

Abstract
Cryptosporidium is recognized as one of the main causes of childhood diarrhea worldwide. However, the current treatment for cryptosporidiosis is suboptimal. Calcium flux is essential for entry in apicomplexan parasites. Calcium-dependent protein kinases (CDPKs) are distinct from protein kinases of mammals, and the CDPK1 of the apicomplexan Cryptosporidium lack side chains that typically block a hydrophobic pocket in protein kinases. We exploited this to develop bumped kinase inhibitors (BKIs) that selectively target CDPK1. We have shown that several BKIs of Cryptosporidium CDPK1 potently reduce enzymatic activity and decrease parasite numbers when tested in vitro. In the present work, we studied the anticryptosporidial activity of BKI-1517, a novel BKI. The half maximal effective concentration for Cryptosporidium parvum in HCT-8 cells was determined to be approximately 50 nM. Silencing experiments of CDPK1 suggest that BKI-1517 acts on CDPK1 as its primary target. In a mouse model of chronic infection, 5 of 6 SCID/beige mice (83.3%) were cured after treatment with a single daily dose of 120 mg/kg BKI-1517. No side effects were observed. These data support advancing BKI-1517 as a lead compound for drug development for cryptosporidiosis.
Keywords: BKI, kinase inhibitors, siRNA, CDPK, CDPK1, cryptosporidium, cryptosporidiosis
Cryptosporidiosis, caused by protozoan parasites of the genus Cryptosporidium, is often considered to be a major problem only in immune compromised patients, such as those with AIDS [1]. However in a recent multicenter study from Africa and South Asia, Cryptosporidium was second to rotavirus as a cause of childhood diarrhea morbidity and mortality [2]. Thus, it has emerged as a major contributor to childhood malnutrition, diarrheal disease, and death worldwide [2–5]. Despite its emerging public health importance, there is no effective vaccine to prevent disease and only limited options for treatment. The only Food and Drug Administration–approved therapy for cryptosporidiosis, nitazoxanide, was shown in a randomized trial to lead to 2 fewer days of diarrhea from cryptosporidiosis in treated patients, compared with untreated patients [6], but it does not effectively treat cryptosporidiosis in patients with AIDS [7]. Studies of malnourished children revealed that nitazoxanide yielded a response rate of only about 30%, compared with placebo [7]. Clearly, the development of more-effective drugs is urgently needed for therapy of cryptosporidiosis.
Calcium-dependent protein kinases (CDPKs) are being used as targets for drug development against apicomplexan parasites [8, 9]. The structures of apicomplexan type 1 CDPKs (CDPK1s) are distinct from those of mammalian enzymes in that they lack amino acid side chains that block a hydrophobic pocket near the adenosine triphosphate binding site [8, 10]. This difference has been exploited to design specific inhibitors for parasite CDPK1s, including those of Cryptosporidium, called bumped kinase inhibitors (BKIs), that do not interact with the host enzymes [11]. In vitro experiments confirmed potent inhibition of novel pyrazole-4-carboxamide compounds against Cryptosporidium CDPK1 [11–13]. In this work, we characterized the anti-Cryptosporidium activity of a novel BKI-1517 (5-amino-1-tert-butyl-3-[7-ethoxyquinolin-3-yl]-1H-pyrazole-4-carboxamide; Figure 1A) in vitro and also in a mouse model of chronic infection.
Figure 1.

In vitro anti-Cryptosporidium activity of bumped kinase inhibitor 1517 (BKI-1517). A, Chemical structure of BKI-1517. B, Anticryptosporidial activity of BKI-1517 in vitro. A range of concentrations was used to quantify the inhibitory dose of BKI-1517 needed to reduce the quantity of Cryptosporidium parvum in HCT-8 cells. The results are presented as mean values (±SD) of 3 independent experiments.
