]
- 10.Cuervo AM, Dice JF. Age-related decline in chaperone-mediated autophagy. J Biol Chem 275: 31505–31513, 2000. [DOI] [PubMed] [Google Scholar]
- 11.Dasari S, Chambers MC, Codreanu SG, Liebler DC, Collins BC, Pennington SR, Gallagher WM, Tabb DL. Sequence tagging reveals unexpected modifications in toxicoproteomics. Chem Res Toxicol 24: 204–216, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dudek EJ, Lampi KJ, Lampi JA, Shang F, King J, Wang Y, Taylor A. Ubiquitin proteasome pathway-mediated degradation of proteins: effects due to site-specific substrate deamidation. Invest Ophthalmol Vis Sci 51: 4164–4173, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eshima H, Poole DC, Kano Y. In vivo Ca2+ buffering capacity and microvascular oxygen pressures following muscle contractions in diabetic rat skeletal muscles: fiber-type specific effects. Am J Physiol Regul Integr Comp Physiol 309: R128–R137, 2015. [DOI] [PubMed] [Google Scholar]
- 14.Finoulst I, Pinkse M, Van Dongen W, Verhaert P. Sample preparation techniques for the untargeted LC-MS-based discovery of peptides in complex biological matrices. J Biomed Biotechnol 2011: 245291, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fishovitz J, Li M, Frase H, Hudak J, Craig S, Ko K, Berdis AJ, Suzuki CK, Lee I. Active-site-directed chemical tools for profiling mitochondrial Lon protease. ACS Chem Biol 6: 781–788, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gelfand RA, Barrett EJ. Effect of physiologic hyperinsulinemia on skeletal muscle protein synthesis and breakdown in man. J Clin Invest 80: 1–6, 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Geyelin HR, Harrop G, Murray MF, Corwin E. The use of insulin in juvenile diabetes. J Metab Res 767–791, 1922. [Google Scholar]
- 18.Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature 426: 895–899, 2003. [DOI] [PubMed] [Google Scholar]
- 19.Goldberg AL, Tischler M, DeMartino G, Griffin G. Hormonal regulation of protein degradation and synthesis in skeletal muscle. Fed Proc 39: 31–36, 1980. [PubMed] [Google Scholar]
- 20.Herasse M, Ono Y, Fougerousse F, Kimura E, Stockholm D, Beley C, Montarras D, Pinset C, Sorimachi H, Suzuki K, Beckmann JS, Richard I. Expression and functional characteristics of calpain 3 isoforms generated through tissue-specific transcriptional and posttranscriptional events. Mol Cell Biol 19: 4047–4055, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Holm L, Kjaer M. Measuring protein breakdown rate in individual proteins in vivo. Curr Opin Clin Nutr Metab Care 13: 526–531, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Irving BA, Lanza IR, Henderson GC, Rao RR, Spiegelman BM, Nair KS. Combined training enhances skeletal muscle mitochondrial oxidative capacity independent of age. J Clin Endocrinol Metab 100: 1654–1663, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jaleel A, Short KR, Asmann YW, Klaus K, Morse D, Ford GC, Nair KS. In vivo measurement of synthesis rate of individual skeletal muscle mitochondrial proteins. Am J Physiol Endocrinol Metab 295: E1255–E1268, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson ML, Irving BA, Lanza IR, Vendelbo MH, Konopka AR, Robinson MM, Henderson GC, Klaus KA, Morse DM, Heppelmann C, Bergen HR 3rd, Dasari S, Schimke JM, Jakaitis DR, Nair KS. Differential Effect of Endurance Training on Mitochondrial Protein Damage, Degradation, and Acetylation in the Context of Aging. J Gerontol A Biol Sci Med Sci 70: 1386–1393, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karakelides H, Asmann YW, Bigelow ML, Short KR, Dhatariya K, Coenen-Schimke J, Kahl J, Mukhopadhyay D, Nair KS. Effect of insulin deprivation on muscle mitochondrial ATP production and gene transcript levels in type 1 diabetic subjects. Diabetes 56: 2683–2689, 2007. [DOI] [PubMed] [Google Scholar]
- 26.Kay R, Barton C, Ratcliffe L, Matharoo-Ball B, Brown P, Roberts J, Teale P, Creaser C. Enrichment of low molecular weight serum proteins using acetonitrile precipitation for mass spectrometry based proteomic analysis. Rapid Commun Mass Spectrom 22: 3255–3260, 2008. [DOI] [PubMed] [Google Scholar]
- 27.Kimball SR, Vary TC, Jefferson LS. Regulation of protein synthesis by insulin. Annu Rev Physiol 56: 321–348, 1994. [DOI] [PubMed] [Google Scholar]