METHODS
Invasion Model and Drug Activity Assays
HCT-8 (ATCC) cells suspended in 500 µL of complete medium (Roswell Park Memorial Institute [RPMI] medium with 10% fetal bovine serum and 1% antibiotic-antimycotic solution containing penicillin/streptomycin/amphotericin B [Life Technologies, Grand Island, New York]) were seeded in 24-well cell culture plates and incubated at 37°C in 5% CO2 overnight as described before [14]. For infection experiments, we used sporozoites obtained from Cryptosporidium parvum oocysts (Iowa strain, maintained at the University of Arizona). Sporozoites were prepared as follows: oocysts were centrifuged at 500 × g for 5 minutes, and the pellet was washed 3 times with 250 µL of phosphate-buffered saline (PBS). After washing, the pellet was resuspended in 50 µL of acidic water (pH 2–3) and incubated for 10 minutes on ice. Excystation medium (complete medium supplemented with 0.8% taurocholate) was then added to the sample, which was then incubated for 1 hour at 37°C to induce sporozoite excystation. The sporozoites were quantified by microscopy and then used for infection experiments.
To test for anticryptosporidial activity of BKI on sporozoites, a stock solution of BKI-1517 was diluted with infection medium (RPMI medium plus 1% antibiotic antimycotic solution) at final concentrations of 0.01, 0.05, 0.1, 1, and 10 µM. Then, 500 µL of infection medium containing the drug was mixed with 5 × 105 sporozoites for 15 minutes at 37°C (5% CO2). After incubation, the treated sporozoites were used to infect HCT-8 cells for 2 hours. Controls included infected and uninfected HCT-8 cells treated only with the infection medium. After infection, the infection medium (containing dead and noninfective sporozoites) was removed, and 500 µL of fresh infection medium was added. Plates were incubated for 18 hours at 37°C with 5% CO2. After incubation, medium was removed, and cells were washed by adding 500 µL of phosphate-buffered saline (PBS) and then gently removing the supernatant by pipetting. Attached cells were lysed and harvested, and 350 µL of Buffer RLT (from the RNeasy Plus kit, Qiagen. Valencia, California) with β-mercaptoethanol was added directly to each well. Samples were transferred to 1-mL Eppendorf tubes and stored frozen (−20°C) until subsequent RNA extraction.
Quantitation of Cryptosporidium in HCT-8 Cells by Quantitative Reverse-Transcription Polymerase Chain Reaction (qRT-PCR) Analysis
Quantitation of Cryptosporidium was performed by qRT-PCR as previously described [12]. Briefly, RNA was isolated using a commercial kit (RNeasy Plus kit). The final RNA concentration was determined by spectrophotometry with the Nanodrop 1000 (Thermo Scientific, Wilmington, Delaware). The parasite numbers were monitored by RT-PCR using the Applied Biosystems 7500 Real-Time PCR System (Life Technologies). For all of the reactions, we used the 1-step RT-PCR Super Script III with approximately 100 ng of each sample and specific primers for Cryptosporidium parvum Cp-F (CAA TCA GCA ACC AAG CTC AA) and Cp-R (TTG TTG AGC AGC AGG TTC AG). Quantitation was normalized to host 18s RNA (5′-CCG ATA ACG AAC GAG ACT CTG G-3′ [forward] and 5′-TAG GGT AGG CAC ACG CTG AGC C-3′ [reverse]). The conditions for qRT-PCR analysis were as follows: 1 cycle of 5 minutes at 95°C, 1 cycle of 15 seconds at 95°C, and 40 cycles of 1 minute at 65°C. The specificity of the primers was confirmed by melting-curve analysis. For parasite quantitation, a standard curve with serial dilutions of RNA from a known number of parasites was included in each reaction plate. Total numbers of parasites were calculated with the AB7500 software SDS v1.4.
Mouse Infection and Treatment with BKI-1517
Five-week-old SCID/beige mice (Jackson Laboratory, Sacramento, California) were infected by gavage with 1 × 106 C. parvum oocysts (Iowa strain) contained in 100 µL of PBS as described before [14]. Four days after infection, mice were treated with BKI-1517 suspended in 100 µL of vehicle (7% Tween 80, 3% ethanol, and 90% normal saline) or a vehicle control (placebo) by oral gavage daily for 5 days. Dosages tested included 60 mg/kg once daily (for 6 mice), 120 mg/kg once daily (for 6 mice), and 20 mg/kg at 7:30 am and 40 mg/kg at 4:30 pm (for 7 mice). Approximately 25 mg of stool was collected biweekly up to day 28 for quantification by qPCR, and all samples were stored at −20°C until DNA extraction. Experiments were performed in accordance with the Institutional Animal Care and Use Committee of the University of Texas Medical Branch.
DNA Extraction From Stools and qPCR Assays
DNA was extracted from 25 mg (approximately 3 pellets) of stool from each mouse. Extraction and purification of DNA samples was performed using the QIAamp Fast DNA Stool Mini Kit (Qiagen). The concentration of DNA in samples was determined by spectrophotometry. The samples were stored at −20°C until subsequent analysis. The parasite burden was determined by qPCR (Applied Biosystems 7500 Real-Time PCR System), using the iTaq Universal SYBR Green Supermix Kit (Bio-rad, Hercules, California) with primers for C. parvum as described above. The qPCR assay was conducted under the following conditions: 1 cycle of 20 minutes at 55°C, 1 cycle of 5 minutes at 95°C and 15 seconds at 95°C, 40 cycles of 1 minute at 60°C. An additional dissociation stage was added at the end of the reaction to test the specificity of the amplicons via dissociation curve analysis. A standard curve was generated from serial dilutions of DNA extracted from a known number of parasites spiked in mouse stool. Total numbers of parasites were calculated with AB7500 software SDS v1.4.
Silencing CDPK1 Experiments
We silenced Cryptosporidium CDPK1 (GenBank accession number XM_001388059.1) and GP900 (GenBank accession number AF068065) by RNA interference, using the method described before [15]. Briefly, for assembling silencer complexes (siCDPK1 and siGP900), we used 250 ng of hAgo2 and 1 µM of CDPK1 single-stranded RNA (5′Phos-CAC UUC CUC UCU CUC CUC CC dTdT-3′) or GP900 single-stranded RNA (5′Phos-CUG AAG GGA GAG AUG GGA UU dTdT-3′; control) suspended in assembly buffer (30 mM HEPES [pH 7.4], 150 mM KOAc, and 2 mM MgCl2). We incubated the sample (20 µL) for 90 minutes at room temperature. Next, 35 µL of protein transfection reagent Pro-Jet (Thermo-Fisher, Rockford, IL) was added to the hAgo2/ssRNA-CDPK1 or hAgo2/ssRNA-GP900 complex (siCDPK1 and siGP900). The samples were incubated for 5 minutes at room temperature to allow encapsulation. For transfection, encapsulated complexes were added to oocysts and incubated for 16 hours at 4°C. For infection experiments, we induced excystation of the transfected parasites as described above. The viability of sporozoites was confirmed by microscopy as described before [15]. For silencing experiments, sporozoites were incubated as described before with only transfection reagent, 1 µM of silencer complex siCDPK, or BKI-1517 (0.1 µM) alone; for BKI treatment, sporozoites were incubated as described before with BKI-1517 (0.1 µM) plus siCDPK, BKI-1294 (0.1 µM) alone, or BKI-1294 (0.1 µM) plus siCDPK. Treated parasites were used to infect HCT-8 cells as described before. Finally, RNA was extracted and numbers of parasites were calculated by RT-PCR analysis as described before.
RESULTS
BKI-1517 Reduces Infection in HCT-8 Cells
The anticryptosporidial activity of BKI-1517 was tested in an invasion model. We observed a reduction in the amount of parasites by 90% on infected HCT-8 cells when BKI-1517 was added to sporozoites at concentrations of 10 µM, in these experiments we observed a half-maximal effective concentration between 10–50 nM in experiments performed in triplicate (Figure 1B). Further, no anticryptosporidial activity was observed on parasites treated with siGP900 (Supplementary Figure 1), and no cellular damage or signals of cytotoxicity were observed in HCT-8 cells, as assessed by microscopy.