- 28.Kusminski CM, Chen S, Ye R, Sun K, Wang QA, Spurgin SB, Sanders PE, Brozinick JT, Li WH, Unger RH, Scherer PE. MitoNEET-Parkin Effects in Pancreatic α- and β-Cells, Cellular Survival, and Intrainsular Cross Talk. Diabetes 65: 1534–1555, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lanza IR, Zabielski P, Klaus KA, Morse DM, Heppelmann CJ, Bergen HR 3rd, Dasari S, Walrand S, Short KR, Johnson ML, Robinson MM, Schimke JM, Jakaitis DR, Asmann YW, Sun Z, Nair KS. Chronic caloric restriction preserves mitochondrial function in senescence without increasing mitochondrial biogenesis. Cell Metab 16: 777–788, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li M, Gray W, Zhang H, Chung CH, Billheimer D, Yarbrough WG, Liebler DC, Shyr Y, Slebos RJ. Comparative shotgun proteomics using spectral count data and quasi-likelihood modeling. J Proteome Res 9: 4295–4305, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ljungqvist OH, Persson M, Ford GC, Nair KS. Functional heterogeneity of leucine pools in human skeletal muscle. Am J Physiol Endocrinol Metab 273: E564–E570, 1997. [DOI] [PubMed] [Google Scholar]
- 32.Ma ZQ, Dasari S, Chambers MC, Litton MD, Sobecki SM, Zimmerman LJ, Halvey PJ, Schilling B, Drake PM, Gibson BW, Tabb DL. IDPicker 2.0: Improved protein assembly with high discrimination peptide identification filtering. J Proteome Res 8: 3872–3881, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 15: 1101–1111, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy 3: 542–545, 2007. [DOI] [PubMed] [Google Scholar]
- 35.Nair KS, Ford GC, Ekberg K, Fernqvist-Forbes E, Wahren J. Protein dynamics in whole body and in splanchnic and leg tissues in type I diabetic patients. J Clin Invest 95: 2926–2937, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Robinson MM, Soop M, Sohn TS, Morse DM, Schimke JM, Klaus KA, Nair KS. High insulin combined with essential amino acids stimulates skeletal muscle mitochondrial protein synthesis while decreasing insulin sensitivity in healthy humans. J Clin Endocrinol Metab 99: E2574–E2583, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rooyackers OE, Adey DB, Ades PA, Nair KS. Effect of age on in vivo rates of mitochondrial protein synthesis in human skeletal muscle. Proc Natl Acad Sci USA 93: 15364–15369, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Singh R, Cuervo AM. Autophagy in the cellular energetic balance. Cell Metab 13: 495–504, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sorimachi H, Kimura S, Kinbara K, Kazama J, Takahashi M, Yajima H, Ishiura S, Sasagawa N, Nonaka I, Sugita H, Maruyama K, Suzuki K. Structure and physiological functions of ubiquitous and tissue-specific calpain species. Muscle-specific calpain, p94, interacts with connectin/titin. Adv Biophys 33: 101–122, 1996. [DOI] [PubMed] [Google Scholar]
- 40.Stump CS, Short KR, Bigelow ML, Schimke JM, Nair KS. Effect of insulin on human skeletal muscle mitochondrial ATP production, protein synthesis, and mRNA transcripts. Proc Natl Acad Sci USA 100: 7996–8001, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tabb DL, Fernando CG, Chambers MC. MyriMatch: highly accurate tandem mass spectral peptide identification by multivariate hypergeometric analysis. J Proteome Res 6: 654–661, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vainshtein A, Desjardins EM, Armani A, Sandri M, Hood DA. PGC-1α modulates denervation-induced mitophagy in skeletal muscle. Skelet Muscle 5: 9, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Witt CC, Burkart C, Labeit D, McNabb M, Wu Y, Granzier H, Labeit S. Nebulin regulates thin filament length, contractility, and Z-disk structure in vivo. EMBO J 25: 3843–3855, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zabielski P, Ford GC, Persson XM, Jaleel A, Dewey JD, Nair KS. Comparison of different mass spectrometry techniques in the measurement of l-[ring-(13)C6]phenylalanine incorporation into mixed muscle proteins. J Mass Spectrom 48: 269–275, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zabielski P, Lanza IR, Gopala S, Heppelmann CJ, Bergen HR 3rd, Dasari S, Nair KS. Altered Skeletal Muscle Mitochondrial Proteome As the Basis of Disruption of Mitochondrial Function in Diabetic Mice. Diabetes 65: 561–573, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang B, Chambers MC, Tabb DL. Proteomic parsimony through bipartite graph analysis improves accuracy and transparency. J Proteome Res 6: 3549–3557, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.