BKI-1517 Reduced C. parvum Infection in a Chronic Mouse Model
SCID/beige mice were infected with 1 × 106 C. parvum oocysts and then treated with BKI-1517. Cure of infection was determined by qPCR analysis of stool samples. In an initial experiment, we compared a fixed concentration of compound given once daily (60 mg/kg) or twice daily (20 mg/kg during the day and 40 mg/kg during the night) for 5 days. It was thought that twice-daily administration would be optimal owing to the short serum half-life of BKI-1517 in mice. Both treatments were effective in reducing the prevalence of Cryptosporidium infection during the acute phase at day 8, as assessed by quantifying the number of parasites shed per 25 mg of stool (Figure 2A). However, 5 of 7 mice treated with the split dose experienced relapse at day 28. By contrast, 4 of 6 mice treated with the once-daily therapy were cured, and only 2 mice shed parasites in stools at day 28. This demonstrated that the twice-daily split dose was inferior to the same dose given once daily in completely clearing C. parvum from the mice.
Figure 2.
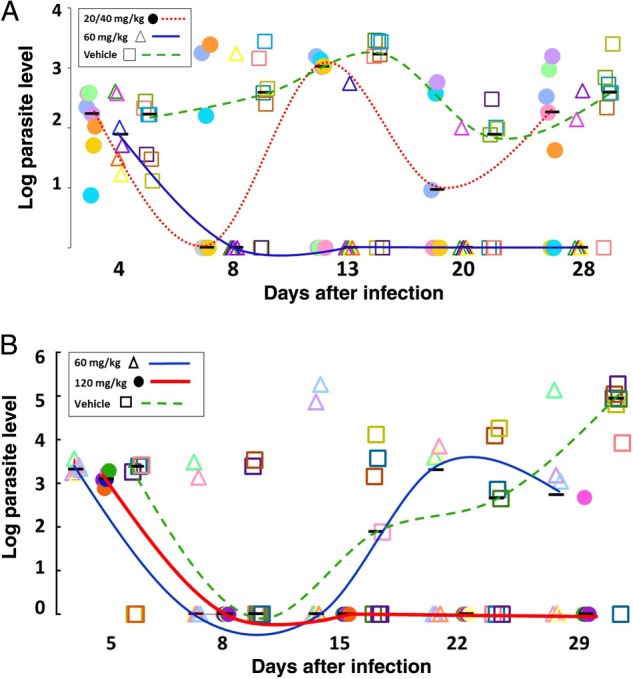
The effects of bumped kinase inhibitor 1517 (BKI-1517) in SCID/beige mice infected with Cryptosporidium parvum. The number of parasites shed per 25 mg of stool in experimental groups was quantified using real-time polymerase chain reaction analysis, normalized to a standard curve. Samples were quantified at different days after infection, with the initial time points indicating oocyst levels before treatment. The median number of oocysts shed by each experimental group are indicated with solid black bars. Curves illustrate infection levels. A, Mice were treated with 60 mg/kg BKI-1517 once daily (solid triangles; n = 6), with 20 mg/kg morning and 40 mg/kg evening daily (solid circles; n = 7), or with vehicle control (hollow squares; n = 7). B, Mice were treated with BKI-1517 at doses of 60 mg/kg once daily (solid circles; n = 6), 120 mg/kg once daily (solid triangles; n = 6), or vehicle control (hollow squares; n = 6).
In a second experiment, we tested the same total daily dose (60 mg/kg) and a higher dose (120 mg/kg) of BKI-1517 given once-daily for 5 days (Figure 2B). The results showed that 50% of mice were completely cured by day 29 when treated with 60 mg/kg, but when we treated the mice with 120 mg/kg we did not detect the infection in any of the 6 mice at day 22 and only 1 of 6 mice shed parasites in stool by day 28 (Figure 2B). During this experiment, both doses (60 and 120 mg/kg) were well tolerated by all the mice, and we did not observe adverse reactions due the treatment.
CDPK1 Silencing and Antiparasitic Activity of BKI-1517
BKI-1517 was designed as an inhibitor of Cryptosporidium CDPK1. To study whether CDPK1 inhibition was responsible for the observed antiparasitic activity, we knocked down CDPK1, using RNA interference (Figure 3A and Supplementary Figure 2A and 2B), and tested for additional activity of BKI-1517 (Figure 3B). When we performed RNA interference with the siCDPK1 complex, the messenger RNA (mRNA) expression of CDPK1 in transfected parasites was reduced by about 90% (Figure 3A), but we did not observe reduction when the unrelated siGp900 complex was used (Figure 3A). This result confirmed that CDPK1 was silenced specifically in parasites treated with siCDPK1. We confirmed this reduction at the protein level by Western blot (Supplementary Figure 3A and 3B), although the reduction was only a little more than 50% at the protein level.
Figure 3.

Effects of bumped kinase inhibitor 1517 (BKI-1517) on CDPK1 inhibition. A, CDPK1 expression in siCDPK- and siGP900-transfected parasites and wild type parasites treated only with transfection reagent (WT). B, Cryptosporidium oocyst containing wild type parasites were treated with only transfection reagent (WT), 1 µM of silencer complex siCDPK to knock down CDPK1, BKI-1517 (0.1 µM) alone, BKI-1517 plus siCDPK, BKI-1294 (0.1 µM) alone, or BKI-1294 plus siCDPK. Numbers of parasites were evaluated (gray bars). The results are the mean value (±SD) of 3 independent experiments evaluated by reverse-transcription polymerase chain reaction analysis. Asterisks denote statistically significant differences.
We then tested the silenced parasites in our infection model. When CDPK1 was knocked down, there was about a 50% decrease in the number of parasites recovered after infection of HCT-8 cells (Figure 3B). In contrast, we did not observe parasite reduction when we used the unrelated siGp900 complex. These results suggest that CDPK1 is involved in the mechanism of invasion or of growth. Therefore, we tested BKI-1517 with parasites while using siCDPK1 to modulate CDPK1 levels. We observed the same phenotype obtained with silenced parasites, then a approximately 50% of reduction was observed in treated parasites. However, when parasites were treated with BKI-1517 after CDPK1 expression was knocked down, an additional decrease (approximately 23%) was noted in parasite load (Figure 3B).
To confirm the synergistic effect of BKI-1517, we attempted to use paromomycin alone and in combination with the BKI siRNA. However, paromomycin showed cytotoxic effects in HCT-8 cells and also interfered with global RNA expression (data not shown); therefore, it was not considered useful as a control for silencing experiments. Then, BKI-1294, a previously tested compound that has anticryptosporidial activity and is thought to target CDPK1 [14], was used to confirm CDPK1 targeting. Similar to BKI-1517, BKI-1294 alone reduced growth to about 9%, whereas BKI-1294 in combination with siCDPK decreased growth to about 5%. These data suggest that CDPK1 is the main target of BKIs because the growth reduction of the combination decreased 2-fold each time. This fits the observation that siCDPK reduced the growth.
DISCUSSION
In this study, we demonstrated the potent anti-Cryptosporidium activity of BKI-1517, which reduced the prevalence of infection at nanomolar concentrations in vitro. Previous compounds have had a pyrazolo-pyrimidine scaffold, but BKI-1517 has a completely different scaffold, the aminopyrazole-carboxamide scaffold. When tested using a 5-week-old SCID/beige mouse model, BKI-1517 not only suppressed acute infection, it also prevented relapse. This murine model appears to better mimic the persistent and relapsing infections commonly reported for immunocompromised patients. Thus, we have confirmed the anticryptosporidial activity of BKIs in the mouse model, using a new chemical scaffold [14].
CDPK1 has been used as a target for drug development in several apicomplexans, including Cryptosporidium [11, 13], and in previous studies we confirmed the anticryptosporidial activity of several BKIs in vitro [12, 14]. A recent publication by Kuhlenschmidt et al reports the lack of correlation between enzymatic inhibition of CDPK1 and antiparasitic activity against C. parvum in vitro [16]. Nevertheless, most of our BKIs with potent CDPK1 inhibition showed activity when tested in our infection model. Indeed, we noted poor correlation between enzymatic inhibition and in vitro activity in very few compounds. In addition to off-target activity, there are numerous potential explanations for this incomplete correlation, including issues of solubility, protein binding, and inability to cross the parasite membrane. One concern is that the BKI may be acting through a different target. In a recent publication, however, we demonstrated that the silencing of CDPK1 caused a reduction in the parasite burden following infection in vitro, which confirms the potential for CDPK1 to serve as a target for drug development [15].
In the current study, we demonstrated that reducing CDPK1 levels by 50% also reduced the infection prevalence by 50%. Therefore, we hypothesized that if BKI-1517 is targeting CDPK1, then a similar phenotype should be observed for parasites treated with BKI. Our investigation revealed an additional 50% reduction in infection prevalence when Cryptosporidium organisms were treated with both BKI and siCDPK1. The silencing experiments showed that siCDPK reduced mRNA expression of CDPK1 by 90% (Figure 3A). However, the infection prevalence was reduced only by half, with the partial reduction likely associated with the stability of preformed or residual CDPK1 (Supplementary Figure 2A).
We hypothesized that the combination of siCDPK with BKI would have the synergistic effect of reducing the infection prevalence. Thus, when we used siCDPK and BKI, the infection prevalence was reduced by approximately 75% (Figure 3B). If BKIs are targeting CDPK1, then similar effects should be observed with another BKI, with a different scaffold, that also effectively inhibits CDPK1. Therefore, we tested BKI-1294, which has a pyrazolo-pyrimidine scaffold, for anticryptosporidial activity. Similar to BKI-1517, BKI-1294 alone reduced growth by about 91%, whereas in combination with siCDPK, BKI-1294 decreased growth by about 95%. These data appear to confirm that the target is CDPK1, because growth is reduced 2-fold by each BKI combined with siCDPK if we consider that siCDPK alone reduced the growth by just more than 50% (Figure 3B).
We confirmed the anticryptosporidial activity of BKI-1517 in an immunocompromised mouse model. This particular model is a stringent test of antiparasitic activity, since it allows for parasites that are not killed to proliferate and for oocyst shedding to reemerge after drug withdrawal. Models differ with regard to the key pharmacologic measures for drug efficacy. Based on a relatively short serum half-life of BKI-1517, we hypothesized that treatment would require multiple daily doses. However, when we compared 60 mg/kg given once daily or twice daily, the single daily dose was more effective than the split-dose approach. This suggests that high peak plasma or enteral levels may be more important than prolonged exposure. The single dose of 60 mg/kg given once daily reduced the prevalence of infection, especially during the acute phase, but we observed relapse in several mice after the end of treatment. To optimize the treatment, we tested a higher dose, 120 mg/kg, given as a single daily dose. The trend observed in experiment 2, in which only 1 of 6 mice that received 120 mg/kg experienced relapse, compared with 3 of 6 mice that received 60 mg/kg, suggests that the higher drug dose was needed to reduce the chance of relapse. Our pharmacokinetics studies of once-daily treated mice (Supplementary Table 1) indicated that there were not proportional increases in BKI-1517 plasma levels at the peak of exposure among mice that received 60 mg/kg, compared with those that received 120 mg/kg. The observation that increasing the dose does not lead to more plasma exposure suggests that the compound is not well absorbed at the higher dose and that, with a higher dose, the compound may be better delivered to the ileum and colon, where the infection predominates. This may facilitate contact with the parasite and yield a trend toward less frequent relapse seen with the higher dose. Importantly, no adverse effects were noted with BKI-1517 in mice, even with these high plasma exposures.
In summary, in this work we demonstrated that treatment with 120 mg/kg BKI-1517 for 5 days eliminated infection in 5 of 6 mice (83.3%). In extrapolating the effective dose from rodents to humans, allometric scaling of the dose will be used. We anticipate that the dose needed to treat humans will be much lower than 60–120 mg/kg. Thus, BKI-1517 is an important lead compound for drug development that should be further explored as a treatment option for chronic Cryptosporidium infections.
Supplementary Data
Supplementary materials are available at http://jid.oxfordjournals.org. Consisting of data provided by the author to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the author, so questions or comments should be addressed to the author.
Notes
Acknowledgments. We thank Ryan Choi and Lynn Barrett (University of Washington), for their experimental and administrative support.
Financial support. This work was supported by the Institute for Translational Sciences at the University of Texas Medical Branch at Galveston; the National Center for Advancing Translational Sciences, National Institutes of Health (Clinical and Translational Science Award UL1TR000071, Eunice Kennedy Shriver National Institute of Child Health and Human Development grant R01HD080670, and National Institute of Allergy and Infectious Diseases grants R01AI111341, R01AI089441, and 1R21AI12627501); the US Department of Agriculture (grant 2014-06183); and the Sealy Center for Vaccine Development, University of Texas Medical Branch (fellowship to S. N.).
Potential conflicts of interest. W. C. V. V. discloses that he is the president of ParaTheraTech, a company involved in developing BKIs for use in animal health. However, W. C. V. V. did not perform the experiments or analyze the data for the present publication. All other authors report no potential conflicts. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Sparks H, Nair G, Castellanos-Gonzalez A, White AC Jr. Treatment of Cryptosporidium: what we know, gaps, and the way forward. Curr Trop Med Rep 2015; 2:181–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kotloff KL, Nataro JP, Blackwelder WC et al. . Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case-control study. Lancet 2013; 382:209–22. [DOI] [PubMed] [Google Scholar]
- 3.Shirley DA, Moonah SN, Kotloff KL. Burden of disease from cryptosporidiosis. Curr Opin Infect Dis 2012; 25:555–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mondal D, Haque R, Sack RB, Kirkpatrick BD, Petri WA Jr. Attribution of malnutrition to cause-specific diarrheal illness: evidence from a prospective study of preschool children in Mirpur, Dhaka, Bangladesh. Am J Trop Med Hyg 2009; 80:824–6. [PMC free article] [PubMed] [Google Scholar]
- 5.Guerrant RL, DeBoer MD, Moore SR, Scharf RJ, Lima AA. The impoverished gut--a triple burden of diarrhoea, stunting and chronic disease. Nat Rev Gastroenterol Hepatol 2013; 10:220–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rossignol JF, Ayoub A, Ayers MS. Treatment of diarrhea caused by Cryptosporidium parvum: a prospective randomized, double-blind, placebo-controlled study of Nitazoxanide. J Infect Dis 2001; 184:103–6. [DOI] [PubMed] [Google Scholar]
- 7.White AC., Jr Cryptosporidium species. In: Mandell GL, Bennett JE, Dolin R, eds. Principles and practice of infectious diseases. 7th ed Philadelphia, PA: Elsevier, 2009. [Google Scholar]
- 8.Keyloun KR, Reid MC, Choi R et al. . The gatekeeper residue and beyond: homologous calcium-dependent protein kinases as drug development targets for veterinarian Apicomplexa parasites. Parasitology 2014; 141:1499–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cavagnino A, Rossi F, Rizzi M. The potent antiplasmodial calmodulin-antagonist trifluoperazine inhibits plasmodium falciparum calcium-dependent protein kinase 4. Protein Pept Lett 2011; 18:1273–9. [DOI] [PubMed] [Google Scholar]
- 10.Ojo KK, Larson ET, Keyloun KR et al. . Toxoplasma gondii calcium-dependent protein kinase 1 is a target for selective kinase inhibitors. Nat Struct Mol Biol 2010; 17:602–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Z, Ojo KK, Vidadala R et al. . Potent and selective inhibitors of CDPK1 from and based on a 5-aminopyrazole-4-carboxamide scaffold. ACS Med Chem Lett 2014; 5:40–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murphy RC, Ojo KK, Larson ET et al. . Discovery of potent and selective inhibitors of calcium-dependent protein kinase 1 (CDPK1) from C. parvum and T. gondii. ACS Med Chem Lett 2010; 1:331–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Z, Ojo KK, Johnson SM et al. . Benzoylbenzimidazole-based selective inhibitors targeting Cryptosporidium parvum and Toxoplasma gondii calcium-dependent protein kinase-1. Bioorg Med Chem Lett 2012; 22:5264–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Castellanos-Gonzalez A, White AC Jr, Ojo KK et al. . A novel calcium dependent protein kinase inhibitor as a lead compound for treating cryptosporidiosis. J Infect Dis 2013; 208:1342–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Castellanos-Gonzalez A, Perry N, Nava S, White AC Jr. Preassembled single-stranded RNA-argonaute complexes: a novel method to silence genes in Cryptosporidium. J Infect Dis 2016; 213:1307–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuhlenschmidt TB, Rutaganira FU, Long S et al. . Inhibition of calcium-dependent protein kinase 1 (CDPK1) In vitro by pyrazolopyrimidine derivatives does not correlate with sensitivity of Cryptosporidium parvum growth in cell culture. Antimicrob Agents Chemother 2015; 60:570–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.